
Abstract
Background
Over 400 million people worldwide are living with a rare disease. Next Generation Sequencing (NGS) identifies potential disease causative genetic variants. However, many are identified as variants of uncertain significance (VUS) and require functional laboratory validation to determine pathogenicity, and this creates major diagnostic delays.
Methods
In this study we test a rapid genetic variant assessment pipeline using CRISPR homology directed repair to introduce single nucleotide variants into inducible pluripotent stem cells (iPSCs), followed by neuronal disease modelling, and functional genomics on amplicon and RNA sequencing, to determine cellular changes to support patient diagnosis and identify disease mechanism.
Results
As proof-of-principle, we investigated an EHMT1 (Euchromatin histone methyltransferase 1; EHMT1 c.3430C > T; p.Gln1144*) genetic variant pathogenic for Kleefstra syndrome and determined changes in gene expression during neuronal progenitor cell differentiation. This pipeline rapidly identified Kleefstra syndrome in genetic variant cells compared to healthy cells, and revealed novel findings potentially implicating the key transcription factors REST and SP1 in disease pathogenesis.
Conclusion
The study pipeline is a rapid, robust method for genetic variant assessment that will support rare diseases patient diagnosis. The results also provide valuable information on genome wide perturbations key to disease mechanism that can be targeted for drug treatments.
Similar content being viewed by others


Background
Rare diseases are collectively prevalent, with over 80% having genetic basis. Currently, Next Generation Sequencing (NGS) has provided a rapid, and ever more affordable tool for the identification of genetic variants in rare disease patients. However, whilst NGS leads to a diagnosis in up to 50% of patients, a large proportion of identified genetic variants are novel and therefore classified as variants of uncertain significance (VUS) meaning the patient cannot receive a diagnosis [1]. These identified VUS require functional validation to determine disease pathogenicity, a process currently performed using functional assays in highly specialised laboratories, that is undertaken in ad hoc fashion. This requirement for functional validation of VUS leads to a significant delay in rare diseases patient diagnosis of > 5 years, if not decades [2]. Faster and more systematic approaches to genetic variant validation are required. We propose a new and rapid pipeline for VUS assessment using a pipeline of CRISPR single base editing, inducible pluripotent stem cell disease modelling, and functional genomics. To demonstrate this pipeline we assess a known patient genetic variant EHMT1_c.3430C > T (p.Gln1144*) CRISPR edited into human inducible pluripotent stem cells (iPSCs), and determine genome wide changes in neural progenitor cell differentiation by functional genomics.
Kleefstra syndrome is caused by EHMT1 haploinsufficiency [3] that results in partial or complete loss of EHMT1 expression [4]. EHMT1 encodes a histone lysine methyltransferase [5], which forms a complex with EHMT2, for regulation of gene expression by histone 3 lysine 9 (H3K9) mono- and di-methylation (H3K9me1 and H3K9me2) [6]. The highly modulatory nature of histone mono-, di- or tri methylation leads to complex transcriptional regulation, based on the specific residues and the pattern of methylation [7, 8]. Kleefstra Syndrome [9, 10] hallmark symptoms include mental retardation, intellectual disability associated with autistic features, limited or absent speech, seizures, hypotonia, microcephaly, and brachycephaly. Other common symptoms are evident in the face (flat face, prognathism, midface hypoplasia and coarse facies), malformed ears, eyes (hypertelorism, upslanting palpebral fissures), and hands (brachydactyly, single transverse palmar creases), amongst others. Structural brain abnormality, congenital heart defects, seizures, high birth weight and/or childhood obesity are also reported.
In this proof-of-principle study we assess a Kleefstra Syndrome patient’s known pathogenic EHMT1 genetic variant. The patient genetic variant EHMT1_c.3430C > T; p.Gln1144* (herein referred to as EHMT1_SNV) was introduced into iPSCs, followed by neuronal cell differentiation. Differentiation of iPSCs to neural progenitor cells was compared between EHTM1_WT (wild-type) cells and EHMT1_SNV cells using RNA sequencing and transcriptomics analysis. Analysis of differentially expressed genes (DEGs) revealed enrichment of gene sets and transcription factor motifs associated with Kleefstra syndrome, changes in neuronal development, and provided insight into disease mechanisms.
Methods
Patient recruitment
The patient recruitment to this study was initiated by a genetic counsellor based at Genome Services Western Australia, followed by written informed consent. The study is in line with the Declaration of Helsinki and the NHMRC National Statement on Ethical Conduct Human Ethics Research. Approved by the Child and Adolescent Health Services, Human Research Ethics Committee, RGS0000000166.
The patient genetic variant was determined with Massively Parallel Sequencing via Trusight One, and a genetic variant detected NM_024757.4(EHMT1): c.[3430C > T];[ =] p.[(Gln1144*)];[( =)] which concluded EHMT1 Haploinsufficiency, which was considered pathogenic for Kleefstra syndrome [3].
Cell culture
KOLF2-C1 (KOLF2; HipSci) cells were grown in 24-well plates coated with Celladhere Vitronectin XF (STEMcell technologies), maintained in TeSR-E8 media (STEMCELL technologies), and media changed daily. Cells were split with Gentle Cell Dissociation Reagent with 10 µM ROCK Inhibitor (Y-27632, STEMCELL Technologies) for 24 h on re-plating. Cell cryopreservation was in CryoStor CS10 (STEMCELL Technologies). All cultures were grown in a 37 °C humidified CO2 (5%) incubator, unless otherwise stated, routinely checked for mycoplasma contamination.
CRISPR/Cas9 EHMT1_SNV transfection and cloning
KOLF2 cells grown to 30–50% confluence, were dissociated with Gentle Cell Dissociation Reagent and 1 × 105 cells in 400 µl mTeSR1 with 10 µM Y-27632 (STEMCELL technologies) aliquoted in 24 well plates. HDR and crRNA were covalently linked with Click Chemistry [10]. In brief, 10 µM EHMT1_HDR, ssDNA (5’AzideN CTCTACCGGACGCGGGACATGGGCTGGGGCGTGCGGTCTCTCTGAGACATTCCTCCTGGAACCTTTGTCTGCGAGTGAGTGAGT3’), and 10 µM crRNA (5’ Dibenzocyclooctyne N-Hydroxysuccinimide, DBCON, sequence: 5’TCCCTGCAGGACATCCC3’, PAM: AGG; EHMT1 NM_024757.2) were incubated at room temperature overnight to create a gDonor molecule [10]. The gRNA was generated in Duplex Buffer (IDT, USA) with 1 µM gDONOR (Integrated DNA Technologies), 1 µM tracrRNA-ATTO550 (Integrated DNA Technologies), heated to 95 °C for 5 min and cooled to RT. CRISPR ribonucleoprotein (RNP complexes) were formed with 63 nM gRNA and 63 nM high-fidelity Cas9 protein (Integrated DNA Technologies) in OPTIMEM with STEM Lipofectamine Cas Plus reagent (Life Technologies) and OPTIMEM. RNPs were transfected into KOLF2 cells with STEM Lipofectamine (Life Technologies), according to the manufacturer’s instruction. Alternatively, cells were transfected on the Neon Transfection system (1400 V, 20 ms, 1 pulse; Invitrogen). Cells were cultured for at least 7 days prior to cell freeze down or genomic DNA extraction using PureLink™ Genomic DNA Mini Kit (Life Technologies). Percentage frequency of HDR to create the EHMT1_SNV were determined by Amplicon sequencing. Transfected cells were then single cell cloned by limiting dilution in 96 well plates, replica plated, and DNA lysate prepared [11]. Further rounds of amplicon sequencing on DNA lysate determined clonal cell lines. Positive clones were thawed, re-tested and cell stocks prepared. Schematic of EHMT1 genetic variant introduction illustrated in Additional file 1: Fig. S1a. The EHMT1_SNV variant sequence mutalyzer variant description is NM_024757.5 (EHMT1):c.3426_3447delinsTCTCTGAGACATTCCTCCGGGA, with affected transcript NM_024757.5(EHMT1_i001):p.(Gln1144*). The top six off target CRISPR crRNA gene cut sites were confirmed as WT by forward and reverse Sanger sequencing (AGRF, WA; Additional file 2: Table S1. EHMT1 off target).
Amplicon sequencing
Next-generation amplicon sequencing was carried out on the MiniSeq Sequencing System (Illumina©). In brief, a 267 bp EHMT1_Ter site PCR product was amplified, from gDNA or DNA lysates, with EHMT1 pAMPF3 (5’ACACTCTTTCCCTACACGACGCTCTTCCGATCTCTCTCTATTTTTCAGGGCAAGG3’) and EHMT1 pAMPR2 (5’GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTACAGCACGAGCTTGGTTCTC3’), using the CRES-Seq [11] for 150 bp, paired end, > 10,000 reads (MiniSeq, Illumina, Australia) and reads aligned to the HDR or WT amplicon with CRISPResso2 software [12].
Cells were plated at 20 cells/well and wells positive for the EHMT1_SNV genetic variant determined with EHMT1 amplicon sequencing [11]. Cells in positive wells were then single cell cloned followed by EHMT1 amplicon sequencing [11] to identify three heterozygous EHMT1_SNV iPSC clones (EHMT1_WT/SNV; EHMT1_SNV_16, EHMT1_SNV_17, EHMT1_SNV_29) and three matched EHMT1 wild-type clones (EHMT1_WT_100, EHMT1_WT_102, EHMT1_WT_103).
Neural progenitor cell differentiation
KOLF2 iPSCs, genetic variant and normal, were stimulated to differentiate into neural progenitor cells with STEMdiff SMADi Neural Induction Kit (STEMCELL, Vic, Australia). At day 0, and 24 of neural cell differentiation 5 × 105 cells were collected, LIVEDEAD ef780 stained for 10 min (eBioscience, US), fix/permeabilized according to Transcription factor staining buffer set (eBioscience, US) and stained for stem cell expression marker (OCT3/4), and neuronal cell marker (PAX6). Samples were collected using an LSRII X20 flow cytometer (BD, Biosciences), and analysed with FlowJo software (TreeStar Inc, Ashlan, OR, USA). Gating strategy was total cells, with subsequent gating on live cells and then singles, prior to determination of percentage frequency protein marker expression. Statistical analysis of data was with one-way ANOVA and Bonferroni’s correction for multiple testing performed using GraphPad Prism Version 8 software (GraphPad Software Inc., La Jolla, CA, USA). In addition, cells were collected for RNA extraction (RNeasy Minikit, with DNase on column treatment; Qiagen).
EHMT1 Western blot and immunohistochemistry
Proteins were extracted from cells lysed with Pierce’s co-IP protein lysis buffer (ThermoFisher Scientific), quantified (Direct-Detect® Infrared Spectrometer, Merck Millipore), electrophoresed on NuPAGE Bis–Tris 4–12% protein gels (Life Technologies) and blotted onto Polyvinylidene fluoride (PVDF) membranes (Life Technologies). Membranes were blocked with Intercept® (TBS) Blocking Buffer (LI-COR® Biosciences) at 4ºC overnight, stained with rabbit anti-human EHMT1 (1:1000, NBP2-57,166, Novus Biologicals, USA), and/or b-Actin (1:5000; MA5-15,729; Life Technologies, Australia), with secondary stain with goat anti-rabbit IRDye® 800CW (1:10,000; LI-COR® Biosciences) or goat anti-mouse IRDye® 680RD (1:10,000; LI-COR® Biosciences) and imaged on the Odyssey® Infrared Imaging System (LI-COR® Biosciences).
EHMT1_WT and EHMT1_SNV iPSCs were cultured on Matrigel-coated chamber slides (ibidi). One day later, cells were fixed with 3.7% formaldehyde (Sigma-Aldrich) for 20 min at room temperature. Subsequently, cells were permeabilised for 15 min with 0.1% Triton-X-100 (Sigma-Aldrich) and blocked with Intercept® Blocking Buffer (LI-COR) for 1 h at room temperature. Cells were incubated overnight at 4 °C with rabbit polyclonal anti-EHMT1 antibody (1:200; Novus Biologicals), washed with 0.05% Tween-20 (Sigma-Aldrich) and incubated with Alexa-Fluor 488-conjugated anti-rabbit antibody (1:1000; Invitrogen) for 1 h at room temperature. After washing cell nuclei were stained using NucBlue stain (Invitrogen) according to the manufacturer’s instructions. Antibody staining was visualised using a Nikon Eclipse TS2R inverted fluorescence microscope, images captured using a monochrome DS-Qi2 camera (Nikon) and immunofluorescence images processed using NIS-Elements software (v.5.21.00) and Adobe Photoshop (v.22.3.1) to add colour and merge channels. A negative control containing no primary antibody was used to set the background fluorescence.
RNA sequencing
RNA integrity was determined on the bioanalyser (Australian Genomics Research Facility (AGRF, Perth, Western Australia). RNA sequencing was performed according to SureSelect Strand-Specific RNA Library Preparation for Illumina Multiplexed Sequencing, paired-end, 100 bp, 30 M read RNA sequencing on the NOVAseq 6000 platform (Illumina, USA) at Genomics WA (Perth. Australia).
Preprocessing, exploratory data analysis and differential analysis
Data pre-processing
Raw sequencing reads were processed using the ENCODE ‘rna-seq-pipeline’ (https://github.com/ENCODE-DCC/rna-seq-pipeline), via the Cromwell wrapper software ‘caper’ (https://github.com/ENCODE-DCC/caper). Within the pipeline, reads were aligned to GRCh38 using the STAR aligner (v2.5.1b) [13] and known transcripts were quantified using Kallisto (v0.44.0) [14]. We created gene and transcript expression tables for downstream analysis with DESeq2, limma and DRIMSeq [15,16,17] using the package tximport [18].
Exploratory data analysis
Gene counts were read into R 4.0.2 (https://www.R-project.org/) using the DESeq2 package function DESeqDataSetFromTximport() [15]. We filtered the counts to retain genes with a count of 3 or more and normalised the data using the variance stabilising transformation [19]. The plotPCA() function was used to produce a principal component analysis plot based on the top 500 variable genes and no outlying samples were observed.
Differential gene and transcript expression analysis. We used limma to fit linear models to test for differentially expressed genes and transcripts to address the following hypotheses: (i) Expression differs when comparing EHMT1_SNV versus EHMT1_WT in NPCs; (ii) Expression differs when comparing EHMT1_SNV versus EHMT1_WT in iPSCs; (iii) Expression differs during differentiation from iPSCs to NPCs for EHMT1_SNV; (iv) Expression differs during differentiation from iPSCs to NPCs for EHMT1_WT; and, (v) Changes in expression during differentiation from iPSCs to NPCs differs when comparing EHMT1_SNV to EHMT1_WT. Genes were filtered using the filterByExpr() function accounting for the number of samples in each group and limma-trend models were fit to normalised log2 counts per million transformed data, calculated using the cpm() function with a prior count of 3. We determined quality weights for each sample using the arrayWeights() function and adjusted for this in the models. We also adjusted for pairing of samples from the same clone using the duplicateCorrelation() function. Genes and transcripts with Benjamini-Hochberg [20] corrected p-values less than 0.05 and absolute fold-changes of at least 2 were deemed to be differentially expressed (DE).
Functional enrichment analysis
We performed functional gene set enrichment analysis on the differentially expressed genes using Enrichr [21, 22] and we used Benjamini–Hochberg corrected p-values when determining significance. We also performed Gene Set Enrichment Analysis (GSEA) [23, 24] on all genes using the moderated t-statistic from the differential expression analysis for ranking and tested for enrichment in the Molecular Signatures Database v7.1 (MSigDB) hallmark gene sets [25] using a false discovery rate less than 0.05 to determine significance.
Upstream regulatory analysis
Analysis was performed to find enriched DNA motifs in regions upstream of the identified differentially expressed genes. For this analysis, the 500 base pairs (bp) upstream of the start site of the differentially extracted genes were extracted from GRCh38, using the bedtools function getfasta [26]. The resulting two stranded base sequences were analysed with the MEME software (meme-suite.org) to identify motifs upstream of the genes [27]. Motif length search space was between 3 and 20, with a default E-value threshold of 0.05.
Discriminative motif discovery analysis was performed between the over and underexpressed genes, the overexpressed and background genes, and the underexpressed and background genes, using the DREME software [28]. The background genes are defined as genes that passed the filtering step in the limma DE analysis and were not differentially expressed in any of the statistical comparisons. TomTom was used [29] to compare the potential motifs identified by MEME and DREME to the JASPAR 2018 vertebrate redundant database [30] of known motifs.
AME software was used to identify overrepresented known motifs [31]. The sequences 500 bp upstream from the significantly over and underexpressed genes, as well as all differentially expressed genes were contrasted against the background sequences, with the JASPAR 2018 vertebrate redundant database as search database [31]. Significant transcription factors were identified via Fisher's exact test, optimized over motif scores (significance threshold ≤ 0.05).
Results
Kleefstra syndrome EHMT1 patient genetic variant
A Kleefstra Syndrome patient was identified to have the genetic variant EHMT1_c.3430C > T (p.Gln1144*). The patient’s phenotype was characterised using the Human Phenotype Ontology terms as detailed in Additional file 2: Table S2.
Euchromatic lysine histone methyltransferase proteins are characterised by the presence of SET/pre-SET domains that are responsible for methyltransferase activity [32,33,34]. The EHMT1 protein also has 6 ankyrin repeat domains that provide binding specificity for H3K9me1 and H3K9me2 to enable chromatin reading. Two- and three-dimensional protein structures for the EHMT1_WT and EHMT1_SNV protein are illustrated (Fig. 1a, b). The 2D protein structure clearly indicates EHMT1_SNV loss of the SET binding domain necessary for protein–protein interaction, and loss of the two histone H3 binding sites, whereas the PreSET domain and histone 3 lysine 9 (H3K9) binding site remain intact. To visualise the effects of the variant on protein structure, 3D models of EHMT1 wild-type and mutant proteins were superimposed onto the protein data bank (PDB) entry: ‘5TUZ structure of human GLP SET-domain (EHMT1) in complex with inhibitor MS0124 (https://doi.org/10.2210/pdb5TUZ/pdb)’. This highlighted changes in functional domains that may affect function for the variant EHMT1_SNV protein.

Changes in EHMT1 protein expression in EHMT1_SNV cells. a Two-dimensional protein structure of EHMT1_WT and EHMT1_SNV with Ankyrin repeat domain (ANK Repeats), PreSET domain, and SET domain. b Three dimensional structure of EHMT1_WT and EHMT1_SNV indicating Pre-SET domain (light blue), and the SET domain with methyl transferase activity in blue and red, In the EHMT1_WT protein the red colouring indicates the region absent in the EHMT1_pGln1144* mutant protein., and the green indicates the truncated protein stop codon. c Western blot of EHMT1 and b-actin protein expression in EHMT1_WT cells and EHMT1_SNV cells. d Bar graph indicates EHMT1 protein expression normalized to b-actin control (paired t-test, one-tailed, *p < 0.05). e, Immunohistochemistry staining of EHMT1 and NucBlue, and merged image, in EHMT1_WT and EHMT1_SNV iPSCs, white bar indicates 50micron
The EHMT1_SNV variant was introduced into KOLF2 iPSCs using CRISPR_HDR. After transfection polyclonal EHMT1 gene modifications were determined by amplicon sequencing [12]. A HDR rate of 1.1 ± 0.11% was achieved for the introduction of the patient EHMT1 genetic variant in KOLF2 iPSC transfected cells, with a NHEJ frequency of 26.47 ± 2.83%. Our amplicon sequencing and cloning method allowed the rapid identification of EHMT1 alleles and selection of cell clones that were heterozygous for the EHMT1_SNV genetic variant.
Next, we determined the protein and gene expression level of EHMT1 for the EHMT1_WT and EHMT1_SNV cells. EHMT1 expression may be reduced by degradation of the truncated protein, or degradation of RNA due to the premature STOP codon [35, 36]. The expression of EHMT1 protein was significantly reduced in the EHMT1_SNV iPSCs compared to the EHMT1_WT iPSCs (Fig. 1c, d), and the truncated form of the protein was not detected. EHMT1 protein expression was localised to the nucleus on immunohistochemistry analysis of EHMT1_SNV and EHMT1_WT cells (data not shown). The RNA-seq EHMT1 gene expression for EHMT1_SNV compared to EHMT1_WT iPSCs also showed reduced expression, however, did not meet our p-value and fold change threshold for DEGs (log2FC = − 0.49, p = 0.23).
This data indicates a reduction in EHMT1 protein expression in EHMT1_SNV cells compared to the EHMT1_WT cells that may affect cell function and differentiation.
EHMT1_WT and EHMT1_SNV iPSCs differentiate into neural progenitor cells
The EHMT1_SNV, and EHMT1_WT, iPSCs were induced for neural differentiation to form neural progenitor cells (NPCs). Principal component analysis (PCA) and hierarchical clustering of the normalised RNA-seq data (Additional file 1: Fig. S1b, c) indicated clear separation between EHMT1_SNV and EHMT1_WT cells. In keeping with expected changes in gene expression for neuronal cell differentiation we determined decreased expression of the OCT3/4 stem cell marker, with a significant increase in expression of the neural cell marker PAX6 by flow cytometry analysis (Fig. 2a, b), and this was similar in the EHMT1_WT and EHMT1_SNV, iPSC and NPCs, respectively. In addition, NPCs with radial alignment and bipolar morphology for the EHMT1_WT and EHMT1_SNV NPCs were visually determined (Fig. 2c). At the RNA expression level EHMT1 showed reduced expression in EHTM1_SNV versus EHMT1_WT NPCs, however, did not meet our fold change threshold for DEGs (log2FC = − 0.47, p = 0.0034).

EHMT1_WT and EHMT1_SNV neural cell differentiation. Cells were subject to neural progenitor cell differentiation with cells harvested at day 0 and day 24 for flow cytometry analysis. a Histogram plots indicate cellular staining for pluripotent stem cell marker, OCT3, and neural marker, PAX6. b Bar graphs indicate mean fluorescence intensity (MFI) for OCT3 and PAX6. (n = 3 experiments with paired WT and SNV clones. *p ≤ 0.05, **p ≤ 0.01, Kruskal–Wallis, Two stage linear set-up of Benjamini, Krieger and Yekutieli). c NPC light microscopy images, black bar indicates 100 micron. d Venn diagram indicates number of DEGs for EHMT1_WT and EHMT1_SNV during iPSC neural differentiation
DEG analysis for the iPSCs compared to NPCs confirmed neural differentiation for EHMT1_WT and EHMT1_SNV cells. There were 7815 DEGs in EHMT1_WT iPS cells during differentiation, and 8026 DEGs in EHMT1_SNV cells, with 5924 DEGs common to both (Fig. 2d). We performed gene set enrichment analysis on the DEGs using the EnrichR software. We compared our DEGs against gene sets associated with tissues (ARCHS4_Tissues), and cell function (GO_Biological Process). ARCHS4_Tissues indicated significant enrichment for the EHMT1_WT iPS cell to NPC DEGs during neuronal differentiation in the following top five genesets associated with nerve and brain: motor neuron (p = 1.14 × 10−18), spinal cord (p = 1.47 × 10−14), spinal cord-bulk (p = 1.47 × 10−14), Brain bulk (p = 2.10 × 10−12), and superior frontal gyrus (p = 8.79 × 10−12). Similarly, for the EHMT1_SNV iPS cell to NPC DEGs, the top enriched ARCHS4_Tissues genesets were: motor neuron (p = 4.33 × 10−37), spinal cord (p = 9.61 × 10−32), spinal cord-bulk (p = 9.61 × 10−32), Superior frontal gyrus (p = 7.03 × 10−22), and brain-bulk (p = 1.84 × 10−20). Furthermore, the top enriched GO_Biological_Process for both EHMT1_WT and EHMT1_SNV was nervous system development (p = 0.00412 and p = 1.17 × 10−4, respectively).
This data indicated the capacity for EHMT1_WT and EHMT1_SNV iPSCs to differentiate into NPCs.
EHMT1_SNV differences in neural differentiation reflect Kleefstra syndrome
Next, we determined the genes that change expression during neural differentiation that are different between the EHMT1_SNV and EHMT1_WT cells (ie. [NPC_SNV – iPSC_SNV] – [NPC_WT – iPSC_WT]; Fig. 3a). We determined 109 significant DEGs, including 89 genes with increased expression, 19 genes with decreased expression, and 58 genes that were brain and/or neuronal related. The top up-regulated protein coding gene was the homeobox transcription factor NKX2.1, which is involved in establishment of a permissive chromatin state for the formation of medial ganglionic eminence progenitors and production of GABAergic neurons [37]. The top down-regulated gene was DMRT3. The loss of this gene induces massive production of GABAergic neurons [38].

Differences in gene expression during differentiation for EHMT1_WT and EHMT1_SNV cells. iPSC clones for EHMT1_WT and EHMT1_SNV, were differentiated to NPCs and DEGs determined for the differences in expression during differentiation. a Volcano plot for EHMT1_SNV compared to EHMT1_WT for the differences in expression during differentiation. DEGs are shown in red. Grey dot adjusted p ≥ 0.05 and |log2FC|≤ 1; green dot adjusted p ≥ 0.05 and |log2FC|> 1; blue dot adjusted p < 0.05 and |log2FC|≤ 1; red dot adjusted p < 0.05 and |log2FC|> 1. b Dot plots of significantly enriched gene sets associated with differences in expression during differentiation identified with Enrichr. c Enrichment plots for GSEA hallmark significant gene sets for genes showing upregulation during differentiation for EHMT1_SNV compared to EHMT1_WT. d Enrichment plots for GSEA hallmark significant gene sets for genes showing downregulation during differentiation for EHMT1_SNV compared to EHMT1_WT
Enrichr analysis of the DEGs indicated enrichment in genesets associated with methylation, cell type, tissue type, cell compartment, molecular function, cellular process, pathways, and transcription factors (Additional file 2: Table S3. Enrichr Diff in Diffn EHMT1_SNV v EHMT1_WT), with identification of the rare disease Kleefstra Syndrome (Fig. 3b).
For Epigenomics_Roadmap_HM_ChIP-seq gene sets significant enrichment was seen for H3K27me3 BMP4 derived trophoblast cultured cells (p = 9.29 × 10−5). Further ENCODE_Histone_modifications_2015 top 5 enriched gene sets relevant to human were: H3K27me3 endothelial cell of umbilical vein hg19 (p = 1.62 × 10−5), H3K27me3 BJ hg19 (p = 1.62 × 10−5), H3K27me3 kidney epithelial cell hg19 (p = 1.62 × 10−5), H3K27me3 cardiac mesoderm hg19 (p = 2.06 × 10−4), and H3K27me3 bronchial epithelial cell hg19 (p = 0.0023). This data indicated changes in histone methylation due to the effect of EHTM1_SNV on neural cell differentiation.
Enrichment in tissue and cell associated gene sets were next examined. The top 5 enriched ARCHS4_Tissues gene sets were: spinal cord (p = 2.05 × 10−19), spinal cord-bulk (p = 2.05 × 10−19), brain-bulk (p = 5.04 × 10−18), superior frontal gyrus (p = 5.04 × 10−18), and cingulate gyrus (p = 1.42 × 10−16), with the same top 5 gene sets for the overexpressed DEGs. In Jensen_Compartments the top five significant gene sets were: Axon (p = 1.6 × 10−4), cell projection (p = 5.05 × 10−4), Neuron projection (p = 5.05 × 10−4), somatodendritic compartment (p = 0.0011), and site of polarized growth (p = 0.0011). For GO_Biological_Process_2018 significantly enriched gene sets from all DEGs were: neuron projection development (p = 0.0028), regulation of N-methyl-D-aspartate receptors (NMDAR) activity (p = 0.0126), positive regulation of axogenesis (p = 0.0126), and positive regulation of axon extension (p = 0.0284). Interestingly, GO_Biological_Process enriched gene sets for overexpressed DEGs analysis identified the additional genesets: negative regulation of transferase activity (p = 0.04365), regulation of glutamate receptor signalling pathway (p = 0.0436), regulation of neurotransmitter receptor activity (p = 0.0436), regulation of axon extension (p = 0.0436), and regulation of filopodium assembly (p = 0.0460). This data indicated changes in neural differentiation that affect cell specification and cell function.
Next, we investigated enriched transcription factor and hub proteins. Significantly enriched gene sets in ENCODE_and_CHEA_Consensus_TFs_from_ChIP-X were: REST ENCODE (p = 1.21 × 10−5) and SUZ12 CHEA (p = 0.0252). REST, RE1 Silencing Transcription Factor, is a neural regulator gene that exhibits both positive and negative gene regulation dependent on the stage of cell development [39]. SUZ12 is part of the polycomb repressive complex 2 (PRC2) which is an epigenetic regulator with H3K27me3 histone methyltransferase activity in chromatin modification associated with repression of cell proliferation or self-renewal genes [40].
Strikingly, in investigation of enriched gene sets associated with disease (Enrichr Rare Disease_AutoRIF_ARCHS4_predictions) the Kleefstra Syndrome geneset was significantly enriched (p = 3.61 × 10−8).
To further characterise the changes in gene expression for the difference in differentiation for EHMT1_SNV compared to EHMT1_WT neural cell differentiation, we performed GSEA using the DEGs from the ranked gene list (Fig. 3c and d; Suppl Table 4. GSEA Diff in Diffn EHMT1_SNV v EHMT1_WT). Significantly enriched hallmark gene sets associated with overexpressed genes included: Hedgehog signalling, myogenesis, p53 pathway, and KRAS signalling DN. Hallmark gene sets associated with underexpressed genes included: E2F targets, G2M checkpoint, MYC targets v1, MYC targets v2, mitotic spindle, MTORC1 signalling, epithelial mesenchymal transition, and androgen_response. Relevant to neural cell differentiation is the Hedgehog signalling which is regulated by REST [41] and involved in GABAergic neuron formation, and KRAS (DN) which suppresses neuronal cell differentiation from iPSCs [42]. Further, EHMT1_SNV neural cell differentiation had reduced cell division pathway gene expression for E2F, G2M, mitotic spindle, and MYC; with increased p53 pathway that suppresses cell proliferation.
These data indicate that neural cell differentiation of EHMT1_SNV iPSCs, compared to EHMT1_WT iPSCs, had increased neural gene expression, with increased GABAergic related genes and pathways, as well as repressed cell proliferation/cell division. The genes associated with these differences were enriched for genes associated with Kleefstra Syndrome.
Changes in EHMT1_SNV transcripts and transcription factors identifies key regulators REST and SP1
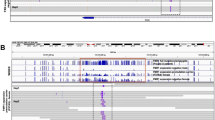
We performed upstream regulatory analysis to identify transcription factors (TFs) driving the observed changes in gene expression. We selected the 500 bp upstream region of annotated gene promoters of differentially expressed genes to search for motifs using MEME/TomTom, DREME/TomTom, and AME to identify key regulatory TFs (Additional file 2: Table S5. Transcription factors Diff in Diffn_MEME DREME AME). MEME/TomTom identifies de novo enriched transcription factors, DREME identifies differential motifs, and AME identifies known transcription factors.
In both the EHMT1_WT, and EHMT1_SNV, iPSC to NPC neural cell differentiation analysis of downregulated DEGs, MEME discovered significant overrepresentation of KLF4 one of the four key Yamanaka Factors for maintaining stem cell integrity, p = 0.00047 and p = 0.0183, respectively [43]. Similarly, in AME analysis of underexpressed DEGs for the EHMT1_WT, and EHMT1_SNV, iPSC to NPC neural cell differentiation there was significant overrepresentation of KLF4, p = 1.14 × 10−9, and p = 0.00047, respectively [43]. This confirmed a downregulation of the KLF4 transcription factor as expected in neural cell differentiation.
For genes that show differences in expression during differentiation for EHMT1_SNV versus EHMT1_WT, MEME analysis of overexpressed genes identified significant transcription factors were ZNF263, EGR1, SP1, and SP2. In MEME analysis of down-regulated genes there were no significantly enriched TF binding sites identified.
ZNF263 is a chromatin binding zinc finger protein involved in chromatin loop formation and is capable of recruiting DNMT co-repressor complexes to silence transcription via H3K27me3 modification [44]. In rodents, EGR1 (Early growth response 1) co-localizes with Nestin expression and is a marker of NPCs [45]. SP1 regulates cell cycle, and in concert with E2F3 controls cell cycle transcription networks and epigenetic modifiers that determines cell fate [46, 47]. SP2 is a regulator of late neurogenic gene expression [48].
AME analysis of up-regulated genes showed enrichment for binding of REST, a regulator of neural differentiation [39], and SP1, involved in cell cycle regulation [46]. Previous studies indicate that the REST TF binds CYDL and recruits EHMT1 to the complex to repress gene transcription [49]. The REST TF motif was discovered upstream of three DEGs: MAPK8IP1, OGDHL and FBLL1. We also examined REST genesets from other sources (ENCODE_and_CHEA_Consensus _TFs_from_ChIP-X and ENCODE_TF_ChIP_seq_2015) and determined a further 28 REST related genes (Fig. 4). This indicates that 31 of the 109 DEGs could be directly regulated by REST. For the AME analysis of up-regulated genes 57 of the 109 DEGs were identified with an SP1 transcription factor motif (Fig. 4).

Genes showing differences in expression during differentiation for EHMT1_SNV compared to EHMT1_WT reveal predominant REST and SP1 modulation. Heatmap of normalised log2 counts per million, where genes have been centred and scaled by the standard deviation for the 109 genes showing differences in expression during differentiation for EHMT1_SNV compared to EHMT1_WT. Genes with REST and SP1 motifs are highlighted. List of genes that were identified with REST motif (green highlight) or in REST gene sets (pale orange highlight), and with SP1 motif (purple highlight)
We further investigated whether there were differences at the level of individual transcripts by performing differential transcript expression (DTE) analysis (Suppl Table 6. DTE Diff of Diffn). Interestingly, however, in the DTE for EHMT1_WT NPCs versus iPSCs there was a significant change in the expression of transcript SP1-204 (log2FC = 1.41, p = 0.03). This shorter SP1-204 variant, of 162 amino acids, is a stronger activator of transcription than SP1-201 [50], where higher SP1-204 transcripts levels are found in G phase cells compare to S phase cells. This indicates increased SP1-204 expression in the EHMT1_WT cells may contribute to changes in gene expression relevant to cell cycle and neural cell differentiation.
Discussion
Rare disease diagnosis is often difficult and determining the relevance of genetic variants of uncertain significance leads to significant delays in patient diagnosis. In this study a patient with a genetic variant in the EHMT1 gene is assessed with relevance to Kleefstra Syndrome [36]. CRISPR SNV gene editing in KOLF2 iPSCs, neuronal disease modelling, and functional genomics identified Kleefstra disease and associated changes in neural differentiation. Importantly, the study combination of CRISPR with click chemistry and high throughput genetic variant screening methodology enabled the rapid identification of cells containing the genetic variant. In addition, the RNA-seq expression data confirms successful neural cell differentiation of EHMT1_SNV iPSCs, including relevant neural expression markers and transcription factors. However, despite successful differentiation, changes in cell function, epigenetics, neurotransmitters, cell proliferation, and neural differentiation were identified in the EHMT1_SNV, Kleefstra-like cells. In addition, the study linked transcription factors REST and SP1 to disease specific changes relevant to Kleefstra Syndrome that portend pivotal cellular changes underpinning disease mechanism.
The study of rare genetic diseases identifies single nucleotide variants in the genome that may or may not be disease causative. Current methods for the rapid introduction of these genetic variants in cells is poor. Whilst CRISPR gene editing for knock out genes has been robustly developed, the introduction of single nucleotide changes in DNA requires HDR which occurs at frequencies of < 1%. In this study we employed CRISPR click chemistry and amplicon sequencing to identify iPSC clones that were heterozygous for the EHMT1_c.3430C > T (p.Gln1144*) patient variant. Historically iPSCs have been difficult to edit with CRISPR however our methodology rapidly identified functional iPSCs that were stimulated for neuronal modelling to determine cellular changes relevant to Kleefstra Syndrome and identify novel disease mechanisms.
Importantly, when examining the genes that show differences in expression during differentiation for EHMT1_SNV cells compared to EHMT1_WT cells, findings were consistent with the patient disease phenotype, Kleefstra Syndrome, were identified. In this study there were no observed differences in neural marker expression and cell morphology between the EHMT1_WT cells and EHMT1_SNV iPSCs or NPCs. Similarly, others have determined no significant phenotypic differences in neurons derived from patients with Kleefstra Syndrome and healthy controls [51]. We were able to determine reduced EHMT1 gene and protein expression in the EHMT1_SNV iPSCs and NPCs in keeping with findings in Kleefstra patient cells [51,52,53]. In addition, in this study DEG analysis of EHMT1_WT and EHMT1_SNV iPSC to NPC differentiation indicated similar neural differentiation profiles. Strikingly, however DEG analysis that tested for differences in gene expression during differentiation for EHTM1_SNV compared to EHMT1_WT revealed distinct changes related to histone methylation, cell proliferation/cell cycling, and NPC differentiation/maintenance.
Epigenetic modification of chromatin in neurons has a role in cognitive impairment and intellectual disability [54] and EHMT1 is long established in H3K9me1 and me2, for heterochromatin formation and gene repression [5, 6]. Dysregulation of H3K9me1/me2 is thought to lead to alternative histone methylation by allowing access of other methyl transferases. Genes with significantly different changes in expression during differentiation when comparing EHMT1_SNV to EHMT1_WT were associated with strong dysregulation of H3K27 methylation. Interestingly, we observed TFs involved in H3K27me3 including EZH2 methyltransferase [55], and ZNF263 via SIX3 modulation [44]. Reportedly, ZNF263 also plays a role in chromatin restructuring via the CCCTC-binding factor (CTCF) chromatin loop formation [44].
Dysregulation of cell cycle length has been indicated as a driver of premature differentiation of neural progenitor cells [46], and siRNA EHMT1 knockdown experiments indicate reduced cell proliferation and increased cell differentiation [56]. Furthermore, TFs play a crucial role in control of the cell cycle [57], here, in EHMT1_SNV neural differentiation we find decreased activity of E2F, and SP1, with increased activity of the cell proliferation regulator p53. We also identified reduced cellular proliferation profiles in EHMT1_SNV during differentiation via reduced expression of genes involved in G2/M [58], mTORC1 pathways, and KRAS signalling [42] and conversely increased expression of genes involved in Hedgehog signalling [41].
Functional genomic analysis of EHMT1_SNV neural differentiation identified increased neurotransmitter receptor activity. Similarly, others identified differences in action potential decay, with lower frequency network bursts, with longer burst and inter-burst duration in Kleefstra Syndrome patient derived cells [51]. We demonstrate a further change in EHMT1_SNV neural differentiation indicative of increased GABAergic neurons via downregulation of DMRT3. Similarly, others observed increased GABAergic neurons in DMRT3 knockout experiments in mice [38], whilst in Ehmt1 ± mice dysregulation of GABAergic interneurons reportedly leads to increased neuron firing [51]. Further, EHMT1_SNV overexpression of NKX2.1 was observed, where reportedly NKX2.1 establishes a permissive chromatin state for the production of GABAergic interneurons [37].
Neural differentiation of EHMT1_SNV iPSC cells indicated a change in both stem cell and neural associated transcription factors including MYC, REST and SP1. A negative association with the MYC targets likely indicates loss of stemness in these cells as they are induced for neural cell differentiation [59, 60].
REST regulates neural cell differentiation and governs neuronal cell phenotype and specification [39]. The REST transcription factor forms a complex with EHMT1 via its interaction with CDYL [49], for histone methylation and chromatin modulation in the regulation of gene expression. Accordingly, during neural cell differentiation a consequence of the EHMT1 truncated protein may be disruption of the REST/EHMT1 complex, which leads to the perturbations in gene expression observed in Kleefstra Syndrome. Potentially REST binds DNA yet is unable to effectively recruit EHMT1 p.Gln1144* protein, or EHMT1 p.Gln1144* is recruited but does not function.
The TF motif analysis further identified SP1 as being associated with genes showing differences in expression during differentiation for EHMT1_SNV versus EHMT1_WT. We also identified differential expression of a short SP1-204 transcript during EHMT_WT iPSC differentiation that is a strong activator of cell cycle gene expression. Potentially loss of SP1-204 expression in EHMT1_SNV cells contributes to decreased cell cycle activity and altered gene expression. Alternatively, SP1 recruitment to the OCT4 promoter, via PML (promyelocytic leukemia 1), maintains open chromatin for OCT4 gene expression in stem cells [61], where induction of neural differentiation leads to SP1 dissociation and subsequent recruitment of an EHMT2 complex leading to heterochromatin formation and gene suppression [61]. We observed similar downregulation of OCT4 (POU5F1) in neural differentiation for both the EHMT1_SNV and EHMT1_WT iPSCs, potentially indicating that in this scenario EHMT2 works as a homodimer, or the EHMT1/EHMT2 complex remains functional in this capacity. In other work, in viral infection SP1 regulates HIST1H1C which subsequently modulates EHMT1/EHMT2 complex formation and gene expression [62]. Here the genes showing differences in expression during differentiation for EHMT1_SNV compared to EHMT1_WT similarly provide a link between SP1 and EHMT1/EHTM2 in the regulation of gene expression.
Interestingly there is interplay between SP1 and REST gene expression in adult neurons where REST binding to chromatin can suppress SP1 gene expression, alternatively, in other neural states/conditions SP1 has been shown to positively modulate REST expression [63]. Additionally, in neural cells SP1 directly binds the promoter of gene SYN1 (Synapsin I) leading to gene expression, whilst REST binding of the SYN1 promoter suppresses gene expression [64]. SYN1 plays a crucial role in neurogenesis, including axonogenesis and synaptogenesis, and has a role in synaptic transmission and mature neuron plasticity [64]. Here, we demonstrate that REST and SP1 interplay to co-ordinately regulate gene expression of a specific subset of genes associated with Kleefstra Syndrome.
The use of bioinformatics in this study has highlighted significant pathways relevant to Kleefstra syndrome. A caveat of the bioinformatics methods is the reliance on available genesets to determine changes in cell function. As more genetic variants in disease are identified and molecular pathways delineated these genesets will however become more expansive.
Conclusions
CRISPR edited EHMT1_SNV iPSC with neural disease modelling and functional genomics analysis clearly validated the patient genetic variant in Kleefstra syndrome. EHMT1 haploinsufficiency, a hallmark of Kleefstra syndrome, was confirmed in the EHMT1_SNV cells by decreased EHMT1 protein expression. Many genetic diseases do not have a specific functional test and this study argues for the utility of patient genetic variant analysis with our CRISPR HDR gene editing, disease modelling and functional genomics analysis pipeline. In addition, transcription factors, REST and SP1, key to the etiology of Kleefstra syndrome were identified providing insight into disease mechanism which may facilitate the identification of drug targets for treatment. Importantly, the pipeline can be modified to assess genetic variants relevant to many genes relevant to rare, or other, disease as iPSCs can be modelled to neuronal, cardiac, lung or other tissue types.
Availability of data and materials
Raw FASTQ files and processed count data are available at the Gene Expression Omnibus repository under accession number GSE178646.
Abbreviations
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- crRNA:
-
Cripsr RNA
- DEG:
-
Differentially expressed gene
- DTE:
-
Differentially expressed transcript
- gDNA:
-
Genomic deoxyribonucleic acid
- HDR:
-
Homology directed repair
- iPSC:
-
Inducible pluripotent stem cells
- NGS:
-
Next generation sequencing
- NPC:
-
Neural progenitor cell
- PAM:
-
Protospacer adjacent motif
- RNA:
-
Ribonucleic acid
- SNV:
-
Single nucleotide variant
- ssDNA:
-
Single stranded DNA
- TF:
-
Transcription factor
- tracrRNA:
-
Trans-acting crRNA
- WT:
-
Wildtype
- VUS:
-
Variant of uncertain significance
References
Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019;12(1):3.
Baynam G, Broley S, Bauskis A, Pachter N, McKenzie F, Townshend S, et al. Initiating an undiagnosed diseases program in the Western Australian public health system. Orphanet J Rare Dis. 2017;12(1):83.
Kleefstra T, van Zelst-Stams WA, Nillesen WM, Cormier-Daire V, Houge G, Foulds N, et al. Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J Med Genet. 2009;46(9):598–606.
Willemsen MH, Vulto-van Silfhout AT, Nillesen WM, Wissink-Lindhout WM, van Bokhoven H, Philip N, et al. Update on Kleefstra Syndrome. Mol Syndromol. 2012;2(3–5):202–12.
Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, et al. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat Struct Mol Biol. 2008;15(3):245–50.
Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3–K9. Genes Dev. 2005;19(7):815–26.
Park JH, Yang SW, Park JM, Ka SH, Kim JH, Kong YY, et al. Positive feedback regulation of p53 transactivity by DNA damage-induced ISG15 modification. Nat Commun. 2016;7:12513.
Spannhoff A, Hauser AT, Heinke R, Sippl W, Jung M. The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors. ChemMedChem. 2009;4(10):1568–82.
Medlineplus. Genetics home reference. Kleefstra syndrome medline plus: bethesda (MD): National Library of Medicine; 2018 [updated 8 Sept 2020. Available from: https://medlineplus.gov/genetics/condition/kleefstra-syndrome/.
Lee K, Mackley VA, Rao A, Chong AT, Dewitt MA, Corn JE, et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. Elife. 2017;6:e25312.
Veeranagouda Y, Debono-Lagneaux D, Fournet H, Thill G, Didier M. CRISPR-Cas9-Edited site sequencing (CRES-Seq): an efficient and high-throughput method for the selection of CRISPR-Cas9-Edited clones. Curr Protoc Mol Biol. 2018;121:31–14.
Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol. 2019;37(3):224–6.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34(5):525–7.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Nowicka M, Robinson MD. DRIMSeq: a Dirichlet-multinomial framework for multivariate count outcomes in genomics. F1000Res. 2016;5:1356.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Ser B (Methodol). 1995;1995(57):289–300.
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013;14:128.
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–7.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–50.
Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–25.
Quinlan AR. BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinform. 2014;47:11–2.
Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36.
Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27(12):1653–9.
Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biol. 2007;8(2):R24.
Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018;46(D1):D1284.
McLeay RC, Bailey TL. Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinform. 2010;11:165.
Herz HM, Garruss A, Shilatifard A. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem Sci. 2013;38(12):621–39.
Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25(8):781–8.
Trievel RC, Beach BM, Dirk LM, Houtz RL, Hurley JH. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell. 2002;111(1):91–103.
Baker KE, Parker R. Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol. 2004;16(3):293–9.
Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet. 2006;79(2):370–7.
Sandberg M, Flandin P, Silberberg S, Su-Feher L, Price JD, Hu JS, et al. Transcriptional networks controlled by NKX2-1 in the development of forebrain GABAergic neurons. Neuron. 2016;91(6):1260–75.
Konno D, Kishida C, Maehara K, Ohkawa Y, Kiyonari H, Okada S, et al. Dmrt factors determine the positional information of cerebral cortical progenitors via differential suppression of homeobox genes. Development. 2019;146(15):174243.
Baldelli P, Meldolesi J. The transcription repressor REST in adult neurons: physiology, pathology, and diseases. eNeuro. 2015;2(4).
Oppel F, Ki DH, Zimmerman MW, Ross KN, Tao T, Shi H, et al. suz12 inactivation in p53- and nf1-deficient zebrafish accelerates the onset of malignant peripheral nerve sheath tumors and expands the spectrum of tumor types. Dis Model Mech. 2020;13(8):042341.
Gates KP, Mentzer L, Karlstrom RO, Sirotkin HI. The transcriptional repressor REST/NRSF modulates hedgehog signaling. Dev Biol. 2010;340(2):293–305.
Kubara K, Yamazaki K, Ishihara Y, Naruto T, Lin HT, Nishimura K, et al. Status of KRAS in iPSCs impacts upon self-renewal and differentiation propensity. Stem Cell Reports. 2018;11(2):380–94.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76.
Yu Z, Feng J, Wang W, Deng Z, Zhang Y, Xiao L, et al. The EGFR-ZNF263 signaling axis silences SIX3 in glioblastoma epigenetically. Oncogene. 2020;39(15):3163–78.
Wells T, Rough K, Carter DA. Transcription mapping of embryonic rat brain reveals EGR-1 induction in SOX2 neural progenitor cells. Front Mol Neurosci. 2011;4:6.
Pauklin S, Madrigal P, Bertero A, Vallier L. Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes Dev. 2016;30(4):421–33.
Tapias A, Lazaro D, Yin BK, Rasa SMM, Krepelova A, Kelmer Sacramento E, et al. HAT cofactor TRRAP modulates microtubule dynamics via SP1 signaling to prevent neurodegeneration. Elife. 2021;10:e61531.
Johnson CA, Ghashghaei HT. Sp2 regulates late neurogenic but not early expansive divisions of neural stem cells underlying population growth in the mouse cortex. Development. 2020;147(4):dev186056.
Mulligan P, Westbrook TF, Ottinger M, Pavlova N, Chang B, Macia E, et al. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol Cell. 2008;32(5):718–26.
Infantino V, Convertini P, Iacobazzi F, Pisano I, Scarcia P, Iacobazzi V. Identification of a novel Sp1 splice variant as a strong transcriptional activator. Biochem Biophys Res Commun. 2011;412(1):86–91.
Frega M, Linda K, Keller JM, Gumus-Akay G, Mossink B, van Rhijn JR, et al. Neuronal network dysfunction in a model for Kleefstra syndrome mediated by enhanced NMDAR signaling. Nat Commun. 2019;10(1):4928.
Bock I, Nemeth K, Pentelenyi K, Balicza P, Balazs A, Molnar MJ, et al. Targeted next generation sequencing of a panel of autism-related genes identifies an EHMT1 mutation in a Kleefstra syndrome patient with autism and normal intellectual performance. Gene. 2016;595(2):131–41.
Mitra AK, Dodge J, Van Ness J, Sokeye I, Van Ness B. A de novo splice site mutation in EHMT1 resulting in Kleefstra syndrome with pharmacogenomics screening and behavior therapy for regressive behaviors. Mol Genet Genomic Med. 2017;5(2):130–40.
Kleefstra T, Kramer JM, Neveling K, Willemsen MH, Koemans TS, Vissers LE, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet. 2012;91(1):73–82.
Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16(22):2893–905.
Chen ES, Gigek CO, Rosenfeld JA, Diallo AB, Maussion G, Chen GG, et al. Molecular convergence of neurodevelopmental disorders. Am J Hum Genet. 2014;95(5):490–508.
Watson ZL, Yamamoto TM, McMellen A, Kim H, Hughes CJ, Wheeler LJ, et al. Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin Epigenet. 2019;11(1):165.
Lian G, Wong T, Lu J, Hu J, Zhang J, Sheen V. Cytoskeletal associated filamin A and RhoA affect neural progenitor specification during mitosis. Cereb Cortex. 2019;29(3):1280–90.
Mossahebi-Mohammadi M, Quan M, Zhang JS, Li X. FGF signaling pathway: a key regulator of stem cell pluripotency. Front Cell Dev Biol. 2020;8:79.
Wang XL, Ma YX, Xu RJ, Ma JJ, Zhang HC, Qi SB, et al. c-Myc controls the fate of neural progenitor cells during cerebral cortex development. J Cell Physiol. 2020;235(4):4011–21.
Chuang YS, Huang WH, Park SW, Persaud SD, Hung CH, Ho PC, et al. Promyelocytic leukemia protein in retinoic acid-induced chromatin remodeling of Oct4 gene promoter. Stem Cells. 2011;29(4):660–9.
Liu X, Yang C, Sun X, Lin X, Zhao L, Chen H, et al. Evidence for a novel mechanism of influenza A virus host adaptation modulated by PB2-627. FEBS J. 2019;286(17):3389–400.
Ravache M, Weber C, Merienne K, Trottier Y. Transcriptional activation of REST by Sp1 in Huntington’s disease models. PLOS ONE. 2010;5(12):e14311.
Paonessa F, Latifi S, Scarongella H, Cesca F, Benfenati F. Specificity protein 1 (Sp1)-dependent activation of the synapsin I gene (SYN1) is modulated by RE1-silencing transcription factor (REST) and 5’-cytosine-phosphoguanine (CpG) methylation. J Biol Chem. 2013;288(5):3227–39.
Acknowledgements
This study makes use of cell lines generated by the HipSci Consortium (Human Induced Pluripotent Stem Cell Initiative (WT098503; http://www.hipsci.org) funded by Wellcome and the MRC. Library preparation and Sequencing was conducted in the Genomics WA Laboratory in Perth, Australia. This facility is supported by funding from Bioplatforms Australia, State Government of Western Australia, Harry Perkins Institute of Medical Research, Telethon Kids Institute, and the University of Western Australia.
Funding
McCusker Charitable Foundation. TL is supported by a Fellowship from the Feilman Foundation.
Author information
Authors and Affiliations
Contributions
VF and TL designed the research. VF, CF, GS, and NS performed the laboratory experiments. VF, TL, DA, and SR analysed the data. VF wrote the paper in conjunction with TL, DA and SR. GB provided, the patient for recruitment and, clinical characterization of disease. VF, SJ and MW were responsible for patient recruitment. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The patient recruitment to this study was initiated by a genetic counsellor based at Genome Services Western Australia, followed by written informed consent. The study is in line with the Declaration of Helsinki and the NHMRC National Statement on Ethical Conduct Human Ethics Research. Approved by the Child and Adolescent Health Services, Human Research Ethics Committee, RGS0000000166.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
. Differential gene expression in iPS cells and NPCs with EHMT1_WT and EHMT1_SNV. a, schematic of CRISPR gene edit in EHMT1. C > T mutation indicated in yellow and capital letters indicate silent mutations. The iPS cell clones for EHMT1_WT and EHMT1_SNV were differentiated to NPCs EHMT1_WT, and EHMT1_SNV. b Principal component analysis. c Euclidean distance between samples
Additional file 2: Table S1
. EHMT1 off target. Table S2. HPO Terms for patient. Table S3. Enrichr Diff in Diffn EHMT1_SNV v EHMT1_WT. Table S4. GSEA Diff in Diffn Suppl Tables_EHMT1_SNV v EHMT1_WT. Table S5. Transcription factor tables Diff in Diffn_AME DREME MEME. Table S6. DTE Diff in Diffn.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fear, V.S., Forbes, C.A., Anderson, D. et al. CRISPR single base editing, neuronal disease modelling and functional genomics for genetic variant analysis: pipeline validation using Kleefstra syndrome EHMT1 haploinsufficiency. Stem Cell Res Ther 13, 69 (2022). https://doi.org/10.1186/s13287-022-02740-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-022-02740-3