
Abstract
Background
Ewing sarcoma/primitive neuroectodermal tumor is a family of highly malignant proliferation of neuroectodermal origin, most often skeletal, adrenal localization is extremely rare. Only few cases have been reported in the literature. Classical management includes radical surgery with adjuvant chemotherapy or radiotherapy or both. This case report is the only one where recurrence was surgically removed, and it confirms the importance of adjuvant treatment, and the efficacy of neoadjuvant chemotherapy.
Case presentation
We report the case of a 23-year-old Moroccan woman presenting with flank pain. An abdominal computed tomography scan showed a large and enhancing left suprarenal mass. After radical nephrectomy, histologic examination revealed a small round cell proliferation. The diagnosis of Ewing sarcoma was confirmed by molecular analysis; time to final diagnosis was 5 months due to financial and coordination issues. Computed tomography (on an asymptomatic patient) revealed a locoregional recurrence, our patient received 12 cycles of the vincristine, doxorubicin and cyclophosphamide/ifosfamide and etoposide protocol used in an alternating schedule, with partial radiologic response (62%) and pathologic complete response, then underwent adjuvant radiotherapy of 45 Gy. The young women is still in remission after 36 months of follow-up.
Conclusions
Our patient had an early recurrence due to absence of adjuvant treatment, but did respond well to neoadjuvant chemotherapy with a pathologic complete response. Management of adrenal Ewing sarcoma could be extrapolated from skeletal one with good outcomes even in locoregional recurrence.
Similar content being viewed by others


Background
Ewing sarcoma/primitive neuroectodermal tumors (ES/PNET) was first described in 1921 as a family of tumors characterized by small round cell proliferation and characteristic translocation t(11;22). It is the second most common bone tumor in children and adolescents. The prevalence of extraskeletal ES ranges between 15 and 20%; primitive adrenal ES tumor is very rare. To date, there have been only 28 cases reported in the literature; here we report another case with a review of the literature.
Most of the cases reported in the literature were managed with initial surgery followed by adjuvant chemotherapy or radiotherapy or both. Here, we present the case of a young woman with classical presentation of adrenal ES, but who did not receive adjuvant therapy after radical nephrectomy. Evolution confirmed the importance of adjuvant treatment, but also the efficacy of the vincristine, doxorubicin and cyclophosphamide/ifosfamide and etoposide (VAC/IE) protocol that allowed a pathologic complete response. This case report is the only one where recurrence was surgically removed.
Case presentation
Our patient is a young Arab needlewoman of 23 years old, unmarried, of middle socioeconomic status, nonalcoholic, a nonsmoker without relevant past medical history, no neoplasm was reported in the family.
Her symptoms included progressive abdominal pain and left flank distension with a conserved general status and regular menstrual cycle, without exophthalmia or any sign of hypertension or flush syndrome. Our patient had lost 1 kg in 1 month, she was taking nonsteroidal anti-inflammatory drugs (NSAIDS) with moderate pain improvement and consulted 26 days after her first symptom.
Clinical examination found a performance status (PS) 1 patient, weighing 60 kg and 158 cm tall, her temperature at admission was 37.3°, with normal blood pressure values (125/80 mmHg) and cardiac frequency (87 beats/min), without neurological abnormality or any sign of Cushing syndrome.
Computed tomography (CT) scans revealed an invasive suprarenal mass of 14 × 11 cm moderately enhanced after contrast administration, with regional adenomegaly, without venous thrombosis or distant abnormality.
Endocrinal analysis was negative: eliminating first a pheochromocytoma (normetanephrine blood level: 1.55 μmol/24 h, metanephrine: 0.32 μmol/24 h), and a corticosurrenaloma (urinary free cortisol at 370 μg/L, and two negative dexamethasone suppression tests). Cervical ultrasound found thyroid nodules of the Thyroid Imaging Reporting and Data System (TIRADS) 4A classification, with negative fine-needle-aspiration cytology, and normal thyroid function analysis.
A complete blood count showed a hypochromic anemia of 11 g/dL, with normal hepatic and renal function.
The young woman underwent an open radical nephrectomy.
Pathological analysis showed a well-limited, whitish mass of 14 × 10 cm with a central fibrous zone, replacing the entire adrenal gland and infiltrating the superior renal pole and pararenal fat with negative margins, consisting of undifferentiated monomorphic small cells (Fig. 1). One lymph node among the 13 removed was metastatic, an immunochemical profile revealed an intense expression of CD99 (Fig. 2) and vimentin, and focal expression of synaptophysin and CD56, Ki67 Antigen expression was 70%.

Malignant small cell proliferation in hematoxylin-eosin-saffron staining, magnification ×20

Diffuse CD99 positive staining
The diagnosis of ES was confirmed by molecular study, in situ hybridization revealed the EWSR1 rearrangement gene (Fig. 3).

Dual-color, break-apart fluorescence in situ hybridization shows split signals typical of the EWS (22q;12) gene rearrangement. Arrows are pointing to probes (red and green spots) and fusion signal (yellow spots)
The final diagnosis was obtained 5 months after surgery due to financial and coordination issues, (Fig. 4), a new imaging in the asymptomatic patient revealed a locoregional recurrence (Figs. 5 and 6).

Timeline of patient’s medical history

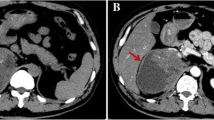
Axial computed tomography performed 5 months after radical nephrectomy showing a heterogeneously enhanced mass of 6 cm in the nephrectomy bed

Axial computed tomography performed 5 months after radical nephrectomy showing adenopathies encompassing the left primitive iliac vessels
The young woman received 12 cycles of vincristine, doxorubicin and cyclophosphamide (VAC)/ifosfamide and etoposide (IE) alternating every 3 weeks, with partial response estimated to be 62% in the primitive site and a complete response in lymph nodes (Figs. 7 and 8), then underwent a second surgery: resection of recurrent tumor, revealing a totally necrotic 2 cm mass [pathologic complete response (pCR)].

Post-chemotherapy axial computed tomography showing a partial response in the primitive site estimated to be 62%

Post-chemotherapy axial computed tomography showing a complete response in lymph nodes
Our patient received adjuvant radiotherapy; 45 Gy was delivered to the tumor bed, para-aortic and left iliac areas, in three-dimensional conformal technique.
After 36 months of follow-up, the young woman is still in remission without any sign of late chemotherapy- or radiotherapy-related toxicity.
Discussion
In our case report, the patient underwent radical surgery without adjuvant therapy due to financial and interdepartmental coordination issues, diagnosed after 5 months with locoregional recurrence in imaging while she was asymptomatic, than benefited from a neoadjuvant chemotherapy with pathologic complete response, our case report is the only one where recurrence was surgically removed, which gives a precious opportunity to evaluate the pathologic response to chemotherapy. This case confirms the importance of adjuvant treatment and the efficacy of the VAC/IE protocol in this rare localization by extrapolation from osseous Ewing sarcoma management.
Ewing sarcoma of the adrenal gland (ES/PNET) is an extremely rare and aggressive malignancy, only few cases have been reported. It’s worth mentioning that diagnosis of primitive ES in this location is quite difficult, indeed; in case of adrenal mass the metastatic nature must be eliminated, thus an exhaustive imaging should be undergone.
In imaging, ES of adrenal gland presents as a large, well-limited mass, heterogeneously enhanced with areas of hemorrhage and necrosis [1].
The final diagnosis of ES/PNET is not always evident, histologic differential diagnosis of ES could include any small round cell tumor. A typical ES could be diagnosed on the basis of morphologic and immunochemical analysis only; however, cytogenetic confirmation could be needed for final diagnosis. NKX2.2 is considered the best ES marker with a sensitivity of 80% and a specificity of 84%, [2] and so the positivity of CD99 and NKX2.2 is highly specific of ES [2].
Our review of the literature found 28 published cases [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27], the first case was reported by Marina et al. in 1989 [3], our observation is the second case in Morocco to be reported.
Median age at diagnosis was 23.4 years, only three patients were older than 50 years, with a clear female predominance (19 female, 8 male), abdominal pain was the most common revealing symptom. The tumor is usually large, and endocrinologically nonfunctioning. Although there is no standard of care or consensus regarding management of adrenal ES, treatment is extrapolated from osseous ES/PNET management. Current strategy includes local control (surgery and/or radiotherapy) with adjuvant radiotherapy or chemotherapy or both. All patients but two underwent surgery (one patient committed suicide with post-mortem diagnosis, one patient died after neoadjuvant chemotherapy).
Follow-up duration ranged between 6 months and 5 years, four patients had local recurrence with a median progression-free survival (PFS) period of 16 months (1 month in two cases, 9 months in one case and 4.5 years in one case). Five patients had metastatic recurrence involving most often lungs, liver and brain, with a median PFS of 7 months (2 months in one case, 5 months in two cases and 13 months in one case). The patient in the report of Zhang et al. [20] with metastatic disease, who did not receive any treatment, died 6 months later.
As shown in the different case reports, adrenal ES/PNET is a rapidly extensive tumor with aggressive behavior. Surgical resection is considered, for the majority of authors, the mainstay of local control. Considering the radio- and chemo-sensitivity of this tumor; adjuvant radiotherapy and chemotherapy are expected to be associated with better disease control by extrapolation to osseous ES, but there is no consensus regarding these adjuvant treatments.
The most common chemotherapy regimen used was VAC/IE for both localized and metastatic disease, other regimens used (for metastatic disease) were: vincristine, actinomycin-D (VAC), adriamycin, cyslophosphamide (AC), platinium, teniposide (PT), prednisolone, cyclophosphamide, vincristine, adriamycin, methotrexate (Pd CVAM), platinium, cyclophosphamide, adriamycin, teniposide (PCAT), vincristine, dacarbazine (VD) and etoposide (E).
None of the studies that has used adjuvant radiotherapy has given details concerning dose and modality but one, the case reported by Yoon et al. [11], of a 17-year-old girl who underwent resection with adjuvant sequential chemoradiation. She received 55.8 Gy of proton beam therapy (36 Gy to the abdomen even without capsular rupture, and 19.8 Gy to the tumor bed), with a follow-up of 2 years without any sign of recurrence or sequela.
Conclusions
Ewing sarcoma of the adrenal gland is an extremely rare and aggressive tumor. Current management includes surgical resection followed by radiotherapy or chemotherapy or both.
A unified therapeutic protocol should be suggested for future cases to point out the best therapeutic sequencing, the appropriate chemotherapy regimen, and radiotherapy volume, dose and timing.
Abbreviations
- A:
-
Adriamycin;
- Ac:
-
Actinomycin-D
- C:
-
Cyclophosphamide
- CT:
-
Computed tomography
- D:
-
Dacarbazine
- E:
-
Etoposide
- ES:
-
Ewing sarcoma
- I:
-
Ifosfamide
- M:
-
Methotrexate
- NSAIDs:
-
nonsteroidal anti-inflammatory drugs
- pCR:
-
Pathologic complete response
- PFS:
-
Progression-free survival
- PNET:
-
Primitive neuroectodermal tumor
- PS:
-
Performance status
- T:
-
Teniposide
- TIRADS:
-
Thyroid Imaging Reporting and Data System
- V:
-
Vincristine
References
Zhang Y, Cai P, Chen M, et al. Imaging findings of adrenal primitive neuroectodermal tumors: a series of seven cases. Clin Transl Oncol. 2017;19(5):641–9.
Shibuya R, Matsuyama A, Nakamoto M, et al. The combination of CD99 and NKX2.2, a transcriptional target of EWSR1-FLI1, is highly specific for the diagnosis of Ewing sarcoma. Virchows Arch. 2014;465(5):599–605.
Marina NM, Etcubanas E, Parham DM, et al. Peripheral primitive neuroectodermal tumor (peripheral neuroepithelioma) in children. A review of the St. Jude experience and controversies in diagnosis and management. Cancer. 1989;64:1952–60.
Matsuoka Y, Fujii Y, Akashi T, et al. Primitive neuroectodermal tumour of the adrenal gland. BJU Int. 1999;83:515–6.
Sasaki T, Onishi T, Yabana T, Hoshina A. Ewing’s sarcoma/primitive neuroectodermal tumor arising from the adrenal gland: a case report and literature review. Tumori. 2013;99(3):104–6.
Kato K, Kato Y, Ijiri R, et al. Ewing’s sarcoma family of tumors arising in the adrenal gland-possible diagnostic pitfall in pediatric pathology: histologic, immunohistochemical, ultrastructural, and molecular study. Hum Pathol. 2001;32:1012–6.
Saboo SS, Krajewski KM, Jagannathan JP, Ramaiya N. IVC tumor thrombus: an advanced case of rare extraosseous Ewing sarcoma of the adrenal gland. Urology. 2012;79(6):e77–8.
Ahmed AA, Nava VE, Pham T, et al. Ewing sarcoma family of tumors in unusual sites: confirmation by rt-PCR. Pediatr Dev Pathol. 2006;9:488–95.
Stephenson J, Gow KW, Meechan J, et al. Ewing sarcoma/primitive neuroectodermal tumor arising from the adrenal gland in an adolescent. Pediatr Blood Cancer. 2011;57:691–2.
Raad RA, Manetti GJ, Colberg JW, et al. Ewing sarcoma/primitive neuroectodermal tumor arising in the adrenal gland. Pathol Int. 2013;63:283–6.
Yoon JH, Kim H, Lee JW, et al. Ewing sarcoma/primitive neuroectodermal tumor in the adrenal gland of an adolescent: a case report and review of the literature. J Pediatr Hematolo. Oncol. 2014;36:e456–9.
Zahir MN, Ansari TZ, Moatter T, et al. Ewing’s sarcoma arising from the adrenal gland in a young male: a case report. BMC Res Notes. 2013;6:533.
Gonin J, Larousserie F, Dousset B, et al. An unusual adrenal gland tumor: Ewing tumor. Ann Pathol. 2011;31:28–31.
Lim SH, Lee JY, Lee JY, et al. Unusual presentation of Ewing sarcoma in the adrenal gland: a secondary malignancy from a survivor of Burkitt lymphoma. Jpn J Clin Oncol. 2013;43:676–80.
Komatsu S, Watanabe R, Naito M, et al. Primitive neuroectodermal tumor of the adrenal gland. Int J Urol. 2006;13:606–7.
Phukan C, Nirmal TJ, Kumar RM, Kekkre NS. Peripheral primitive neuroectodermal tumor of the adrenal gland: a rare entity. Indian J Urol. 2013;29:357–9.
Dutta D, Shivaprasad KS, Das RN, et al. Primitive neuroectodermal tumor of adrenal: clinical presentation and outcomes. J Cancer Res Ther. 2013;9:709–11.
Pirani JF, Woolums CS, Dishop MK, Herman JR. Primitive neuroectodermal tumor of the adrenal gland. J Urol. 2000;163:1855–6.
Mohsin R, Hashmi A, Mubarak M, et al. Primitive neuroectodermal tumor/Ewing’s sarcoma in adult uro-oncology: a case series from a developing country. Urol Ann. 2011;3:103–207.
Zhang Y, Li H. Primitive neuroectodermal tumors of adrenal gland. Jpn J Clin Oncol. 2010;40:800–4.
Sato M, Miyazato M, Yamada S, et al. Retroperitoneal extraskeletal Ewing’ s sarcoma difficult to differentiate from adrenocortical carcinoma : a case report. Hinyokika Kiyo. 2011;57(6):303–7.
Yamamoto T, Takasu K, Emoto Y, et al. Latent adrenal Ewing sarcoma family of tumors: a case report. Leg Med (Tokyo). 2013;15:96–8.
Ait Batahar S, Elidrissi S, Berrada S, et al. Extraskeletal Ewing’s sarcoma: an adrenal localization. Int J Case Rep Images. 2016;7(11):762–5.
Zhang L, Yao M, Hisaoka M, et al. Primary Ewing sarcoma/primitive neuroectodermal tumor in the adrenal gland. APMIS. 2016;124:624–9.
Pal DK, Chandra V, Ranjan KR, et al. Ewing’s sarcoma of the adrenal gland. APSP J Case Rep. 2016;7(3):20.
Blas J-VV, Smith ML, Wasif N, et al. Ewing sarcoma of the adrenal gland: a rare entity. BMJ Case Rep. 2013; https://doi.org/10.1136/bcr-2012-007753
Tsang YP, Lang BHH, Tam SC, et al. Primitive neuroectodermal adrenal gland tumour. Hong Kong Med J. 2014;20(5):444–6.
Acknowledgements
Not applicable.
Funding
None.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
EH recorded history from the diagnosis to last follow-up, wrote the manuscript and carried out the literature review. MK contributed to the literature review. RB recorded the history of endocrine analysis. ETG interpreted the endocrine analysis and orientated the diagnosis pathway. EN interpreted the endocrine analysis and orientated the diagnosis pathway. BR decided the therapeutic strategy, EA decided on the therapeutic and surveillance strategy, reviewed the manuscript, and approved the final version of the manuscript. KM decided on the therapeutic and surveillance strategy. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The patient gave her consent to participate knowing that her identity would be removed.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Eddaoualline, H., Mazouz, K., Rafiq, B. et al. Ewing sarcoma of the adrenal gland: a case report and review of the literature. J Med Case Reports 12, 69 (2018). https://doi.org/10.1186/s13256-018-1601-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-018-1601-7