
Abstract
Background
Adipose tissue-derived mesenchymal stromal cells (ASCs) have been shown to exhibit some promising properties of their use in regenerative medicine as advanced therapy medicinal products (ATMP). However, different sources of their origin, methods of isolation, and expansion procedures cause the laboratory and clinical results difficult to compare.
Methods
ASCs were isolated from lipoaspirates and cultured in three different medium formulations: αMEM and DMEM as a basal medium supplemented with 10% of human platelet lysate (hPL) and DMEM supplemented with 20% fetal bovine serum (FBS) and bFGF as a gold standard medium. Subsequently, the impact of culture media on ASCs growth kinetics, their morphology and immunophenotype, ability to differentiate, clonogenic potential, and secretion profile was evaluated.
Results
All cultured ASCs lines showed similar morphology and similar clonogenic potential and have the ability to differentiate into three lines: adipocytes, osteoblasts, and chondroblasts. The immunophenotype of all cultured ASCs was consistent with the guidelines of the International Society for Cell Therapy (ISCT) allowing to define cells as mesenchymal stromal cell (MSC) (≥ 95% CD105, CD73, CD90 and ≤ 2% CD45, CD34, CD14, CD19, HLA-DR). The immunophenotype stabilized after the second passage and did not differ between ASCs cultured in different conditions. The exception was the ASCs grown in the presence of FBS and bFGF, which expressed CD146 antigens. The secretion profile of ASCs cultured in different media was similar. The main secreted cytokine was IL-6, and its level was donor-specific. However, we observed a strong influence of the medium formulation on ASCs growth kinetics. The proliferation rate of ASCs in medium supplemented with hPL was the highest.
Conclusions
Culture media that do not contain animal-derived antigens (xeno-free) can be used to culture cells defined as MSC. Xeno-free medium is a safe alternative for the production of clinical-grade MSC as an advanced therapy medicinal product. Additionally, in such culture conditions, MSC can be easily expanded in accordance with the Good Manufacturing Process (GMP) requirements to a desired amount of cells for clinical applications.
Similar content being viewed by others


Background
Mesenchymal stromal cells (MSC) have been isolated from various tissues and organs throughout the human body [1]. The exceptional ease of their isolation and ex vivo expansion additional to their ascribed regeneration potential led to the extremely broad application of MSC in a clinical trial [2] and subsequently led them to clinical medicine as advanced therapy medicinal products (ATMP) [3, 4]. However, many concerns and controversies arising from their clinical application in the so-called cell therapy are still not solved [5, 6]. The major issue is the heterogeneity of MSC population [7]. Despite meeting all the criteria of the International Society for Cellular Therapy (ISCT) [8], namely (1) plastic adherence in standard culture conditions, (2) defined expression of specific markers (CD73, CD90, CD105) along with lack of expression of hematopoietic markers (CD14, CD34, CD45, HLA-DR) and (3) differentiation potential into adipocytes, osteoblasts, and chondroblasts in vitro, MSC may differ in secretome and/or transcriptome profile depending on the donor of the tissue or the site of collection [5, 9, 10]. An additional drawback is the isolation and expansion protocols varying between laboratories. Different medium formulations used to culture MSC, especially animal-derived supplements used in MSC manufacturing, may lead to clinical complication and data misinterpretation. Accordingly, to consider MSC as an ATMP, precise thorough characteristics, large-scale quality, and relatively low-cost production of clinical-grade MSC need to be developed in xeno-free media.
In the study, MSC derived from human adipose tissues (in the paper, ASCs acronym will be used [11]) obtained by liposuction procedure were cultured in three different medium formulations: (1) αMEM supplemented with 10% human platelet lysate (hPL), (2) DMEM supplemented with 10% hPL, and (3) DMEM supplemented with 20% fetal bovine serum (FBS) and basic fibroblast growth factor (bFGF). Subsequently, we evaluated the effect of culture media on ASCs growth kinetics, their morphology and phenotype, ability to differentiate, clonogenic potential, and secretome profile.
No differences were found in the morphology, phenotype, differentiation potential, ability to clone forming, and secretome profile in all cultured ASCs. However, secretion of the most abundant cytokine (IL-6) was donor-specific. The growth kinetics was affected by medium formulation. The highest proliferation rate was observed in ASCs cultured in αMEM supplemented with 10% hPL medium.
Methods
Biological material used in the study
The material used in the study comprised of liposuction aspirates (n = 6) obtained from the Hospital for Minimally Invasive and Reconstructive Surgery in Bielsko-Biała. All female donors (mean age = 45 (range from 27 to 60 years old), BMI = 24.68 ± 2.45) undergo tumescent liposuction under general anesthesia performed by the same plastic surgeon. The area of liposuction in all cases was the abdomen. The donor site was previously infiltrated with Klein solution (1000 ml normal saline, 50 ml 1% lignocaine, 1 ml 1:1000 epinephrine, 12.5 ml sodium bicarbonate) and left for 10 min. Liposuction was performed using 3.0-mm 3-hole blunt tip cannula under low pressure (below 0.5 bar). Aspirate was collected directly to a sterile container. The experiments were performed in accordance with the Declaration of Helsinki, with the approval of the Institutional Review Board and Bioethical Committee (KB/430-62/13). All patients provided written informed consent for the collection of liposuction aspirates and subsequent analysis.
ASCs isolation and culture
The obtained liposuction aspirates were washed with PBS− (Gibco BRL, Paisley, UK) supplemented with 1% FBS (Gibco) until the remaining blood was removed. Fat tissue was digested with collagenase NB4 solution (0.39 U/ml, Serva Electrophoresis, Heidelberg, Germany) for 1 h at 37 °C. After digestion, the cell suspension was filtered through 70-μm and 40-μm strainers. The suspension of single cells (16.6 × 103 cells/cm2) was seeded on plastic culture plates in 3 different culture media: (1) αMEM medium (Macopharma, Tourcoing, France), supplemented with 10% human platelet lysate (Macopharma); heparin (2 U/ml, Polfa, Warszawa, Poland); and penicillin-streptomycin (Sigma Aldrich, St. Louis, MO, USA); (2) DMEM supplemented with 10% human platelet lysate (Macopharma), heparin (2 U/ml, Polfa), and penicillin-streptomycin (Sigma Aldrich); and (3) DMEM supplemented with 20% FBS (Gibco), basic fibroblast growth factor (bFGF, 10 ng/ml, eBioscience, San Diego, CA, USA), and penicillin-streptomycin (Sigma Aldrich) [12]. After 72 h, the plates were rinsed with a fresh culture medium. Cell cultures were grown in an incubator under standard culture conditions (at 37 °C in 5% CO2). Cells were passaged at 80–90% confluency using 0.25% trypsin solution (Sigma Aldrich). ASCs from passage 2 were used for subsequent experiments.
ASCs immunophenotypic analysis and differentiation analysis
The phenotype of ASCs was determined directly after cell isolation and in the subsequent passages (from 0 to 4 passages) by flow cytometer (BD FACSCanto™, BD, Franklin Lakes, NJ, USA) using specific antibodies. The cells were incubated with appropriate antibodies directed against the following human antigens: CD29-FITC (eBioscience), CD105-APC, CD73-PE, CD90-PE-Cy7, CD44-FITC, CD34-PE-Cy7, CD31-FITC, CD146-PE, KDR-PE, LIN-FITC, CD45-PE, and HLA-DR-PE-Cy7 (BD Biosciences, San Jose, CA, USA) or isotype-matched control antibodies were used. 7-Amino-actinomycin D (7AAD) (eBioscience) was used to stain non-viable cells just before running the flow analysis.
The Human Mesenchymal Stem Cell Functional Identification Kit (R&D Systems, Minneapolis, USA) was used to differentiate ASCs into adipocytes, osteoblasts, and chondroblasts. The procedure was performed in accordance with the manufacturer’s instructions. Adipocytes were stained with FABP4 antibody (Abcam, Cambridge, UK), and a secondary antibody linked to FITC fluorochrome (Fluorescent anti-rabbit IgG Kit, Vector Laboratories, Burlingame, CA, USA). Fluorescence imaging of stained cells was performed using a LSM710 confocal microscope (Carl Zeiss Microscopy GmbH). The ability of the cells to differentiate into chondroblasts and osteoblasts was assessed by histochemical staining using Safranin O and Alizarin Red, respectively (Sigma Aldrich). The stained cells were visualized using an Eclipse 80i microscope (Nikon Instruments Inc., Melville, NY, USA).
Assessment of ASCs clonogenic potential
ASCs from 3 different culture media were treated with 0.25% trypsin solution and placed in a corresponding culture medium in plates at a density of 1 cell/3 cm2. ASCs cultures were grown in an incubator under standard culture conditions. After 10 days, the plates were washed twice with PBS− buffer and stained with Giemsa solution for 15 min at room temperature. Arising clones were visualized using Eclipse 80i microscope. Clones consisting of more than 50 cells were counted. Clonogenic potential of cells was calculated using the formula:
Analysis of ASCs secretome
ASCs (second passage) were plated at high density in a complete medium. On the next day of the culture, the medium was replaced with a medium without serum or hPL. Following 48 h, the medium was collected. The type and quantity of cytokines and growth factors secreted by ASCs were assessed using the Human Cytokine Antibody Array C5 kit (RayBiotech, Norcross, GA, USA). The analysis was conducted in accordance with the manufacturer’s instructions. Densitometry calculations were performed using an ImageJ 1.48y software (NIH) to analyze the quantity of cytokines and growth factors secreted by ASCs.
The determination of interleukin 6 secreted by ASCs in vitro
The concentration of IL-6 secreted by ASCs was determined by ELISA (eBioscience) in accordance with the manufacturer’s instruction. The total protein in the homogenates was determined by the Bradford method.
Results
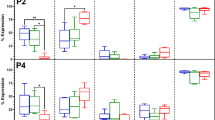
Adipose tissue collected by liposuction was obtained from the Hospital for Minimally Invasive and Reconstructive Surgery, Bielsko-Biała, Poland. For the purpose of this paper, only the abdominal fat was used. We isolated cells from six healthy female donors under 60 years. After isolation, before seeding on plates, cells were immunophenotyped by flow cytometry using the specific antibodies. Flow cytometry analysis showed that freshly isolated adipose-derived cells, following collagenase digestion, did not possess phenotype of mesenchymal stromal/stem cells according to the guidelines of the International Society of Cell Therapy. Freshly isolated, uncultured cells expressed low level of mesenchymal markers such as CD105 (only 32% of cells), CD73 (47% of cells), CD90 (67% of cells), CD29 (69% of cells), and CD44 (89% of cells) and possessed antigen CD34 (over 60% of cells), HLA-DR (21% of cells), CD31 (15%), and Lin (14%) (Fig. 1). Subsequently, isolated cells were seeded into three different culture media: αMEM+10%hPL, DMEM+10%hPL, and DMEM+20%FBS+bFGF. Morphologically, plastic adherent cells, cultured in FBS-supplemented medium revealed more flattened shapes, whereas in media supplemented with hPL, the cells were more elongated and spindle-shaped (Fig. 2).

ASCs surface markers expression. The plots show the phenotype of ASCs obtained directly after ASCs isolation (before seeding on plates) and in the subsequent passages. Passage “0” indicates cells seeded after isolation that were trypsinizated when obtained 80–90% confluency. The cell phenotype stabilizes from passage 2 (expression of surface markers according to the guidelines of ISCT). The graphs summarized the surface marker expression on cells obtained from six isolation from different donors (n = 6)

The morphology of ASCs cultured in three different medium formulations. a Phase-contrast images of ASCs at passage 1 (40% of confluency, 3–5 days of culture) parallel cultivated in either FBS- or hPL-supplemented basal medium. b Slight differences in the morphology between ASCs cultured in three different medium formulations were confirmed by flow cytometry gating of forward vs side scatter (FSC vs SSC). ASCs cultured in media supplemented with hPL were smaller and exhibited less complexity (granularity)
The phenotype of cultured cells changed with time and passages. Seven to 10 days after plating, when ASCs reached 80–90% confluency (passage “0,” Fig. 1), the cells were harvested and immunophenotyped again. ASCs still possessed CD34 antigens (over 50% of cells in each culture), but the expression of all mesenchymal markers increased (over 90% of cells in each culture). At passage “1,” ASCs in each culture still expressed low level of CD34 marker (~ 5% of cells) and more than 95% of cells expressed mesenchymal markers. Starting from passage “2,” phenotype of ASCs stabilizes, exhibiting mesenchymal cells features. The unique observation was the expression of CD146 antigen in cells cultured in DMEM+20%FBS+bFGF medium. The percentage of CD146+ cells varied from 4.5 to 55.4% and remained at a similar level throughout cell culture.
The ability of ASCs cultured in three different medium formulations to differentiate into adipocytes, osteoblasts, and chondroblasts in vitro was tested on ASCs harvested from the second passage. After 21 days in adipogenic medium, characteristic lipid droplets inside the cells were observed and almost all ASCs were differentiated into adipocytes (FABP4-positive cells; Fig. 3). After 21 days in osteogenic medium, cultured cells showed typical cuboidal and flattened osteoblastic morphology and extracellular calcium deposits were stained with Alizarin Red. After 21 days in chondrogenic medium, we could detect cartilaginous proteoglycans stained by Safranin O. We did not observe differences among tested medium formulations in terms of differentiation towards adipocytes, osteocytes, and chondroblasts.

Differentiation analysis of ASCs. ASCs cultured in three different media were harvested and plated in differentiation media. ASCs differentiated in vitro into adipocytes (FABP4, green, n = 4, magnification × 10), osteoblasts (Alizarin Red, red, n = 4, magnification × 4), and chondroblasts (Safranin O, dark red, n = 4, magnification × 20)
In the study, we determined the clonogenic potential of ASCs cultured in three different medium formulations. The medium formulation did not influence the potential of ASCs to create clones. However statistically insignificant, the highest rate of cloning capacity showed ASCs cultured in αMEM medium supplemented with 10% hPL. On average, 26% of these cells were able to form clones (Fig. 4).

Clonogenic potential of ASCs. ASCs cultured in three different medium formulations exhibit similar clonogenic potential (n = 4). However statistically insignificant, more clones were obtained from single cells cultured on αMEM+10% hPL medium
ASCs cultured in three different media exhibited similar secretion profile. ASCs cultured for 48 h in media deprived of serum or platelet lysate secreted mainly interleukin 6, RANTES, osteoprotegerin, MCP-1, TGF-β2, and GRO particles (Fig. 5a). Nevertheless, IL-6 was the dominant cytokine secreted by ASCs in all culture conditions. However, the secretion of IL-6 was highly associated with a donor from whom adipose tissue was obtained. The graph (Fig. 5b) depicted the amount of secreted IL-6 in respective culture conditions depending on the tissue donor. It is clearly visible that medium formulation does not influence IL-6 secretion and is only donor-specific. Additionally, some of the ASCs did not secrete detectable amounts of IL-6 (data not shown) or secrete only a little.

Secretome of ASCs. a During 48 h culture, ASCs secreted into the culture medium mainly IL-6 and small amounts of RANTES, osteoprotegerin, MCP-1, and GRO particles. The graph shows an averaged level of secreted cytokines by ASCs (n = 3). b The secretion of the most abundant cytokine—IL-6—was donor-specific, and the medium formulation did not influence its secretion. The amount of secreted IL-6 based on the ELISA assay (n = 4) performed in respective cell culture
Obtaining a sufficient number of cells adequate for cellular therapies is a crucial task in the stem cell research field. The key point is to yield a large number of cells starting with the smallest volume of tissue as possible. We evaluated the proliferation rate of cultured ASCs in three different medium formulations. Based on the number of cells obtained during cell cultures, from the initial number of one million seeded ASCs—it is approximately 2 ml of adipose tissue—we may obtain almost 50 million of ASCs in αMEM medium supplemented with 10% hPL within approximately 4 weeks. Indeed, the highest proliferation rate was observed in αMEM medium supplemented with 10% hPL, lower was in DMEM medium supplemented with 10% hPL, and the lowest in DMEM supplemented with 20% FBS and bFGF (Fig. 6). Interestingly, the proliferation rate in each culture condition was similar in the first passages; just in passage “2” the proliferation accelerated in αMEM+10% hPL medium and consequently the number of yield ASCs was the highest.

The evaluated number of cells that may be obtained in three different culture conditions. The highest yield of seeded cells may be achieved in cell culture carried out in αMEM supplemented with 10% hPL. After approximately 4 weeks, from 1 million of isolated ASCs, almost 50 million of cells may be obtained
Discussion
In this work, we have tested three different medium formulations for adipose-derived mesenchymal stromal cell (ASCs) culture. The first one consisted of α-modified Minimum Essential Medium (αMEM) as the basal medium supplemented with 10% human platelet lysate (hPL). Basal medium and hPL supplementation were obtained from Macopharma® who developed clinical-grade serum-free and xeno-free culture media dedicated to specific stem cell populations such as MSC. GMP-grade hPL is required for expanding cells for clinical use [13]. Further, hPL is currently the most promising supplement replacing FBS [13, 14]. The second medium formulation consisted of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% hPL. The last tested medium contained DMEM supplemented with 20% FBS and 10 ng/ml bFGF [12]. The above basal media are the most often used for MSC culture [15,16,17,18,19]. It was also shown that culture media based on αMEM are more suitable for both isolation and expansion of MSC [20]. We aimed to select the most effective medium formulation for ASCs expansion. Additionally, we intended to check ASCs growth condition in xeno-free culture media in order to obtain a large number of clinical-grade ASCs. FBS is typically used to expand MSC, and there were conducted successful clinical trials involving MSC expanded in FBS-containing media [14] although it is critically rated by European Medicines Agency [14]. There are severe concerns about the use of FBS for clinical applications. Growth medium consisting of animal-derived serum may lead to the introduction of xenogeneic antigens with MSC transplant and host immune rejection [21,22,23,24], possible contamination with non-human pathogens (viruses, prions, mycoplasma) and endotoxins [25,26,27]. FBS has also lot-to-lot variability—the concentration of growth factors may differ [27]. An ideal FBS alternative for clinical GMP production should possess well-defined composition, a reduced degree of contaminants and no risk of xenogeneic compound transmission, low production costs, easy availability, and no ethical issues [17]. Human platelet lysate meets most of these requirements and additionally contains a high level of growth factors (such as bFGF, EGF, HGF, IGF-1, PDGF, TGFβ1, VEGF) [14, 28]. Existing composition variability, which is donor-related, may be reduced by pooling different donations [29]. Possible transmission of human diseases may be overcome by virus inactivation through short-wave ultraviolet light (UV-C) [30].
By now, many reports have been published evaluating the use of hPL or other xeno-free supplements for MSC ex vivo expansion according to GMP-grade protocols [31,32,33,34]. In such studies, FBS-supplemented media represent a gold standard medium for MSC culture. It has been shown that substitution of FBS by human platelet lysate increases cell proliferation without affecting MSC immunophenotype, immunomodulatory potential, differentiation potential, and relative telomere length [34]. Indeed, we observed the highest proliferation rate of ASCs cultured in αMEM supplemented with 10% hPL; the lower rate was in DMEM supplemented with 10% hPL. Hence, the substitution of FBS by hPL accelerates the production of clinical-grade MSC in therapeutically relevant numbers [35, 36]. According to our isolation and expansion procedure, in 4 weeks, 50 million of ASCs may be obtained from a small volume of adipose tissue (2 ml). It seems also important for clinical use to not affect the immunological profile of cultured MSC. We did not observe the induction of HLA-DR antigens in all culture condition, but Sotiropoulou et al. indicated that bFGF supplementation may increase HLA class I expression [20]. There are also discrepancies between researches on immunosuppressive properties of MSC cultured in hPL-supplemented media. Some indicating that hPL-supplemented media abolish their immunosuppressive features [16], and other claims that hPL maintain these immunosuppressive properties [17]. Thus, the effect of platelet lysate on immunomodulatory characteristics of MSC remains controversial and needs to be further investigated.
Since the therapeutic potential of MSC relies on their ability to secrete a variety of soluble factors [37], we evaluated ASCs secretion profile cultured in three different medium formulations. We examine a panel of 80 secreted cytokines and growth factors. ASCs secreted into the culture medium mainly IL-6 and small amounts of chemokine CCL5 (RANTES), osteoprotegerin (OPG), MCP-1, and GRO particles. Secretion profile was unaffected by culture conditions. The most abundant cytokine was IL-6. We described earlier that MSC which secreted IL-6 through macrophages M1 ➔ M2 polarization facilitated fast functional recovery of ischemic limb and post-infarcted heart and promoted angiogenesis [12, 38]. Further, OPG cytokine (a member of the tumor necrosis factor receptor (TNFR) superfamily) stimulates the secretion of IL-6 cytokine [39].GRO particles are associated with angiogenesis promotion [40]. Therefore, secreted cytokines stimulate mainly the formation of reparative M2 macrophages and angiogenesis. However, the observed ASCs secretion profile was different than described by others [41]. It was presented that ASCs secrete mainly proangiogenic or immunomodulatory factors, such as VEGF, HGF, TGFβ, PGE2, and bFGF [42, 43]. Thus, recently, MSC has gained another moniker: medicinal signaling cells [44]. According to Caplan, medicinal signaling cells home to the sites of injured tissues and secrete factors that modulate the immune response, reduce inflammation, promote wound healing, and inhibit cell death [44, 45].
We and others [46] have shown that cytokine and growth factor secretion is donor-specific. The level of secreted IL-6 in some cases was almost at the limit of detection or even beyond, in the other was much higher. Additionally, Serra et al. have shown that cellular passaging does not influence significantly ASCs secretome properties [47]. It seems that the donor of the tissue, tissue collection method, and what is more isolation and expansion protocols influence secretion and transcription profile of isolated MSC, causing a difficult issue to compare the results between different laboratories. Additionally, Stępniewski et al. recently revealed that cells expressing the same mesenchymal markers and isolated from human heart tissue (right ventricle and epicardial fat) have distinct transcriptomes [10]. Taken together our observations concerning donor-specific secretion profile, further treatments based on mesenchymal cells need to be reevaluated.
Subcutaneous fat has become an alternative tissue source for stromal cells for regenerative medicine [48]. Adipose-derived cell therapy has shown potential in almost every preclinical animal model. It resulted in a robust clinical application often without adequate analysis of ASCs properties [6, 9]. US National Library of Medicine (clinicaltrials.gov) identified ongoing or completed 242 clinical trials with keywords “adipose derived stem cells” (905 studies found for: “mesenchymal stem cell”). To date, the majority of these clinical trials are at early phase 1 and phase 1 (only 4 trails currently at phase 4). Adipose-derived cell therapy has so far shown a favorable safety profile, but the safety assessment description has been of poor quality [49]. Safety concerns include the risk of thromboembolic complications, the use of allogeneic cells and possible rejection, and in the setting of previous cancer therapy. Subsequently, most of the studies may be classified as levels 3–4 based on the criteria according to the Center for Evidenced Based Medicine (poorly controlled case series—individual case-controlled study). In general, it is recognized as low-quality studies with increased risk of bias, which leads to an increasing chance of findings that do not represent reality [49, 50]. Therefore, it is important to design well-conducted randomized controlled trials with adequate blinding, including a placebo/sham treatment, and the safety assessment together with the application of clinical-grade ASCs [51].
Conclusions
Taken together, our results indicate that xeno-free media do not alter the typical for mesenchymal cell expression pattern, multipotentiality, and secretion profile. However, the proliferation rate is higher when compared with conventionally used medium formulation containing xenogeneic serum. Thus, the media containing hPL as a serum xeno-free substitute are suitable for clinical use when in a relatively short time, the large-scale expansion is required.
Nevertheless, there is a great need to improve the understanding of the biology of cells currently termed MSC. The main concern is the severe difference in the gene expression, transcription and secretion profile, and differentiation potential between cells isolated from different tissues, different donors, or even different regions of the same tissue. Still, despite their heterogeneity, indisputably is their ability to secrete cocktails of proteins that modulate the immune response, reduce inflammation, promote wound healing, and inhibit cell death. What we need to do is to resolve the true nature of MSC and conditions which will allow to culture MSC as a precise and established cell product suitable for clinical use.
Availability of data and materials
Not applicable.
Abbreviations
- ASCs:
-
Adipose tissue-derived mesenchymal stromal cells
- ATMP:
-
Advanced therapy medicinal products
- bFGF:
-
Basic fibroblast growth factor
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- FABP4:
-
Fatty acid-binding protein 4
- FBS:
-
Fetal bovine serum
- GMP:
-
Good Manufacturing Practice
- GRO:
-
Growth-regulated protein
- hPL:
-
Human platelet lysate
- IL-6:
-
Interleukin 6
- ISCT:
-
International Society for Cell Therapy
- MCP-1:
-
Monocyte chemoattractant protein-1
- PBS:
-
Phosphate-buffered saline
- RANTES:
-
Regulated on Activation, Normal T cell Expressed and Secreted
- TGF-β2:
-
Transforming growth factor beta 2
- αMEM:
-
Minimum Essential Medium Eagle Alpha Modification
References
Marquez-Curtis LA, Janowska-Wieczorek A, McGann LE, Elliott JA. Mesenchymal stromal cells derived from various tissues: biological, clinical and cryopreservation aspects. Cryobiology. 2015;71:181–97.
Squillaro T, Peluso G, Galderisi U. Clinical trials with mesenchymal stem cells: an update. Cell Transplant. 2016;25:829–48.
Maciulaitis R, D’Apote L, Buchanan A, Pioppo L, Schneider CK. Clinical development of advanced therapy medicinal products in Europe: evidence that regulators must be proactive. Mol Ther. 2012;20:479–82.
Confalonieri D, Schwab A, Walles H, Ehlicke F. Advanced therapy medicinal products: a guide for bone marrow-derived MSC application in bone and cartilage tissue engineering. Tissue Eng Part B Rev. 2018;24:155–69.
Pittenger MF, Le Blanc K, Phinney DG, Chan JKY. MSCs: scientific support for multiple therapies. Stem Cells Int. 2015;2015:280572.
Langrzyk A, Nowak WN, Stępniewski J, Jaźwa A, Florczyk-Soluch U, Józkowicz A, Dulak J. Critical view on mesenchymal stromal cells in regenerative medicine. Antioxid Redox Signal. 2018;29:169–90.
Sacchetti B, Funari A, Remoli C, Giannicola G, Kogler G, Liedtke S, Cossu G, Serafini M, Sampaolesi M, Tagliafico E, Tenedini E, Saggio I, Robey PG, Riminucci M, Bianco P. No identical “mesenchymal stem cells” at different times and sites: human committed progenitors of distinct origin and differentiation potential are incorporated as adventitial cells in microvessels. Stem Cell Reports. 2016;6:897–913.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop DJ, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–7.
Martin I, Galipeau, Kessler, Blanc L, Dazzi. Challenges for mesenchymal stromal cell therapies. Sci Transl Med. 2019;11:eaat2189.
Stępniewski J, Florczyk-Soluch U, Szade K, Bukowska-Strakova K, Czapla J, Matuszczak S, Jarosz-Biej M, Langrzyk A, Tomczyk M, Rumieńczyk I, Kulecka M, Mikuła M, Ostrowski J, Jaźwa-Kusior A, Zembala M, Józkowicz A, Zembala MO, Dulak J. Transcriptomes of human mesenchymal cells isolated from the right ventricle and epicardial fat differ strikingly both directly after isolation and long-term culture. ESC Heart Fail. 2019. https://doi.org/10.1002/ehf2.12397.
Bourin P, Bunnell BA, Casteilla L, Dominici M, Katz AJ, March KL, Redl H, Rubin JP, Yoshimura K, Gimble JM. Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: a joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT). Cytotherapy. 2013;15:641–8.
Czapla J, Matuszczak S, Wiśniewska E, Jarosz-Biej M, Smolarczyk R, Cichoń T, Głowala-Kosińska M, Śliwka J, Garbacz M, Szczypior M, Jaźwiec T, Langrzyk A, Zembala M, Szala S. Human cardiac mesenchymal stromal cells with CD105+CD34- phenotype enhance the function of post-infarction heart in mice. PLoS One. 2016;11:e0158745.
Strunk D, Lozano M, Marks DC, Loh YS, Gstraunthaler G, Schennach H, Rohde E, Laner-Plamberger S, Öller M, Nystedt J, Lotfi R, Rojewski M, Schrezenmeier H, Bieback K, Schäfer R, Bakchoul T, Waidmann M, Jonsdottir-Buch SM, Montazeri H, Sigurjonsson OE, Iudicone P, Fioravanti D, Pierelli L, Introna M, Capelli C, Falanga A, Takanashi M, Lόpez-Villar O, Burnouf T, Reems JA, Pierce J, Preslar AM, Schallmoser K. International forum on GMP-grade human platelet lysate for cell propagation: summary. Vox Sang. 2018;13:80–7.
Bieback K. Platelet lysate as replacement for fetal bovine serum in mesenchymal stromal cell cultures. Transfus Med Hemother. 2013;40:326–35.
Inamdar AA, Inamdar AC. Culture conditions for growth of clinical grade human tissue derived mesenchymal stem cells: comparative study between commercial serum-free media and human product supplemented media. J Regenerative Med Tissue Eng. 2013. https://doi.org/10.7243/2050-1218-2-10.
Oikonomopoulos A, van Deen WK, Manansala AR, Lacey PN, Tomakili TA, Ziman A, Hommes DW. Optimization of human mesenchymal stem cell manufacturing: the effects of animal/xeno-free media. Sci Rep. 2015;5:16570.
Cimino M, Gonçalves RM, Barrias CC, Martins MCL. Xeno-free strategies for safe human mesenchymal stem/stromal cell expansion: supplements and coatings. Stem Cells Int 2017:6597815. doi: https://doi.org/10.1155/2017/6597815.
Fernandez-Rebollo E, Mentrup B, Ebert R, Franzen J, Abagnale G, Sieben T, Ostrowska A, Hoffmann P, Roux PF, Rath B, Goodhardt M, Lemaitre JM, Bischof O, Jakob F, Wagner W. Human platelet lysate versus fetal calf serum: these supplements do not select for different mesenchymal stromal cells. Sci Rep. 2017;7:5132.
Kandoi S, Praveen kumar L, Patra B, Vidyasekar P, Sivanesan D, Vijayalakshmi S, Rajagopal K, Verma RS. Evaluation of platelet lysate as a substitute for FBS in explant and enzymatic isolation methods of human umbilical cord MSCs. Sci Rep. 2018;8:12439.
Sotiropoulou PA, Perez SA, Salagianni M, Baxevanis CN, Papamichail M. Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells. 2006;24:462–71.
Spees JL, Gregory CA, Singh H, Tucker HA, Peister A, Lynch PJ, Hsu SC, Smith J, Prockop DJ. Internalized antigens must be removed to prepare hypoimmunogenic mesenchymal stem cells for cell and gene therapy. Mol Ther. 2004;9:747–56.
Gregory CA, Reyes E, Whitney MJ, Spees JL. Enhanced engraftment of mesenchymal stem cells in a cutaneous wound model by culture in allogenic species-specific serum and administration in fibrin constructs. Stem Cells. 2006;24:2232–43.
Sundin M, Ringdén O, Sundberg B, Nava S, Götherström C, Le Blanc K. No alloantibodies against mesenchymal stromal cells, but presence of anti-fetal calf serum antibodies, after transplantation in allogeneic hematopoietic stem cell recipients. Haematologica. 2007;92:1208–15.
Haque N, Kasim NH, Rahman MT. Optimization of pre-transplantation conditions to enhance the efficacy of mesenchymal stem cells. Int J Biol Sci. 2015;11:324–34.
Wessman SJ, Levings RL. Benefits and risks due to animal serum used in cell culture production. Dev Biol Stand. 1999;99:3–8.
Even MS, Sandusky CB, Barnard ND. Serum-free hybridoma culture: ethical, scientific and safety considerations. Trends Biotechnol. 2006;24:105–8.
van der Valk J, Brunner D, De Smet K, Fex Svenningsen A, Honegger P, Knudsen LE, Lindl T, Noraberg J, Price A, Scarino ML, Gstraunthaler G. Optimization of chemically defined cell culture media--replacing fetal bovine serum in mammalian in vitro methods. Toxicol in Vitro. 2010;24:1053–63.
Burnouf T, Strunk D, Koh MB, Schallmoser K. Human platelet lysate: replacing fetal bovine serum as a gold standard for human cell propagation? Biomaterials. 2016;76:371–87.
Hemeda H, Giebel B, Wagner W. Evaluation of human platelet lysate versus fetal bovine serum for culture of mesenchymal stromal cells. Cytotherapy. 2014;16:170–80.
Viau S, Chabrand L, Eap S, Lorant J, Rouger K, Goudaliez F, Sumian C, Delorme B. Pathogen reduction through additive-free short-wave UV light irradiation retains the optimal efficacy of human platelet lysate for the expansion of human bone marrow mesenchymal stem cells. PLoS One. 2017;12:e0181406.
Corotchi MC, Popa MA, Remes A, Sima LE, Gussi I, Lupu PM. Isolation method and xeno-free culture conditions influence multipotent differentiation capacity of human Wharton’s jelly-derived mesenchymal stem cells. Stem Cell Res Ther. 2013;4:81.
Díez JM, Bauman E, Gajardo R, Jorquera JI. Culture of human mesenchymal stem cells using a candidate pharmaceutical grade xeno-free cell culture supplement derived from industrial human plasma pools. Stem Cell Res Ther. 2015;6:28.
Blázquez-Prunera A, Díez JM, Gajardo R, Grancha S. Human mesenchymal stem cells maintain their phenotype, multipotentiality, and genetic stability when cultured using a defined xeno-free human plasma fraction. Stem Cell Res Ther. 2017;8:103.
Becherucci V, Piccini L, Casamassima S, Bisin S, Gori V, Gentile F, Ceccantini R, De Rienzo E, Bindi B, Pavan P, Cunial V, Allegro E, Ermini S, Brugnolo F, Astori G, Bambi F. Human platelet lysate in mesenchymal stromal cell expansion according to a GMP grade protocol: a cell factory experience. Stem Cell Res Ther. 2018;9:124.
Schallmoser K, Bartmann C, Rohde E, Reinisch A, Kashofer K, Stadelmeyer E, Drexler C, Lanzer G, Linkesch W, Strunk D. Human platelet lysate can replace fetal bovine serum for clinical-scale expansion of functional mesenchymal stromal cells. Transfusion. 2007;47:1436–46.
Bieback K, Hecker A, Kocaömer A, Lannert H, Schallmoser K, Strunk D, Klüter H. Human alternatives to fetal bovine serum for the expansion of mesenchymal stromal cells from bone marrow. Stem Cells. 2009;27:2331–41.
Yin JQ, Zhu J, Ankrum JA. Manufacturing of primed mesenchymal stromal cells for therapy. Nature Biomedical Engineering. 2019;3:90–104.
Pilny E, Smolarczyk R, Jarosz-Biej M, Hadyk A, Skorupa A, Ciszek M, Krakowczyk Ł, Kułach N, Gillner D, Sokół M, Szala S, Cichoń T. Human ADSC xenograft through IL-6 secretion activates M2 macrophages responsible for the repair of damaged muscle tissue. Stem Cell Res Ther. 2019;13(10):93.
Caidahl K, Ueland T, Aukrust P. Osteoprotegerin: a biomarker with many faces. Arterioscler Thromb Vasc Biol. 2010;30:1684–6.
Shintani S, Ishikawa T, Nonaka T, Li C, Nakashiro K, Wong DT, Hamakawa H. Growth-regulated oncogene-1 expression is associated with angiogenesis and lymph node metastasis in human oral cancer. Oncology. 2004;66:316–22.
Salgado AJ, Reis RL, Sousa NJ, Gimble JM. Adipose tissue derived stem cells secretome: soluble factors and their roles in regenerative medicine. Curr Stem Cell Res Ther. 2010;5:103–10.
Rehman J, Traktuev D, Li J, Merfeld-Clauss S, Temm-Grove CJ, Bovenkerk JE, Pell CL, Johnstone BH, Considine RV, March KL. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation. 2004;109:1292–8.
Sadat S, Gehmert S, Song YH, Yen Y, Bai X, Gaiser S, Klein H, Alt E. The cardioprotective effect of mesenchymal stem cells is mediated by IGF-I and VEGF. Biochem Biophys Res Commun. 2007;363:674–9.
Caplan AI. Mesenchymal stem cells: time to change the name! Stem Cells Transl Med. 2017;6:1445–51.
Sipp D, Robey PG, Turner L. Clear up this stem-cell mess. Nature. 2018;561:455–7.
Kalinina N, Kharlampieva D, Loguinova M, Butenko I, Pobeguts O, Efimenko A, Ageeva L, Sharonov G, Ischenko D, Alekseev D, Grigorieva O, Sysoeva V, Rubina K, Lazarev V, Govorun V. Characterization of secretomes provides evidence for adipose-derived mesenchymal stromal cells subtypes. Stem Cell Res Ther. 2015;6:221.
Serra SC, Costa JC, Assunção-Silva RC, Teixeira FG, Silva NA, Anjo SI, Manadas B, Gimble JM, Behie LA, Salgado AJ. Influence of passage number on the impact of the secretome of adipose tissue stem cells on neural survival, neurodifferentiation and axonal growth. Biochimie. 2018;155:119–28.
Gimble JM, Bunnell BA, Chiu ES, Guilak F. Concise review: adipose-derived stromal vascular fraction cells and stem cells: let’s not get lost in translation. Stem Cells. 2011;29:749–54.
Toyserkani NM, Jørgensen MG, Tabatabaeifar S, Jensen CH, Sheikh SP, Sørensen JA. Concise review: a safety assessment of adipose-derived cell therapy in clinical trials: a systematic review of reported adverse events. Stem Cells Transl Med. 2017;6:1786–94.
Bateman ME, Strong AL, Gimble JM, Bunnell BA. Concise review: using fat to fight disease: a systematic review of nonhomologous adipose-derived stromal/stem cell therapies. Stem Cells. 2018;36:1311–28.
Galipeau J, Weiss DJ, Dominici M. Response to Nature commentary “Clear up this stem-cell mess”. Cytotherapy. 2019;21:1–2.
Funding
This work was supported by a grant from the National Science Centre no. UMO-2014/15/B/NZ4/00696 and by the National Centre for Research and Development (STRATEGMED 2/269415/11/NCBR/2015) (Poland). This work was conducted on the equipment bought for the purposes of the project no. POIG.02.01.00–00-166/08: “Silesian BIO-FARMA. Center for Biotechnology, Bioengineering, and Bioinformatics” co-financed by European Regional Development Fund within the framework of Innovative Economy Operational Programme 2007–2013. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
JC, SM, MJB, SS, RS, and TC conceived and designed the experiments. TS, RS, and TC were responsible for the human material collection and delivery. JC, SM, KK, EW, EP, MJB, RS, and TC were responsible for the cell isolation and culture. EP was responsible for the ELISA analysis. SM was responsible for the secretome analysis. JC, SM, and MJB were responsible for the FACS analysis. JC wrote the paper. MOZ, MZ, TS, and TC obtained permission for the use of human tissue. JC, SM, EW, and MJB analyzed the data. TC and SS were responsible for the work coordination and supervision. JC, SM, MJB, KK, EW, RS, and TC contributed reagents. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The experiments were performed in accordance with the Declaration of Helsinki. Researches using human adipose tissue has been accepted by the Institutional Review Board and Bioethical Committee (KB/430-62/13). All patients provided written informed consent for the collection of liposuction aspirates and subsequent analysis.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Czapla, J., Matuszczak, S., Kulik, K. et al. The effect of culture media on large-scale expansion and characteristic of adipose tissue-derived mesenchymal stromal cells. Stem Cell Res Ther 10, 235 (2019). https://doi.org/10.1186/s13287-019-1331-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-019-1331-9