
Abstract
Background
Nitric oxide (NO) plays a role in a number of physiological processes including stem cell differentiation and osteogenesis. Endothelial nitric oxide synthase (eNOS), one of three NO-producing enzymes, is located in a close conformation with the caveolin-1 (CAV-1WT) membrane protein which is inhibitory to NO production. Modification of this interaction through mutation of the caveolin scaffold domain can increase NO release. In this study, we genetically modified equine adipose-derived stem cells (eASCs) with eNOS, CAV-1WT, and a CAV-1F92A (CAV-1WT mutant) and assessed NO-mediated osteogenic differentiation and the relationship with the Wnt signaling pathway.
Methods
NO production was enhanced by lentiviral vector co-delivery of eNOS and CAV-1F92A to eASCs, and osteogenesis and Wnt signaling was assessed by gene expression analysis and activity of a novel Runx2-GFP reporter. Cells were also exposed to a NO donor (NONOate) and the eNOS inhibitor, l-NAME.
Results
NO production as measured by nitrite was significantly increased in eNOS and CAV-1F92A transduced eASCs +(5.59 ± 0.22 μM) compared to eNOS alone (4.81 ± 0.59 μM) and un-transduced control cells (0.91 ± 0.23 μM) (p < 0.05). During osteogenic differentiation, higher NO correlated with increased calcium deposition, Runx2, and alkaline phosphatase (ALP) gene expression and the activity of a Runx2-eGFP reporter. Co-expression of eNOS and CAV-1WT transgenes resulted in lower NO production. Canonical Wnt signaling pathway-associated Wnt3a and Wnt8a gene expressions were increased in eNOS-CAV-1F92A cells undergoing osteogenesis whilst non-canonical Wnt5a was decreased and similar results were seen with NONOate treatment. Treatment of osteogenic cultures with 2 mM l-NAME resulted in reduced Runx2, ALP, and Wnt3a expressions, whilst Wnt5a expression was increased in eNOS-delivered cells. Co-transduction of eASCs with a Wnt pathway responsive lenti-TCF/LEF-dGFP reporter only showed activity in osteogenic cultures co-transduced with a doxycycline inducible eNOS. Lentiviral vector expression of canonical Wnt3a and non-canonical Wnt5a in eASCs was associated with induced and suppressed osteogenic differentiation, respectively, whilst treatment of eNOS-osteogenic cells with the Wnt inhibitor Dkk-1 significantly reduced expressions of Runx2 and ALP.
Conclusions
This study identifies NO as a regulator of canonical Wnt/β-catenin signaling to promote osteogenesis in eASCs which may contribute to novel bone regeneration strategies.
Similar content being viewed by others
Background
Mesenchymal stem cells (MSCs) have been isolated from various tissues such as adipose [1], heart [2], bone marrow [3, 4], and blood [5–9], and have the potential to differentiate into different lineages, including osteoblasts, chondrocytes, and adipocytes [10, 11]. The osteoblast differentiation program of MSCs is switched on by cell recruitment, and timely expression of genes including Runx2, alkaline phosphatase (ALP), type I collagen (ColA1), and osteocalcin (OC) followed by extracellular matrix mineralization [12]. This process can be induced by soluble molecules such as bone morphogenetic proteins (BMPs) [13] or Wnts [14–16] that activate several pathways and other various downstream signals such as protein kinase [17] and growth factors [18] to trigger osteoblast differentiation of mesenchymal stem cells.
Nitric oxide (NO) is a signaling molecule with a short half-life [19, 20]. It can react within the cell where it is produced or penetrate cell membranes to affect adjacent cells [21]. NO exerts a variety of physiological effects such as regulating blood pressure via smooth muscle relaxation [22], mediating immune responses [23], controlling cell proliferation [24], modulating apoptosis [20], promoting growth factor-induced angiogenesis [4, 21], accelerating wound healing [4, 25], and functioning as a neurotransmitter [26]. These responses can be mediated through activating the primary NO effector soluble guanylyl cyclase to produce cGMP [27] by NO-based chemical modifications of proteins through S-nitrosylation [28] or through epigenetic modification [29]. NO is known to play an important role in bone homeostasis. It is generated by many cell types present in the bone environment, most notably the osteoblast [30].
NO is synthesized from l-arginine by three isozymes of nitric oxide synthase (NOS), including neuronal NOS (nNOS), endothelial NOS (eNOS), and cytokine-inducible NOS (iNOS) [31]. Both iNOS [32, 33] and eNOS [34] have been shown to play a role in osteoblast differentiation. Mice lacking eNOS have shown marked bone abnormalities due to impaired osteoblast differentiation resulting in poor maintenance of bone mass [35, 36]. Gene expression data from neonatal calvarial osteoblasts from eNOS–/– mice have shown downregulation of Runx2, Cbfa-1, and osteocalcin [37]. On the other hand, high concentrations of NO released due to the pathological iNOS expression promote bone resorption through induced osteoclastogenesis [38]. Therefore, an optimum level of NO is important to drive osteogenic differentiation of the MSCs.
In contrast with other NOS family members, eNOS is localized mainly in specific intracellular membrane domains, including the Golgi apparatus [39] and plasma membrane caveolae [40, 41]. A previously demonstrated direct interaction of eNOS with wild-type caveolin-1 (CAV-1WT) [42] has proposed that CAV-1WT functions as an endogenous negative regulator of eNOS [43]. In this context, eNOS binds to the caveolin-1 scaffolding domain (CSD; amino acids 82–101) [44] and, furthermore, Thr-90 and Thr-91 (T90 and T91), and Phe-92 (F92) were identified as critical residues for eNOS binding and inhibition [41]. Genetic modification of endothelial cells through overexpression of a mutated version of CAV-1 with a phenylalanine to alanine substitution at the amino acid position 92 (CAV-1F92A) resulted in increased NO production, overcoming the inhibitory effect of CAV-1WT [41].
In the present study, we tested the hypothesis that molecular control of NO synthesis in equine adipose-derived stem cells (eASCs), can promote osteogenic differentiation where endogenous eNOS is not available, by recreating the interaction between eNOS and CAV-1 (CAV-1WT and CAV-1F92A) regulates the osteogenic differentiation of eASCs. Our results indicate that the optimum level of NO induces osteogenic differentiation through activation of the downstream canonical Wnt/β-catenin signaling pathway.
Methods
Cell culture
eASCs were isolated from subcutaneous adipose tissue as previously described [45]. All sampling was carried out using protocols approved by the Charles Sturt University Animal Care and Ethics Committee. Human embryonic kidney 293 T cells (HEK293T) (ATCC, VA, USA) and eASCs were cultured and maintained in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich, MO, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Bovogen, VIC, Australia), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine (Invitrogen) (growth medium) at 37 °C and 5% CO2.
Plasmid constructs
The list of cDNA for the genes of interest used in this study is listed in Table 1, and was used for construction of lentiviral vectors. HF Phusion (New England Biolabs; NEB) DNA polymerase was used for all the polymerase chain reactions (PCRs) and all the restriction endonucleases were purchased from NEB unless indicated otherwise.
To construct the CMV promoter-driven eNOS expressing lentiviral vector, a codon optimized eNOS gene was synthesized [4] and subcloned into the pWPT-GFP lentiviral plasmid (Addgene, MA, USA) using BamH1 and Sal1 restriction endonucleases. Doxycycline (DOX) inducible eNOS construct was prepared by amplifying the eNOS gene using the forward (ATCAGAATTCATGGGCAACCTGAA) and reverse primer (ATCAGAATTCTCATCAGGGGCTGT) by introducing EcoR1 restriction sites (underlined sequences in both forward and reverse primers) at both the 5’ and 3’ ends of the final PCR product, followed by subcloning the PCR product into FUW-TetO vector (Addgene). Human wild-type caveolin-1 (CAV-1WT) expressing lentiviral vector was constructed by inserting the full length CAV-1WT (Addgene) into pLVX-AcGFP1-C1 and pLVX-DsRed-C1 (Clontech, CA, USA) using EcoR1 and BamH1 restriction endonucleases. A mutated caveolin-1 (CAV-1F92A) in which phenylalanine (F) at the amino acid position 92 was replaced with alanine (A) [41] was synthesized (Geneart), amplified by PCR by introducing BamH1 and Sal1 sites at the 5’ and 3’ ends of the PCR product, respectively, via forward primer (ATCAGGATCCATGTCTGGGGGCA) and reverse primer (ATCAGTCGACTTATATTTCTTTCTG) (restriction sites are underlined). The PCR product was then ligated into the pWPT-GFP lentiviral vector (Addgene) at the BamH1 and Sal1 restriction sites replacing GFP. Wnt3a and Wnt5a expressing lentiviral plasmids were purchased from DNAsu plasmid repository.
GFP reporter constructs
A 343‐bp fragment of the Runx2 enhancer region (sequence information was kindly provided by Toshihisa Komori at the Department of Cell Biology, Nagasaki University) [46] was synthesized together with the sequence of the Hsp68 minimal promoter (GenScript, NJ, USA). The entire fragment was then subcloned into pTRIP-eGFP lentiviral vector at the Mlu1 and BamH1 restriction sites replacing an insulin-specific promoter, upstream of the enhanced GFP (eGFP) coding sequence. The Wnt responsive lentiviral TCF/LEF-dGFP reporter system was purchased from Addgene.
Lentiviral vector production and transduction of equine adipose stem cells
Lentiviral vectors used in this study (Table 1) were generated by four plasmid transfection of HEK293T cells. Briefly, each well of a six-well tissue culture plate was coated with 50 μg/mL of dl-lysine (Sigma-Aldrich) in phosphate-buffered saline (PBS) and incubated for 2 h at 37 °C. HEK293T cells were then seeded at a density of 1 × 106 cells per well, 24 h prior to transfection of 6.3 μg of packaging plasmid psPAX2 (Addgene), 3.1 μg of Rev expression plasmid pRSV Rev (Addgene), 3.5 μg of VSV-G envelop pMD2.G (Addgene), and 10 μg of the gene of interest expression transfer vector using a standard calcium phosphate transfection method [47]. Seventeen hours post-transfection, the media was changed and supernatant containing lentiviral vectors were collected at 48 h and 72 h post-transfection, combined, and filtered through a 0.45-μM PVDF filter, and used for eASC transduction in the presence of 4 μg/mL Polybrene (Sigma-Aldrich).
Osteogenic differentiation
eASCs were seeded in a 12-well plate (11,000 cells/cm2) in triplicate in growth medium overnight followed by transduction with eNOS, CAV-1WT, and CAV-1F92A lentiviruses. After 3 days, growth medium was replaced with osteogenic induction medium (OM; growth medium + 0.2 mM 2-phospho-l-ascorbic acid trisodium salt + 10 nM dexamethasone + 10 mM β-glycerol phosphate; Sigma-Aldrich). Non-induced control cells were cultured in growth medium. Medium was changed every 3 days (see Fig. 2a below).
After 11 days incubation in OM or control growth medium, cells were washed with PBS and fixed with 4% (w/v) paraformaldehyde (Sigma-Aldrich) for 20 min, washed with distilled water, and then stained with 2% (w/v) Alizarin Red S (pH 4.2) for 20 min. Stained cells were washed with distilled water prior to assessment by light microscopy using a Nikon Eclipse Ti-S inverted microscope (Nikon, Japan).
Alizarin Red S quantification
Quantification of Alizarin Red S staining was performed as previously described [48]. Briefly, after staining the cells with Alizarin Red S for 20 min, 10% acetic acid was added to the 12-well cell culture plate and incubated for 30 min with shaking. The Alizarin Red S stain was extracted and the absorbance was measured at 405 nm in parallel with Alizarin Red S standards comprising of serial 1:2 dilutions of 50 mM Alizarin Red S (pH 4.2).
Quantitative real-time PCR
Total RNA from transduced and control cells after 11 days of incubation in OM or growth medium was isolated using the PureZol reagent (Bio-Rad, CA, USA) according to the manufacturer’s instructions, and the concentration of isolated RNA was determined using a Nanodrop spectrophotometer (Thermo Fisher Scientific), treated with RQ1 RNase free DNase (1 U/1 μg RNA; Promega, WI, USA). cDNA was synthesized with 1 μg RNA from all samples using a High Capacity Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative real-time PCR assays were performed on a BioRad CFX96 Real-Time system (Bio-Rad) using the SsoFast EvaGreen Supermix (Bio-Rad). Primer sequences used for target gene amplification are described in Table 2. Assays were performed in triplicate and target gene expression was normalized to equine β-actin mRNA levels using the ΔΔCt method.
Immunocytochemistry and confocal microscopy
Immunocytochemical detection of eNOS and caveolin-1 (CAV-1WT and CAV-1F92A) expression in eASCs was performed as follows. Briefly, cells were fixed in 4% paraformaldehyde for 20 min at 37 °C, treated with 0.1% Triton-X100 in PBS for 10 min, and blocked in a 10% FBS in PBS solution for 30 min at room temperature. This was followed by a 2-h incubation with a primary mouse monoclonal anti-eNOS antibody (BD Biosciences, CA, USA) or rabbit polyclonal anti-CAV-1 antibody (Cell Signaling Technology, MA, USA), and subsequently with an anti-mouse IgG secondary antibody conjugated with Alexa 488 (Cell Signaling Technology) or anti-rabbit IgG secondary antibody conjugated with Alexa 555 (Cell Signaling Technology) for 1 h and counterstained with DAPI for nuclear staining (Sigma-Aldrich). eNOS and CAV-1 co-localization was observed by confocal microscopy (Nikon).
To detect β-catenin expression, eNOS transduced cells (with or without DOX treatment) and un-transduced cells were fixed in 4% paraformaldehyde for 20 min at 37 °C, treated with 0.1% Triton-X100 in PBS for 10 min, and blocked in a 10% FBS in PBS solution for 30 min at room temperature. This was followed by an overnight incubation with a primary rabbit monoclonal anti-β-catenin antibody (Cell Signaling Technology) and subsequently with an anti-rabbit IgG secondary antibody conjugated with Alexa 488 (Cell Signaling Technology) for 1 h and counterstained with DAPI.
GFP reporter assays
For GFP-based reporter assays for both TCF/LEF-dGFP and Runx2.Hsp68-eGFP, cells transduced with the TCF/LEF-dGFP and Runx2.Hsp68-eGFP were subjected for reverse transcription quantitative PCR (RT-qPCR) for GFP expression and fluorescence microscopic analysis, respectively.
Nitric oxide detection
Extracellular NO production was measured using the Griess reagent (Promega) according to the manufacturer’s instructions and measurement of absorbance at 540 nm. Triplicates of each sample were measured at each time-point during osteogenic differentiation from day 0 to day 11.
Statistical analysis
All experiments were performed in triplicate and at least three times. Data are presented as mean ± SEM. The statistical significances were determined by one-way analysis of variance (ANOVA) followed by Tukey’s test. All tests were performed using the statistical software GraphPad Prism 6 (GraphPad, CA, USA). p < 0.05 was considered statistically significant.
Results
eASC characterization
eASCs were spindle-shaped and adherent to plastic tissue culture dishes (data not shown). We have previously reported their tri-linage differentiation potentials [49].
eNOS and caveolin-1 expression in eASCs
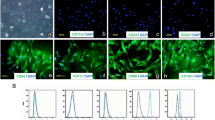
eNOS activation is controlled at the cell plasma membrane significantly by CAV-1, a major structural protein in caveolae [50, 51]. We investigated eNOS and CAV-1 expression in un-transduced and transduced eASCs by immunofluorescence microscopy. Wild-type CAV-1 (CAV-1WT)-transduced eASCs (eASCCAV-1WT) and un-transduced eASCs (eASCWT) expressed CAV-1 protein; notably, the CAV-1 expression was increased in eASCCAV-1WT (Fig. 1d) compared to eASCWT (Fig. 1c). Interestingly, eNOS expression was absent in eASCWT (Fig. 1a), whereas strong eNOS expression was observed in eNOS-transduced cells (eASCeNOS) (Fig. 1b). Next, we examined the localization of eNOS, CAV-1WT, and mutated CAV-1 (CAV-1F92A) in genetically modified eASC by confocal microscopy. As expected, eNOS expression was detected at the cytoplasm (Fig. 1e), whereas both the CAV-1F92A (Fig. 1f) and CAV-1WT (Fig. 1g) expressions were observed at the plasma membrane, confirming that F92A mutation of CAV-1 does not affect its cellular localization. Co-localization of eNOS and CAV-1F92A in co-transduced eASCs with eNOS and CAV-1F92A (eASCeNOS+CAV-1F92A) was examined by confocal microscopy with expression of eNOS in the cytoplasm and CAV-1F92A at the plasma membrane (Fig. 1h). As controls for primary antibodies, immunostaining was carried out in the absence of primary antibodies specific to eNOS and CAV-1 (Additional file 1: Figure S1).

Immunofluorescence localization of eNOS, CAV-1F92A, and CAV-1WT in lentiviral transduced eASCs and un-transduced cells. a Un-transduced eASCs show no endogenous endothelial nitric oxide synthase (eNOS) expression. b eNOS-transduced eASCs (green) show strong cytoplasmic expression. c Un-transduced eASCs show endogenous caveolin expression (red). d Wild-type caveolin-1 (CAV-1 WT)-transduced eASCs show significantly stronger expression. When e eNOS, f mutated caveolin-1 (CAV-1 F92A), and g CAV-1WT were transduced to eASCs using lentiviral vectors, eNOS showed cytoplasm localization (red) whereas both the CAV-1F92A and CAV-1WT showed plasma membrane localization (green). h Confocal microscopy analysis of co-transduction of eASCs with eNOS and CAV-1F92A resulted in cytoplasmic eNOS expression (red) and membrane localization of CAV-1F92A (green)
NO enhances osteogenic differentiation
To examine the role of the NO signaling in eASCs osteogenesis, we first compared the osteogenic differentiation between eNOS transduced (eASCeNOS) and un-transduced eASCs (eASCWT). A greater number of Alizarin Red S-positive nodules were induced in the eASCeNOS cultures compared to eASCWT culture after 11 days (Fig. 2b). NO synthesis was also significantly increased in eASCeNOS compared to eASCWT (Fig. 2c). Quantification of calcium deposition showed increased levels of calcium deposition in eASCeNOS compared to eASCWT (Fig. 2d). Quantitative analysis of Runx2 (Fig. 2e) and ALP (Fig. 2f) gene expression were also significantly upregulated in the eASCeNOS cultures compared to the eASCWT cultures.

Nitric oxide promotes osteogenic differentiation of eASCs. a Schematic of lentiviral transduction and osteogenic induction strategy. b Alizarin Red S staining after 11 days in osteogenic induction medium or growth medium. c Nitric oxide release from cells undergoing osteogenic differentiation through quantification of nitrite by Greiss assay. d Quantification of calcium deposition. Relative mRNA transcripts analysis by qPCR of e Runx2 and f alkaline phosphtase (ALP) osteoblast markers showing increased expressions in endothelial nitric oxide synthase (eNOS) and eNOS + mutated caveolin-1 (eNOS + CAV-1 F92A) transduced cells compared to all other treatments. *p < 0.05, versus eNOS+CAV-1WT, CAV-1WT, CAV-1F92A, eASC (osteogenic induction), eASC (growth medium). ; # p < 0.05 versus eNOS, eNOS+CAV-1WT, CAV-1WT, CAV-1F92A, eASC (osteogenic induction), eASC (growth medium).CAV-1 WT wild-type caveolin-1
NO-mediated osteogenic differentiation was further highlighted by inhibition of eNOS activity. eASCeNOS were treated with 2 mM of the nitric oxide synthase inhibitor, l-NG-nitroarginine methyl ester (l-NAME) for 11 days. l-NAME treatment resulted in a significant downregulation of osteoblast-specific marker expressions, ALP (Fig. 3a) and Runx2 (Fig. 3b), compared to untreated eASCeNOS.

Inhibition of endothelial nitric oxide synthase (eNOS) through l-NG-nitroarginine methyl ester (l -NAME) treatment downregulates eASC osteogenic differentiation. Relative mRNA transcripts analysis by qPCR showing that 2 mM l-NAME treatment decreased the expression of osteoblast markers a alkaline phosphatase (ALP) and b Runx2 in eNOS transduced eASCs compared to untreated eNOS transduced cells. *p < 0.05, versus eNOS (l-NAME), eASC (osteogenic induction), and eASC (growth medium)
Co-expression of eNOS and CAV-1F92A enhances NO production and osteogenic differentiation
Lentiviral vectors expressing eNOS and CAV-1F92A (mutant) or CAV-1WT (wild-type) were co-expressed in eASCs, eASCeNOS+CAV-1F92A, and eASCeNOS+CAV-1WT, respectively. Co-expression of eNOS and CAV-1F92A promoted osteogenesis as evident by Alizarin Red S staining (Fig. 2b) and calcium deposition (Fig. 2d) compared to eNOS alone (eASCeNOS), and NO levels were also significantly increased in the eASCeNOS+CAV-1F92A (Fig. 2c). Co-expression of eNOS with CAV-1WT in eASCs (eASCeNOS+CAV-1WT) reduced NO production (Fig. 2c) and also osteogenesis as evident by Alizarin Red staining (Fig. 2b) and calcium deposition (Fig. 2d). Quantitative real-time PCR analysis revealed that Runx2 (Fig. 2e) and ALP (Fig. 2f) were significantly upregulated in the eASCeNOS+CAVF92A cultures and downregulated in the eASCeNOS+CAV-1WT cultures as compared with the eASCeNOS cultures (Fig. 2e and f).
Exogenous NO donor enhances osteogenesis of eASC in a dose-dependent manner
To confirm the direct role of NO levels on eASC osteogenesis, we treated eASCs with a concentration range of exogenous NO donor (NONOate; Sigma-Aldrich). Treatment with exogenous NO donor promoted osteogenesis from 5 μM to 15 μM but this was reduced with high concentrations of NO donor (20 μM) as evident by Alizarin Red staining (Fig. 4a). NONOate treatment also resulted in a dose-dependent increase in Runx2 (Fig. 4b) and ALP (Fig. 4c) gene expression, in which maximum levels of both were achieved with 15 μM NONOate; notably, significantly lower levels of Runx2 and ALP expression were observed with 20 μM NO donor treatment.

The exogenous nitric oxide donor NONOate promotes osteogenic differentiation in a dose-dependent manner. a Alizarin Red S staining of eASCs after 11 days in osteogenic induction medium or growth medium. Relative mRNA transcript analysis shows a significant increase of osteoblast markers b Runx2 and c alkaline phosphatase (ALP) when the eASCs were treated with 15 μM NONOate. *p < 0.05, versus 0 μM, 5 μM, 10 μM, and 20 μM NONOate, and control (growth medium)
NO promotes endogenous Runx2 expression in differentiating eASCs
To monitor the effect of NO on endogenous Runx2 expression in differentiating eASCs, we generated a GFP lentiviral reporter system under the control of a Runx2 enhancer fused to the Hsp68 promoter (Runx2.Hsp68-eGFP; Fig. 5a) based on a novel Runx2 enhancer. eASCs which were stably transduced with the Runx2 reporter showed low levels of GFP expression in mostly undifferentiated cells (Fig. 5b), whereas GFP expression as a result of Runx2 promoter activity in differentiating eASCs was increased (Fig. 5b). When eASCs were transduced with eNOS (eASCeNOS), the GFP signals were increased compared to eASCs co-transduced with eNOS and CAV-1WT (eASCeNOS+CAV-1WT) and un-transduced control (eASCWT) (Fig. 5b). Interestingly, we observed that endogenous Runx2 activity was remarkably increased when the eASCs were co-transduced with eNOS and CAV-1F92A (eASCeNOS+CAV-1F92A) (Fig. 5b) compared to eASCeNOS, eASCeNOS+CAV-1WT, and eASCWT. Osteogenic nodule formation was significantly increased in the eASCeNOS+CAV-1F92A (Fig. 5c), and these result suggests that endogenous Runx2 expression is less active in undifferentiated eASCs and its expression is significantly increased through NO signaling during osteogenic differentiation.

Activity of a Runx2 reporter during eASC osteogenic differentiation. a Schematic of the Runx2.Hsp68-eGFP lentiviral reporter used to transduce eASCs. The construct contains 343 bp of Runx2 enhancer and Hsp68 minimal promoter upstream to the enhanced green fluorescent protein (eGFP). b Runx2 reporter containing eASCs were transduced with endothelial nitric oxide synthase (eNOS), eNOS + mutated caveolin-1 (eNOS + CAV-1 F92A), and eNOS + wild-type caveolin-1 (eNOS + CAV-1 WT) following osteogenic induction up to 11 days, and GFP signals were detected by fluorescence microscopy and c increased numbers of osteogenic nodules were observed. *p < 0.05, versus eNOS, eNOS + CAV-1WT, eASC (osteogenic induction), and eASC (growth medium). DMEM Dulbecco’s modified Eagle’s medium
NO modulates Wnt signaling to promote osteogenic differentiation
To examine the role of canonical and non-canonical Wnt signaling during NO-mediated osteogenic differentiation, expression of Wnt3a, Wnt8a, and Wnt5a was assessed by quantitative real-time PCR. Non-canonical Wnt5a expression was reduced in eASCeNOS (Fig. 6c), and was significantly further decreased in eASCeNOS+CAVF92A (Fig. 6c). However, expression of canonical Wnt ligands Wnt3a (Fig. 6a) and Wnt8a (Fig. 6b) was upregulated in eASCeNOS and significantly further increased in eASCeNOS+CAVF92A (Fig. 6a and b, respectively). Treatment with 2 mM l-NAME showed downregulation of Wnt3a expression (Fig. 6d) and upregulation of Wnt5a (Fig. 6e) in eASCeNOS, indicating that NO modulates Wnt signaling pathway in eASCs.

Nitric oxide signaling modulates Wnt signaling in eASCs. Relative mRNA transcript analysis by qPCR shows that endothelial nitric oxide synthase (eNOS) and eNOS + mutated caveolin-1 (eNOS + CAV-1 F92A) transduced cells increased the expression of canonical Wnt ligands a Wnt3a and b Wnt8a, whilst downregulating c non-canonical Wnt5a. Relative mRNA transcripts analysis by qPCR shows that treatment with 2 mM l-NG-nitroarginine methyl ester (l -NAME) downregulated d Wnt 3a and upregulated e Wnt5a expression. *p < 0.05 eNOS+CAV-1WT, CAV-1WT, CAV-1F92A, eASC (osteogenic induction), eASC (growth medium). and # p < 0.05 versus eNOS + wild-type caveolin-1 (CAV-1 WT), CAV-1WT, CAV-1F92A, eASC (osteogenic induction), and eASC (in DMEM); ## p < 0.05, versus eNOS (l-NAME) eASC (osteogenic induction) and eASC (growth medium)
Furthermore, treatment with NO donor (NONOate) also resulted in increased expression of canonical Wnt ligands Wnt3a (Fig. 7a) and Wnt8a (Fig. 7b), and downregulation of non-canonical Wnt5a expression (Fig 7c) in a dose-dependent manner from 5 μM to 15 μM of NONOate. Interestingly, when the NO donor concentration was increased up to 20 μM, the effect was completely reversed by downregulating Wnt3a (Fig. 7a) and Wnt8a (Fig. 7b), and upregulating Wnt5a expression (Fig. 7c). Control cells (eASCs in normal growth medium) also showed increased expression of Wnt5a (Fig. 7c), suggesting that induction of osteogenic differentiation of eASCs requires activation of canonical Wnt signaling and suppression of non-canonical Wnt5a expression.

Treatment with nitric oxide donor NONOate modulates Wnt signaling. Relative mRNA transcript analysis by qPCR shows that exogenous NONOate treatment significantly upregulates the expression of canonical Wnt ligands a Wnt3a and b Wnt8a in a dose-dependent manner up to 15 μM with a corresponding downregulation of the non-canonical Wnt ligand c Wnt5a. *p < 0.05, versus 0 μM, 5 μM, 10 μM, 20 μM, and 0 μM (growth medium)
To further analyze the relationship between NO-induced osteogenic differentiation and Wnt signaling, eNOS-transduced eASCs were treated with 20 ng/mL of the Wnt signaling inhibitor, Dickkopf-related protein 1 (Dkk-1). Dkk-1 treatment resulted in a significant downregulation of the osteoblast specific markers ALP (Fig. 8a) and Runx2 (Fig. 8b) compared to untreated eNOS transduced cells.

Inhibition of Wnt signaling through Dickkopf-related protein 1 (Dkk-1) downregulates eASC osteogenic differentiation. Relative mRNA transcript analysis by qPCR shows that Dkk-1 treatment (20 ng/mL) decreased the expression of osteoblast markers a alkaline phosphatase (ALP) and b Runx2 in endothelial nitric oxide synthase (eNOS)-transduced eASCs compared to untreated eNOS-transduced eASCs. *p < 0.05, versus eNOS (Dkk-1), eASC (osteogenic induction with Dkk-1), eASC (osteogenic induction), and eASC (growth medium)
Canonical Wnt3a promotes osteogenesis while non-canonical Wnt5a suppresses osteogenesis
To explore the opposite effects of canonical and non-canonical Wnt signaling pathways on eASC osteogenesis, we generated canonical Wnt3a and non-canonical Wnt5a expressing lentiviral vectors and transduced eASCs. Wnt3a-transduced eASCs (eASCWnt3a) and Wnt5a-transduced eASCs (eASCWnt5a) were incubated in osteogenic induction medium (OM) for 11 days. Interestingly, we found that Wnt3a (eASCWnt3a) promoted osteogenesis as evident by Alizarin Red staining compared to un-transduced eASCs (eASCWT) (Fig. 9a). On the other hand, overexpression of Wnt5a (eASCWnt5a) reduced osteogenic differentiation (Fig. 9a). Quantitative analysis of the mRNA levels by real-time PCR revealed that ALP (Fig. 9b) and Runx2 (Fig. 9c) were upregulated in the eASCWnt3a culture as compared with the ASCWT culture, and downregulated in eASCWnt5a culture, suggesting that lenitiviral vector-mediated Wnt3a expression can promote osteogenesis while expression of non-canonical Wnt5a suppresses osteogenesis.

Lentiviral expression of canonical Wnt3a promotes osteogenesis and non-canonical Wnt5a results in suppressed osteogenesiss. a Alizarin Red S staining after 11 days in osteogenic induction medium or growth medium showing that lentiviral Wnt3a transduction increased the calcium deposition, whereas Wnt5a decreased calcium deposition levels. Relative mRNA transcript analysis by qPCR showing that Wnt3a upregulates the expression of osteoblast markers b alkaline phosphatase (ALP) and c Runx2 expression, whereas Wnt5a resulted in downregulation. *p < 0.05, versus Wnt5a, eASC (osteogenic induction), and eASC (growth medium)
NO promotes the canonical Wnt signaling pathway by promoting nuclear translocation of β-catenin
To further explore mechanisms by which NO promotes canonical Wnt signaling, we used a lentiviral vector expressing GFP reporter under the control of the TCF/LEF promoter (TOPFLASH; Addgene). In the canonical Wnt signaling pathway, β-catenin translocation to the nucleus is promoted by the activation of canonical Wnt signaling [52, 53]. Accordingly, eASCs were introduced with the lentiviral TCF/LEF-dGFP reporter, and those cells were then co-transduced with doxycycline inducible eNOS expressing lentiviral vector (Fig. 10a). As a readout for nuclear translocation of β-catenin, the TCF/LEF-driven GFP mRNA expression levels were measured by quantitative real-time PCR. Under osteogenic induction conditions, increased GFP mRNA expression was demonstrated compared to non-osteogenic induction conditions (cells in growth medium) (Fig. 10b). When doxycycline was added to the OM, GFP mRNA expression was significantly increased in the eNOS-transduced cells (eASCeNOS) (Fig. 10b). Interestingly, when doxycycline was removed from the medium, GFP mRNA expression in eASCeNOS was similar to other osteogenic induction conditions (Fig. 10b).

Nitric oxide signaling promotes activity of a β-catenin reporter. a Schematic of lentiviral transduction of a β-catenin reporter, TCF/LEF (TOP-dGFP), and a doxycycline (DOX) inducible endothelial nitric oxide synthase (eNOS) transduction and assessment of TCF/LEF activity during osteogenesis. b Relative mRNA transcript analysis by qPCR showing eNOS upregulates GFP mRNA expression when treated with DOX. *p < 0.05, versus TOP-dGFP + eNOS (no DOX), TOP-dGFP (osteogenic induction), TOP-dGFP (growth medium), and un-transduced eASC (growth medium)
Using a β-catenin-specific monoclonal antibody, we further investigated the effect of NO on β-catenin nuclear translocation. eASCs were transduced with the doxycycline inducible eNOS lentiviral vector followed by immunostaining with β-catenin-specific monoclonal antibody (Cell Signaling Technology). When doxycycline was added to the OM, the expression of β-catenin was observed in eNOS-transduced cells (eASCeNOS) (Fig. 11a) in both the nucleus and cytoplasm, and when doxycycline was removed from the medium β-catenin expression in eASCeNOS was reduced (Fig. 11a) to that seen in un-transduced control cells (Fig. 11a). Furthermore, we observed nuclear co-localization of β-catenin and DAPI only in doxycycline-treated eASCeNOS suggesting that NO may promote nuclear localization of β-catenin (Fig. 11b). As a control for the primary antibody, immunostaining was carried out in the absence of the primary antibody specific to β-catenin (Additional file 2: Figure S2).

Nitric oxide promotes nuclear translocation of β-catenin. a Immunostaining with a β-catenin-specific monoclonal antibody reveals that the expression of β-catenin in endothelial nitric oxide synthase (eNOS) transduced cells when doxycycline (DOX) is available in the medium. b nuclear localization of beta catenin in eNOS transduced cells in DOX containing medium. Arrows indicate nuclear localisation of β-catenin
Together, these findings support the paradigm that cellular environments rich in bioavailable NO through either genetic modification or exogenous sources can modulate Wnt signaling, by upregulating the canonical and downregulating the non-canonical pathways resulting in increased osteogenic differentiation (Fig. 12).

Proposed signaling mechanism underlying osteogenic differentiation induced by NO in eASCs. Molecular control of NO levels may activate and suppress the expression of endogenous canonical and non-canonical Wnt ligands, respectively, to promote nuclear localization of β-catenin and subsequent activation of osteogenic differentiation through promoting osteoblast-specific gene transcription. CAV-1 F92A mutated caveolin-1, CAV-1 WT wild-type caveolin-1, eNOS endothelial nitric oxide synthase
Discussion
NO plays an important role in osteogensis, bone remodeling, and metabolism [54–56]. It has been reported that both iNOS and eNOS play a role in osteogenesis of embryonic stem cells [57]. We [4] and others [58] have shown that MSCs do not express eNOS. Therefore, in order to investigate the role of eNOS in osteogenic differentiation of eASCs, in this study eASCs were genetically modified by lentiviral vector-based eNOS. ASCs are promising candidates for stem cell-based therapy for bone repair [59], and the role of eNOS-mediated NO synthesis and its downstream effect on osteogenesis of MSCs remains to be explored. We found that eNOS gene transfer by lentiviral vector promoted osteoblast-specific gene expressions (Fig. 2e and f), contributing to the matrix mineralization as visualized by Alizarin Red S staining (Fig. 2b and d). Noteworthy, this osteogenic potential of eASCseNOS was significantly abrogated by l-NAME treatment (Fig. 3), suggesting that NO derived from eNOS plays a major role in enhancing osteogenesis in eASCs.
CAV-1 is a key negative regulator of eNOS activation and thus inhibits the production of NO [41, 60] and, importantly, CAV-1 is expressed endogenously in MSCs [61]. The scaffolding domain (82-101 amino acids) of CAV-1 protein interacts with eNOS at the plasma membrane and this interaction inhibits the eNOS activation reducing NO synthesis [41]. An alanine scanning approach revealed that substitution of phenylalanine at the amino acid position 92 with alanine to produce CAV-1F92A mutant restored the eNOS activation and promoted NO synthesis [41]. Thus, in order to understand the contribution of caveolin-1 on the control of NO synthesis in eASC osteogenesis, we modified eASCs by expressing CAV-1WT (as a negative regulator for eNOS activation) or CAV-1F92A (as a positive regulator for eNOS activation) together with eNOS. Confirming a previous observation [41], we found that co-expression of eNOS and CAV-1F92A increased NO production while eNOS and CAV-1WT co-expressed eASCs showed reduced NO production (Fig. 2c), suggesting that CAV-1 is an important regulator of NO production in eASCs. We further found that these controlled levels of NO synthesis regulate osteogenesis, where eNOS together with CAV-1F92A resulted in increased osteogenic differentiation of eASCs.
To explore the molecular basis of NO-mediated osteogenesis, we investigated the effect of NO on Wnt signaling. Wnt signaling pathways have been shown to regulate osteoblastogenesis [62], in which canonical Wnt ligands promote osteogenesis [63, 64], and non-canonical Wnt5a can inhibit the canonical Wnt signaling [65]. In the canonical Wnt pathway, binding of canonical Wnt ligands such as Wnt3a and Wnt8a to cell surface frizzled receptors results in the nuclear translocation of β-catenin [66], which ultimately binds with the TCF/LEF region to initiate the transcription of osteogenic genes such as Runx2 [62]. On the other hand, binding of non-canonical Wnt5a ligand to the ROR2 member of the Ror-family of RTKs inhibits canonical Wnt signaling by promoting β-catenin degradation, and downregulation of β-catenin reduced osteoblast-specific gene expression [67]. Our results revealed that genetic manipulation of eASCs with eNOS and CAV-1F92A (eASCeNOS+CAV-1F92A) increased canonical Wnt3a and Wnt8a expression, whereas eASCeNOS+CAV-1WT decreased Wnt3a and Wnt8a expression (Fig. 6a and b), suggesting that NO levels may regulate Wnt ligand expression and promote osteogenesis. Confirming the role of Wnt signaling on osteogenesis, inhibition of canonical Wnt signaling through Dkk-1 treatment of eNOS-expressing cells attenuated osteogenesis as evident by downregulation of osteoblast-specific Runx2 and ALP expression (Fig. 8). On the other hand, the effect of non-canonical Wnt5a expression was completely the opposite (Fig. 6c) to the canonical Wnt3a and Wnt8a expression profiles, suggesting that molecular control of NO synthesis through eNOS/CAV-1 interaction or exogenous NO treatment (Fig. 7) results in differential regulation of Wnt ligand expression and their subsequent effect on osteogenic differentiation. Furthermore, we also found that NO modulates Wnt signaling and promotes osteogenesis when a differentiation environment is enhanced with an optimum concentration of exogenous NO (Figs. 4 and 7).
It has been shown that Wnt3a can directly promote osteogenesis [68], whilst Wnt5a plays a role in self-renewal of stem cells [69]. We further investigated the direct effect of canonical Wnt3a and non-canonical Wnt5a on eASC osteogenesis through lentiviral vector overexpression. Interestingly, corroborating our results on NO-mediated Wnt-regulated osteogenesis, Wnt3a promoted osteogenesis (Fig. 9a–c) whereas Wnt5a inhibited osteogenesis (Fig. 9a–c). It was shown that increased levels of β-catenin can promote bone formation through increasing the expression of osteoblast-specific genes [70, 71], whilst abnormal osteoblast differentiation has been observed with β-catenin knockdown [70, 72]. Thus, it is possible that Wnt3a promotes osteogenesis by increasing β-catenin stability and Wnt5a may suppress osteogenesis by degrading β-catenin. NO may regulate this mechanism by increasing Wnt3a and suppressing Wnt5a ligand availability to modulate nuclear localization of β-catenin via the canonical Wnt ligand transduction pathway. In support of this, we observed that eNOS-transduced cells promoted the expression of β-catenin and its nuclear localization (Fig. 11), and a TCF/LEF-dGFP reporter assay demonstrated responsiveness in a NO-rich cellular environment (Fig. 10b), which could be controlled through the expression of DOX-inducible eNOS.
Conclusions
In summary, our findings provide an insight into the role of NO in promoting eASC osteogenic differentiation in a cellular environment of optimum levels of NO through interaction with Wnt signaling pathways. This may lead to the development of novel cell-based therapeutic approaches for bone repair, in particular in vitro modification of MSCs by NO to optimize the endogenous Wnt signaling pathway to promote osteogenic differentiation upon subsequent transplantation.
Abbreviations
- ALP:
-
Alkaline phosphatase
- CAV-1:
-
Caveolin-1
- DOX:
-
Doxycycline
- eASC:
-
Equine adipose-derived stem cell
- eGFP:
-
Enhanced green fluorescent protein
- eNOS:
-
Endothelial nitric oxide synthase
- FBS:
-
Fetal bovine serum
- iNOS:
-
Cytokine-inducible nitric oxide synthase
- l-NAME:
-
l-NG-nitroarginine methyl ester
- MSC:
-
Mesenchymal stem cell
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- OM:
-
Osteogenic induction medium
- PBS:
-
Phosphate-buffered saline
- PCR:
-
Polymerase chain reaction
- RT-qPCR:
-
Reverse transcription quantitative polymerase chain reaction
References
Zannettino ACW, Paton S, Arthur A, Khor F, Itescu S, Gimble JM, et al. Multipotential human adipose-derived stromal stem cells exhibit a perivascular phenotype in vitro and in vivo. J Cell Physiol. 2008;214(2):413–21.
Zhang Y, Sivakumaran P, Newcomb AE, Hernandez D, Harris N, Khanabdali R, et al. Cardiac repair with a novel population of mesenchymal stem cells resident in the human heart. Stem Cells. 2015;33(10):3100–13.
Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic transplants of bone marrow. Transplantation. 1968;6(2):230–47.
Bandara N, Gurusinghe S, Chen H, Chen S, Wang LX, Lim SY, Strappe P. Minicircle DNA-mediated endothelial nitric oxide synthase gene transfer enhances angiogenic responses of bone marrow-derived mesenchymal stem cells. Stem Cell Res Ther. 2016;7:48.
He Q, Wan C, Li G. Concise review: multipotent mesenchymal stromal cells in blood. Stem Cells. 2007;25(1):69–77.
Oh W, Kim DS, Yang YS, Lee JK. Immunological properties of umbilical cord blood-derived mesenchymal stromal cells. Cellular Immunol. 2008;251(2):116–23.
Meng X, Ichim TE, Zhong J, Rogers A, Yin Z, Jackson J, et al. Endometrial regenerative cells: a novel stem cell population. J Transl Med. 2007;5:57.
Hida N, Nishiyama N, Miyoshi S, Kira S, Segawa K, Uyama T, et al. Novel cardiac precursor-like cells from human menstrual blood-derived mesenchymal cells. Stem Cells. 2008;26(7):1695–704.
Patel AN, Park E, Kuzman M, Benetti F, Silva FJ, Allickson JG. Multipotent menstrual blood stromal stem cells: isolation, characterization, and differentiation. Cell Transplant. 2008;17(3):303–11.
Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276(5309):71–4.
Phinney DG, Prockop DJ. Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair—current views. Stem Cells. 2007;25:2896–902.
Lian JB, Javed A, Zaidi SK, Lengner C, Montecino M, van Wijnen AJ, et al. Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Crit Rev Eukaryot Gene Expr. 2004;14:1–41.
Zhang W, Zhu C, Wu Y, Ye D, Wang S, Zou D, et al. VEGF and BMP-2 promote bone regeneration by facilitating bone marrow stem cell homing and differentiation. Eur Cell Mater. 2014;15(27):1–11.
Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem. 2005;280:33132–40.
Choi SY, Song MS, Ryu PD, Lam AT, Joo SW, Lee SY. Gold nanoparticles promote osteogenic differentiation in human adipose-derived mesenchymal stem cells through the Wnt/β-catenin signaling pathway. Int J Nanomedicine. 2015;7(10):4383–92.
Goodnough LH, Dinuoscio GJ, Ferguson JW, Williams T, Lang RA, Atit RP. Distinct requirements for cranial ectoderm and mesenchyme-derived Wnts in specification and differentiation of osteoblast and dermal progenitors. PLoS Genet. 2014;10(2):e1004152.
Ge C, Xiao G, Jiang D, Franceschi RT. Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J Cell Biol. 2007;176(5):709–18.
Kratchmarova I, Blagoev B, Haack-Sorensen M, Kassem M, Mann M. Mechanism of divergent growth factor effects in mesenchymal stem cell differentiation. Science. 2005;308(5727):1472–7.
Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265–9.
Vaughn MW, Kuo L, Liao JC. Effective diffusion distance of nitric oxide in the microcirculation. Am J Physiol. 1998;274:H1705–14.
Lancaster Jr JR. A tutorial on the diffusibility and reactivity of free nitric oxide. Nitric Oxide. 1997;1:18–30.
Tare M, Parkington HC, Coleman HA, Neild TO, Dusting GJ. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71.
Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–16.
Davis KL, Martin E, Turko IV, Murad F. Novel effects of nitric oxide. Annu Rev Pharmacol Toxicol. 2001;41:203–36.
Clark RH, Kueser TJ, Walker MW, Southgate WM, Huckaby JL, Perez JA, et al. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. N Engl J Med. 2000;342:469–74.
Miller MR, Wadsworth RM. Understanding organic nitrates—a vein hope? Br J Pharmacol. 2009;157:565–7.
Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A. 1977;74:3203–7.
Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–66.
Hickok JR, Vasudevan D, Antholine WE, Thomas DD. Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain-containing demethylases. J Biol Chem. 2013;288(22):16004–15.
Evans DM, Ralston SH. Nitric oxide and bone. J Bone Miner Res. 1996;11(3):300–5.
Thomas DD. Breathing new life into nitric oxide signaling: a brief overview of the interplay between oxygen and nitric oxide. Redox Biol. 2015;5:225–33.
Lin TH, Yang RS, Tang CH, Lin CP, Fu WM. PPARgamma inhibits osteogenesis via the down-regulation of the expression of COX-2 and iNOS in rats. Bone. 2007;41(4):562–74.
Hou CH, Lin J, Huang SC, Hou SM, Tang CH. Ultrasound stimulates NF-kappaB activation and iNOS expression via the Ras/Raf/MEK/ERK signaling pathway in cultured preosteoblasts. J Cell Physiol. 2009;220(1):196–203.
Ma P, Gu B, Xiong W, Tan B, Geng W, Li J, et al. Glimepiride promotes osteogenic differentiation in rat osteoblasts via the PI3K/Akt/eNOS pathway in a high glucose microenvironment. PLoS One. 2014;9(11):e112243.
Aguirre J, Buttery L, O’ Shaughnessy M, Afzal F, Fernandez De Marticorena I, Hukkanen, et al. Endothelial nitric oxide synthase gene-deficient mice demonstrate marked retardation in postnatal bone formation, reduced bone volume, and defects in osteoblast maturation and activity. Am J Pathol. 2001;158(1):247–57.
Armour KE, Armour KJ, Gallagher ME, Gödecke A, Helfrich MH, Reid DM, et al. Defective bone formation and anabolic response to exogenous estrogen in mice with targeted disruption of endothelial nitric oxide synthase. Endocrinology. 2001;142(2):760–6.
Afzal F, Polak J, Buttery L. Endothelial nitric oxide synthase in the control of osteoblastic mineralizing activity and bone integrity. J Pathol. 2004;202(4):503–10.
Zhao Z, Hou X, Yin X, Li Y, Duan R, Boyce BF, et al. TNF Induction of NF-κB RelB enhances RANKL-induced osteoclastogenesis by promoting inflammatory macrophage differentiation but also limits it through suppression of NFATc1 expression. PLoS One. 2015;10(8):e0135728.
Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, et al. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci U S A. 2006;103(52):19777–82.
Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci U S A. 1996;93:6448–53.
Bernatchez PN, Bauer PM, Yu J, Prendergast JS, He P, Sessa WC. Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc Natl Acad Sci U S A. 2005;102(3):761–6.
Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem. 1996;271:22810–4.
Maniatis NA, Shinin V, Schraufnagel DE, Okada S, Vogel SM, Malik AB, et al. Increased pulmonary vascular resistance and defective pulmonary artery filling in caveolin-1–/– mice. Am J Physiol Lung Cell Mol Physiol. 2008;94:L865–73.
Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, et al. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the NOS-caveolin binding domain in vivo. J Biol Chem. 1997;272:25437–40.
Petersen GF, Hilbert B, Trope G, Kalle W, Strappe P. Efficient transduction of equine adipose-derived mesenchymal stem cells by VSV-G pseudotyped lentiviral vectors. Res Vet Sci. 2014;97(3):616–22.
Kawane T, Komori H, Liu W, Moriishi T, Miyazaki T, Mori M, Matsuo Y, et al. Dlx5 and mef2 regulate a novel runx2 enhancer for osteoblast-specific expression. J Bone Miner Res. 2014;29(9):1960–9.
Kingston RE, Chen CA, Rose JK. Calcium phosphate transfection. Curr Protoc Mol Biol. 2003;Chapter 9: Unit 9.1. doi:10.1002/0471142727.mb0901s63.
Gregory CA, Gunn WG, Peister A, Prockop DJ. An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem. 2004;329:77–84.
Petersen GF, Hilbert BJ, Trope GD, Kalle WH, Strappe PM. Direct conversion of equine adipose-derived stem cells into induced neuronal cells is enhanced in three-dimensional culture. Cell Reprogram. 2015;17(6):419–26.
Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, et al. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000;6:1362–7.
Michel JB, Feron O, Sacks D, Michel T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2 + -calmodulin and caveolin. J Biol Chem. 1997;272:15583–6.
Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127(3):469–80.
Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303(5663):1483–7.
Evans DM, Ralston SH. Nitric oxide and bone. J Bone Miner Res. 1996;11:300–305.
Taylor BC, Schreiner PJ, Zmuda JM, Li J, Moffett SP, Beck TJ, et al. Association of endothelial nitric oxide synthase genotypes with bone mineral density, bone loss, hip structure, and risk of fracture in older women: The SOF study. Bone. 2006;39:174–80.
Wang CJ, Yang KD, Ko JY, Huang CC, Huang HY, Wang FS. The effects of shockwave on bone healing and systemic concentrations of nitric oxide (NO), TGF-beta1, VEGF and BMP-2 in long bone non-unions. Nitric Oxide. 2009;20(4):298–303.
Ehnes DD, Geransar RM, Rancourt DE, Zur Nieden NI. Exogenous nitric oxide enhances calcification in embryonic stem cell-derived osteogenic cultures. Differentiation. 2015;89(3–4):97–103.
Gomes SA, Rangel EB, Premer C, Dulce RA, Cao Y, Florea V, et al. S-nitrosoglutathione reductase (GSNOR) enhances vasculogenesis by mesenchymal stem cells. Proc Natl Acad Sci U S A. 2013;10:2834–9.
Saeed H, Ahsan M, Saleem Z, Iqtedar M, Islam M, et al. Mesenchymal stem cells (MSCs) as skeletal therapeutics—an update. J Biomed Sci. 2016;23:41. doi:10.1186/s12929-016-0254-3.
Bernatchez P, Sharma A, Bauer PM, Marin E, Sessa WC. A noninhibitory mutant of the caveolin-1 scaffolding domain enhances eNOS-derived NO synthesis and vasodilation in mice. J Clin Invest. 2011;121(9):3747–55.
Baker N, Shon J, Tuan RS. Promotion of human mesenchymal stem cell osteogenesis by PI3-kinase/Akt signaling and the influence of caveolin-1/cholesterol homeostasis. Stem Cells Res Ther. 2015;6:238.
Cai T, Sun D, Duan Y, Wen P, Dai C, Yang J, He W. WNT/β-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp Cell Res. 2016;345(2):206–17.
Liu G, Vijayakumar S, Grumolato L, Arroyave R, Qiao H, Akiri G, Aaronson SA. Canonical Wnts function as potent regulators of osteogenesis by human mesenchymal stem cells. J Cell Biol. 2009;185(1):67–75.
Zhou C, Zou J, Zou S, Li X. INO80 is Required for Osteogenic Differentiation of Human Mesenchymal Stem Cells. Sci. Rep. 2016;6:35924.
Topol L, Jiang X, Choi H, Garrett-Beal L, Carolan PJ, Yang Y. Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3 independent beta catenin degradation. J Cell Biol. 2003;162(5):899–908.
Voloshanenko O, Erdmann G, Dubash TD, Augustin I, Metzig M, et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun. 2013;4:2610.
Yuan Y, Niu CC, Deng G, Li ZQ, Pan J, Zhao C, et al. The Wnt5a/Ror2 noncanonical signaling pathway inhibits canonical Wnt signaling in K562 cells. Int J Mol Med. 2011;27(1):63–9.
Cho YD, Yoon WJ, Kim WJ, Woo KM, Baek JH, Lee G, Ku Y, van Wijnen AJ, Ryoo HM. Epigenetic modifications and canonical wingless/int-1 class (WNT) signaling enable trans-differentiation of nonosteogenic cells into osteoblasts. J Biol Chem. 2014;289(29):20120–8.
Yeh JR, Zhang X, Nagano MC. Wnt5a is a cell-extrinsic factor that supports self-renewal of mouse spermatogonial stem cells. J Cell Sci. 2011;124(Pt 14):2357–66.
Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8(5):739–50.
Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, MacDougald OA. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005;102(9):3324–9.
Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8(5):727–38.
Acknowledgements
The authors are grateful for the assistance of staff at the Veterinary Clinical Centre (Charles Sturt University) and the O’Brien Institute Department, St. Vincent’s Institute of Medical Research.
Funding
This work was supported by an Australian Government International Post-Graduate Research Scholarship (IPRS) and Australian Post-Graduate Award (APA) to NB. LXW, SC, and HW acknowledge funding from the National Natural Science Foundation of China (N. 81270104).
Availability of supporting data
Two supplementary figures are available.
Authors’ contributions
NB conceived this study, collected data, performed data analysis, and prepared the manuscript. SG and BH collected data and performed data analysis, and prepared the manuscript. HC, DW, and SC collected data and prepared the manuscript. LXW performed data analysis and prepared the manuscript. SYL prepared and revised the manuscript. PS conceived this study, performed data analysis, and prepared and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors gave consent for publication.
Ethical approval and consent to participate
Isolation of equine adipose-derived mesenchymal stem cells was approved by the Charles Sturt University Animal Care and Ethics Committee (Ethics Number 09/111).
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Figure S1.
Primary antibody controls for Fig. 1. As a control for specific primary antibody binding, immunostaining was performed in the absence of primary antibodies, with only fluorophore tagged secondary antibody present. We confirm no florescence signal was detected these controls for eNOS and CAV-1. (a) Detection of nuclear staining by Dapi and (b) immunostaining performed without mouse monoclonal anti-eNOS antibody but with an anti-mouse IgG secondary antibody conjugated with Alexa 488. (c) Detection of nuclear staining by Dapi and (d) immunostaining performed without rabbit polyclonal anti-CAV-1 antibody but with an anti-rabbit IgG secondary antibody conjugated with Alexa 555. (e) Detection of nuclear staining by Dapi and (d) fluorescence detection with no mouse monoclonal anti-eNOS antibody with an anti-mouse IgG secondary antibody conjugated with Alexa 555. (f) Detection of nuclear staining by Dapi and (g) fluorescence detection with no rabbit polyclonal anti-CAV-1 antibody with an anti-rabbit IgG secondary antibody conjugated with Alexa 488. (PPT 182 kb)
Additional file 2: Figure S2.
Primary antibody controls for Fig. 11. As a control for specific β-catenin primary antibody binding, immunostaining was carried out in the absence of the primary antibody with no florescence signal detected. (a) Detection of nuclear staining by Dapi and (b) fluorescence detection without rabbit monoclonal anti-β-catenin antibody with an anti-mouse IgG secondary antibody conjugated with Alexa 488. (PPT 102 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bandara, N., Gurusinghe, S., Lim, S.Y. et al. Molecular control of nitric oxide synthesis through eNOS and caveolin-1 interaction regulates osteogenic differentiation of adipose-derived stem cells by modulation of Wnt/β-catenin signaling. Stem Cell Res Ther 7, 182 (2016). https://doi.org/10.1186/s13287-016-0442-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-016-0442-9