
Abstract
Background
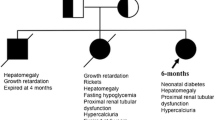
The p.R63W mutation in hepatocyte nuclear factor-4 alpha (HNF4A) leads to a heterogeneous group of disorders with various clinical presentations. Recently, patients with congenital hyperinsulinism and Fanconi syndrome due to the p.R63W mutation in HNF4A have been described. Although other clinical variations such as liver dysfunction have been associated with HNF4A mutations, hearing impairment has not previously been associated. We report the case of a patient with Fanconi syndrome and hyperinsulinemic hypoglycemia caused by the mutation of HNF4A presenting with additional auditory phenotypes.
Case presentation
We present a case report of a 10-year-old girl of Chinese Han ethnicity who presented with renal Fanconi syndrome, infantile hyperinsulinemic hypoglycemia, and transient cholestasis. In addition, she presented with bilateral severe hearing loss. Gene analysis showed a heterozygous p.R63W mutation in the HNF4A gene that is responsible for Fanconi syndrome and hyperinsulinemic hypoglycemia.
Conclusions
This is the first case of HNF4A mutation associated with an auditory phenotype. It expands the clinical phenotypes and supports speculation in the literature that HNF4A may be a candidate gene for deafness. In conclusion, hearing loss may be found in children with HNF4A-related Fanconi syndrome, and auditory function should be assessed.
Similar content being viewed by others

Background
Renal Fanconi syndrome (FS) is a consequence of decreased solute and water reabsorption in the proximal tubule of the kidney. The common clinical manifestation is polydipsia and polyuria with phosphaturia, glycosuria, and aminoaciduria. In children, FS often occurs as part of a multisystem metabolic disease originating from heterogeneous genetic deficiencies. Recently, FS caused by mutations in the hepatocyte nuclear factor-4 alpha (HNF4A) gene was reported by Hamilton et al. [1], as a clinical syndrome accompanied by hyperinsulinemic hypoglycemia and early-onset diabetes.
Here, we report a case of FS caused by HNF4A mutation that presented with hearing loss. To the best of our knowledge, this is the first report of HNF4A-related FS associated with an additional auditory phenotype, adding to the scope of the disease.
Case presentation
A 10-year-old girl was referred to the Children’s Hospital of Fudan University because of polydipsia and polyuria. She was born to non-consanguineous healthy parents of Chinese Han ethnicity and good socioeconomic status. She was the only child of the family, and there was no family history of FS. Newborn hearing screening failed. There was no history of birth defects. She was not receiving any medication and did not take alcohol or smoke tobacco. She was in Grade 4 of primary school and was not good at studying.
At 3 months of age, she presented with jaundice, hepatomegaly (3.5 cm below the costal margin), and splenomegaly (4 cm below the costal margin). She was admitted to our hospital. Laboratory findings revealed: elevation of direct bilirubin (DB), that is, total bilirubin (TB) 66.1 μmol/L (normal range, 0–6 μmol/L) and DB 61.4 μmol/L (normal range, 5.1–17.6 μmol/L); and almost normal transaminases, that is, alanine aminotransferase (ALT) 24 IU/L (normal range, 0–40 IU/L) and aspartate aminotransferase (AST) 46 IU/L (normal range, 0–40 IU/L)). Laboratory tests for hepatotropic viruses were negative. Magnetic resonance cholangiopancreatography excluded bile duct obstruction. After treatment with ursodiol, the jaundice resolved gradually. During the follow-up years, her liver functions were normal. Hypoglycemia was initially noticed during hospitalization, and fasting blood glucose ranged from 1.4 to 2.8 mmol/l. Prior to this, there was no record of a hypoglycemic episode. At the time of hypoglycemia (blood glucose 1.4 mmol/l), an inappropriate glycemic response to glucagon (increase of 4.3 mmol/l) was consistent with excess insulin action, confirming hyperinsulinism. Frequent feeding combined with intravenously administered glucose (6–7 mg/kg per minute) was required to maintain normoglycemia. She was discharged from the nursery with stable glucose levels (4.3–6.5 mmol/l) on the condition of frequent feeds. At 1 year of age, she experienced an episode of hypoglycemia, and the symptoms resolved after feeding. Subsequently, no symptoms of hypoglycemia appeared again.
Short stature was noticed (height and weight were below the third percentile) by routine physical examination at the age of 5. Laboratory investigations revealed proteinuria and glycosuria, mild acidosis, and hypophosphatemic rickets. Prior to this, she had recurrent urinary tract infections, suggesting an earlier onset of abnormal urine. Based on the clinical impression of FS, she was treated with calcitriol, phosphorus, and potassium citrate. Over the ensuing years, sodium citrate and other medications were used to maintain acid-base balance, but there was no improvement in her short stature, and her serum creatinine remained elevated by age 10.
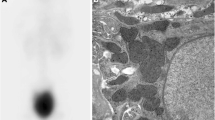
At 10 years of age, she was admitted to our clinic because of symptoms of FS and elevated serum creatinine: 75 μmol/L (normal range, 21–65 μmol/L). On this admission, her height was 126.5 cm (below the third percentile) and her weight was 28 kg (25th percentile). Laboratory investigations revealed: renal glycosuria in the absence of hyperglycemia; proteinuria with a urinary protein to creatinine ratio (pro/Cr) of 2.17 (normal range, 0–0.2); hypercalciuria with a urinary calcium to creatinine ratio (U-Ca/Cr) of 0.31 (normal range, < 0.21); and hypouricemia with 66 μmol/L (normal range, 90–420 μmol/l). Fasting glucose and post-prandial glucose were normal. No known cause for FS was identified. Glomerular filtration rate (GFR) was 55.7 ml/minute/1.73 m2 measured by 99mTc-diethylenetriaminepentacetate (DTPA) renal dynamic imaging. Renal ultrasonography showed nephrocalcinosis (Fig. 1). A radiological examination showed complete recovery of rickets. Pure tone audiometry revealed a bilateral hearing loss of more than 50 dB. Because of multisystem involvement, she underwent whole exome sequencing and mutational analysis, revealing a heterozygous p.R63W mutation in the HNF4A gene. Both parents tested negative for the mutation. No genes associated with deafness phenotypes were found.

Renal ultrasonographic images show increased reflectivity of the renal pyramids, which demonstrate nephrocalcinosis
Discussion
We reported the case of a patient carrying the HNF4A p.R63W mutation, who presented with FS, infantile hyperinsulinemic hypoglycemia, and transient cholestasis, similar to reported cases in the literature [1,2,3,4,5,6]. In addition, our patient had bilateral severe hearing loss.
The link between HNF4A p.R63W mutation and FS was first described by Hamilton et al. in 2014 [1]. The authors proposed that the HNF4A p.R63W mutation was a mutation specific for renal FS. Subsequently, more than 14 patients from 11 families have been described worldwide [1,2,3,4,5,6]. Most of these patients presented with hyperinsulinism, hypoglycemia, renal proximal tubular dysfunction, and liver involvement. Hyperinsulinism, hypoglycemia, and FS are frequently reported clinical phenotypes with this mutation. Almost all the patients carried the p.R63W mutation and had the pancreatic β-cell phenotype as well as the renal phenotype [1,2,3,4,5,6]. Phenotypic characteristics of our patient were similar to those reported in the literature except for hearing loss. A total of 14 patients with the p.R63W mutation have been described, and their clinical phenotypes are summarized in Table 1.
HNF4A mutations can cause hyperinsulinemic hypoglycemia and macrosomia in the neonatal period, as well as diabetes in adolescence or early adulthood [7, 8] caused by an impaired insulin secretory response to glucose in pancreatic β-cells [9]. Increased insulin secretion in neonates and decreased insulin secretion in later life might explain the hypoglycemia and diabetes at various stages of the disease, but the mechanism of the differential aspects of the HNF4A defect on β-cell function is unclear. In these reported case series, 100% (14/14) of the patients with HNF4A p.R63W mutations had infantile hyperinsulinemic hypoglycemia and 50% (7/14) were macrosomic (Table 1). The severity and duration of hyperinsulinemic hypoglycemia was variable, with some patients requiring diazoxide therapy for several years, while some required intravenously administered glucose for days. Most of the patients experienced hypoglycemia in the neonatal period, suggesting that it is essential to monitor glucose in neonates. Two of the 14 patients developed diabetes at 12 and 20 years of age [1]. To date, diabetes has not developed in our patient. Oral glucose tolerance testing is necessary for HNF4A mutation carriers during follow-up.
According to Hamilton et al. [1], FS is a mutation-specific phenotype associated with the p.R63W mutation, and the penetrance of FS in patients with the mutation is complete. The mechanism of the mutation-specific phenotype is unclear; it was hypothesized that the mutant HNF4A altered DNA binding and influenced the interaction with major regulatory genes; however, the details of target genes are not known [1, 9]. All 14 patients who were reported to be carrying this particular mutation presented with typical or atypical FS. The youngest onset age of HNF4A-related FS reported was the neonatal period, with 85% (12/14) diagnosed by age 4. The usual complications of FS are growth retardation and rickets in children. In these case series, 64% (9/14) had growth retardation or short stature, and 42% (6/14) had rickets. Supplementation of substances lost in the urine (primarily phosphate and bicarbonate) is the primary treatment and can induce rickets remission and catch-up growth. Short stature and rickets also occurred in our patient, and sodium citrate, potassium, phosphate, and calcitriol treatment led to remission of the rickets. The prognosis for this disease is not optimistic, with half of the patients progressing to stage II–IV chronic kidney disease (CKD). Renal function impairment was also observed in our patient, with a GFR of 55.7 ml/minute/1.73 m2. Further follow-up is necessary to assess the renal function and monitor for progression of CKD in those patients.
Liver phenotypic variability and incomplete penetrance are seen in patients with the R63W mutation. Approximately 35.7% (5/14) of the patients with this mutation presented with variable liver phenotypes, including hepatomegaly, jaundice, elevated transaminase, and increased glucose stores. Our patient presented with hepatomegaly and jaundice, similar to the case reported by Improda et al. [4]. Most of the liver dysfunction is transient. It is obvious that this mutation does not lead to severe liver disease.
Our patient presented with severe hearing loss. To date, 11 patients have been reported to be carrying the p.R63W mutation in the HNF4A gene, and in none of them was auditory status mentioned. The association of HNF4A defects with hearing loss has not been previously described. Stamatiou and Stankovic [10] identified a number of molecules including HNF4A that are likely to be key mediators of genetic hearing loss through network and pathway analyses. According to their report, HNF4A may be a new candidate gene for deafness. Our hypothesis is that mutant HNF4A may play an important role in hearing loss. On the one hand, HNF4A is a member of the steroid hormone receptor superfamily of ligand-dependent transcription factors that regulates hundreds of target genes. Based on the fact that the R63 residue is in the DNA-binding domain and directly contacts the DNA response element, we speculated that the heterozygous p.R63W mutation in our patient reduced DNA binding activity and altered expression of some target genes related to otic capsule development or associated hearing loss; on the other hand, according to Stamatiou and Stankovic [10], HNF4A appeared to be a central player in the network and was an attractive gene for hearing loss based on network analysis of human deafness. Further studies will be needed to clarify the association between HNF4A defects and auditory phenotypes.
Taken together, the specific mutation in HNF4A caused a group of disorders, including hypoglycemia, renal FS, and liver dysfunction, similar to Fanconi–Bickel syndrome, a rare type of glycogen storage disease caused by solute carrier family 2 member 2 (SLC2A2) defects with which FS is easily confused. The most noticeable difference between them is that the hypoglycemia of the former is due to hyperinsulinemia, whereas that of the latter is due to impaired hepatic release of glucose [11].
Conclusions
To the best of our knowledge, this is the first case of an HNF4A mutation associated with auditory phenotype, expanding the range of clinical phenotypes and supporting speculation in the literature that HNF4A may be a candidate gene for deafness.
In conclusion, in a case of clinically diagnosed FS with a history of neonatal hyperinsulinemic hypoglycemia, the p.R63W mutation in HNF4A should be suspected. Considering multisystem involvement, close observation of liver function, auditory condition, and blood glucose levels is suggested for these patients, all of which may occur in various courses of the disease.
References
Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, Oram RA, Shields BM, Shepherd M, Inward CD, Hamilton-Shield JP, Kohlhase J, Ellard S, Hattersley AT. The HNF4A R76W mutation causes atypical dominant Fanconi syndrome in addition to a beta cell phenotype. J Med Genet. 2014;51(3):165–9. https://doi.org/10.1136/jmedgenet-2013-102066.
Stanescu DE, Hughes N, Kaplan B, Stanley CA, De Leon DD. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. J Clin Endocrinol Metab. 2012;97(10):E2026–30. https://doi.org/10.1210/jc.2012-1356.
Numakura C, Hashimoto Y, Daitsu T, Hayasaka K, Mitsui T, Yorifuji T. Two patients with HNF4A-related congenital hyperinsulinism and renal tubular dysfunction: a clinical variation which includes transient hepatic dysfunction. Diabetes Res Clin Pract. 2015;108(3):e53–5. https://doi.org/10.1016/j.diabres.2015.03.005.
Improda N, Shah P, Guemes M, Gilbert C, Morgan K, Sebire N, Bockenhauer D, Hussain K. Hepatocyte nuclear Factor-4 Alfa mutation associated with Hyperinsulinaemic Hypoglycaemia and atypical renal Fanconi syndrome: expanding the clinical phenotype. Horm Res Paediatr. 2016;86(5):337–41. https://doi.org/10.1159/000446396.
Clemente M, Vargas A, Ariceta G, Martinez R, Campos A, Yeste D. Hyperinsulinaemic hypoglycaemia, renal Fanconi syndrome and liver disease due to a mutation in the HNF4A gene. Endocrinol Diabetes Metab Case Rep. 2017;2017 https://doi.org/10.1530/EDM-16-0133.
Walsh SB, Unwin R, Kleta R, Van’t Hoff W, Bass P, Hussain K, Ellard S, Bockenhauer D. Fainting Fanconi syndrome clarified by proxy: a case report. BMC Nephrol. 2017;18(1):230. https://doi.org/10.1186/s12882-017-0649-8.
Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, Ellard S, Ferrer J, Hattersley AT. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007;4(4):e118. https://doi.org/10.1371/journal.pmed.0040118.
Kapoor RR, Locke J, Colclough K, Wales J, Conn JJ, Hattersley AT, Ellard S, Hussain K. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes. 2008;57(6):1659–63. https://doi.org/10.2337/db07-1657.
Wang H, Maechler P, Antinozzi PA, Hagenfeldt KA, Wollheim CB. Hepatocyte nuclear factor 4alpha regulates the expression of pancreatic beta-cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J Biol Chem. 2000;275(46):35953–9. https://doi.org/10.1074/jbc.M006612200.
Stamatiou GA, Stankovic KM. A comprehensive network and pathway analysis of human deafness genes. Otol Neurotol. 2013;34(5):961–70. https://doi.org/10.1097/MAO.0b013e3182898272.
Santer R, Steinmann B, Schaub J. Fanconi-Bickel syndrome--a congenital defect of facilitative glucose transport. Curr Mol Med. 2002;2(2):213–27.
Acknowledgements
The authors acknowledge the contribution of Bing-bing Wu for gene analysis.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Author information
Authors and Affiliations
Contributions
JL wrote the manuscript; GL carried out molecular genetic testing for FS; QS was involved in management of case; and HX provided critical comments. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Ethics Committee of Children’s Hospital of Fudan University. Informed consent was obtained for blood collection and genetic analysis from the parents.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian(s) for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, J., Shen, Q., Li, G. et al. HNF4A-related Fanconi syndrome in a Chinese patient: a case report and review of the literature. J Med Case Reports 12, 203 (2018). https://doi.org/10.1186/s13256-018-1740-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-018-1740-x