
Abstract
Introduction
Ewing’s sarcoma belongs to a spectrum of neoplastic diseases known as Ewing’s family of tumors. This family of tumors is usually seen in osseous sites. Ewing’s sarcoma of the cervix is extremely rare, with only 18 cases reported in the English literature. The immunohistochemical profile of Ewing’s sarcoma overlaps with other malignancies like small cell carcinoma. The rarity and complex pathologic picture of Ewing’s sarcoma of the cervix creates the potential for misdiagnosis. Hence, we believe this case needs to be reported to add to the available literature.
Case presentation
A 49-year-old white Caucasian woman presented with vaginal bleeding. A pelvic examination revealed a cystic lesion arising from her cervix. Examination of a biopsy specimen revealed a poorly differentiated neoplasm, with sheets of small hyperchromatic cells, staining weakly for neuroendocrine markers. She was diagnosed with small cell carcinoma and started on concurrent chemotherapy and radiation. However, additional positive immunostaining for CD99 was strongly suggestive of Ewing’s sarcoma. Fluorescence in situ hybridization revealed ESWR1 gene rearrangement, confirming Ewing’s sarcoma. Our patient underwent surgery, which confirmed stage IIB Ewing’s sarcoma. She received adjuvant chemotherapy but died from progressive metastatic disease after four cycles.
Conclusion
With early diagnosis and appropriate treatment, Ewing’s sarcoma of the cervix can be a potentially curable disease. However, owing to overlapping clinical and histopathological features, the diagnosis poses a challenge to oncologists and pathologists. This article guides pathologists to consider Ewing’s sarcoma in the differential diagnosis of small cell carcinoma with weak staining for neuroendocrine markers. This literature review will benefit oncologists encountering this rare entity.
Similar content being viewed by others


Introduction
Ewing’s sarcoma and peripheral neuroectodermal tumor (PNET) are the same entity, displaying varying degrees of neuroectodermal differentiation. They arise from mesenchymal progenitor cells and are part of a spectrum of neoplastic diseases, known as Ewing’s family of tumors (EFT) [1]. EFT are characterized by reciprocal translocation between chromosomes 11 and 22, t (11; 22), and are usually seen in osseous sites, both axial and appendicular. Extra-osseous presentations are uncommon, with specifically PNET of the female genital tract being very rare. The most common site of PNET in the female genital tract is the ovary, with the uterine corpus being the second most common. Primary PNET of the cervix and vulva are extremely rare. In this article, we present a case of primary PNET tumor of the cervix. The rarity of this entity can lead to diagnostic difficulties. In our case, it was initially diagnosed as a neuroendocrine small cell carcinoma of the cervix. We also summarize a literature review of all the cases reported in the English language.
Case presentation
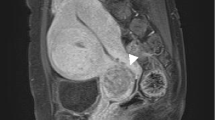
A 49-year-old gravida 2, para 2, perimenopausal woman presented with vaginal bleeding. A pelvic examination revealed a cystic lesion arising from her cervix. Examination of a biopsy specimen revealed a poorly differentiated neoplasm involving the cervical stroma, with sheets of small hyperchromatic cells with slightly irregular nuclei, stippled chromatin, inconspicuous nucleoli, minimal cytoplasm, necrosis, and numerous mitotic figures (Figs. 1 and 2). Immunostaining showed the samples were weakly positive for neuron-specific enolase, CD56, and synaptophysin, and negative for pancytokeratin, CK7, CK20, and CD45. Magnetic resonance imaging (MRI) of the pelvis showed a 5.3 × 4.8 × 6.6 cm irregular, enhancing mass arising from her cervix, with involvement of the adjacent vagina and parametrium (Fig. 3). A positron emission tomography–computed tomography (PET/CT) scan revealed that the mass was hypermetabolic with a standard uptake value of 5.5, and did not demonstrate any distant metastatic disease.

Medium power (200×) hematoxylin and eosin stain showing sheets of small tumor cells, focal necrosis, and a delicate vascular network

High power (400×) hematoxylin and eosin stain showing small cells with focal necrosis and mitotic activity

Magnetic resonance imaging of the pelvis: sagittal view. An enhancing mass is visible in the anterior cervix
The pathologic diagnosis was invasive malignant small round blue cell tumor most consistent with small cell carcinoma. She completed two cycles of cisplatin and etoposide and a planned course of 5,040 cGy over 28 fractions of 180 cGy each, which led to resolution of the vaginal bleeding. However, owing to the only weak staining for the neuroendocrine markers, further immunostaining was performed. Stains for S-100, CD3, CD20, TTF1, and desmin were negative whereas that for CD99 was strongly positive (Fig. 4). Diffuse membranous positivity for CD99 led to a diagnosis of Ewing’s sarcoma/PNET and ruled out small cell carcinoma. Fluorescence in situ hybridization revealed ESWR1 gene rearrangement in 90 % of cells, confirming the diagnosis of Ewing’s sarcoma. Subsequently, chemotherapy was discontinued and our patient underwent total hysterectomy and bilateral salpingo-oophorectomy, upper vaginectomy, and resection of the parametrium. Surgical pathology showed stage IIB Ewing’s sarcoma involving her cervix and extending into the right parametrium. Her endometrium, ovaries, and fallopian tubes were unremarkable and margins were free of tumor.

High power (400×) immunohistochemical diffuse and membranous CD99 positivity
She was started on chemotherapy with vincristine (2 mg/m2 on day 1), doxorubicin (Adriamycin; 75 mg/m2), and cyclophosphamide with mesna (1,200 mg/m2 on day 1), alternating with ifosfamide plus mesna (1,800 mg/m2 days 1–5) and etoposide (100 mg/m2) every 3 weeks. A PET scan done after two cycles demonstrated no recurrence of the tumor. After four cycles, 9 months from initial diagnosis, she developed acute renal failure. CT scans showed evidence of metastases to her lumbar spine, pelvis, and bladder. Nephrostomy tubes were placed, which improved renal function. Two weeks later she presented with distal colonic obstruction secondary to an extensive pelvic tumor. Exploratory laparotomy and a diverting loop colostomy were done. One week later her disease progressed with new lung metastases, which led to clinical deterioration and death, with an overall survival of 10 months.
Discussion
Ewing’s sarcoma/PNET of the cervix is an extremely rare entity. Our review shows that there have been only 18 cases reported so far in the English literature; we report the 19th case (Table 1). In two cases, PNET was reported in association with another malignancy. Horn et al. reported PNET with squamous cell carcinoma of the cervix, and Tsao et al. reported carcinoma in addition to PNET, and described their case as a “carcinosarcoma” [2, 3]. The ages at presentation ranged between 19 and 60 years, with the mean age being 39.
The most common symptom reported was irregular vaginal bleeding. Other symptoms included dysuria, lower abdominal pain, vaginal discharge, and in one case, urinary frequency. The most common physical findings were nodular lesions extending into the anterior vaginal wall and enlarged uterus. The vaginal bleeding with enlarged uterus led to the preliminary diagnosis of fibroid in two patients [3, 4]. One patient had vaginal stenosis and necrotic tissue on the cervix [5].
Stage was not reported in three cases. Ten (62.5 %) patients were stage IB1 or IB2, one (6.25 %) stage IIA, two (12.5 %) stage IIB, one (6.25 %) stage IIIB, and two stage IV (12.5 %).
In the reported cases, multiple imaging modalities including ultrasound, CT, and MRI were used for diagnosis and staging. Our review shows that PNET tumors are highly fludeoxyglucose avid. PET scan may be a useful imaging modality in diagnosis, staging, and monitoring response to therapy.
The diagnosis of EFTs is difficult by routine microscopy because they have small blue cell morphology that can be seen in several malignancies. On histology examination, there are sheets of small blue cells with “stippled salt and pepper chromatin” in the nuclei and absence of nucleoli [6]. Necrosis and nuclear molding of adjacent cells is common. Additionally, the cells easily become crushed during processing of the specimen, producing smudged and streaked extra nuclear chromatin (crush artifact). Small round blue cells can be seen in a wide variety of malignancies which can be remembered using the pneumonic “LEMON” (lymphoblastic lymphoma, Ewing’s sarcoma, medulloblastoma, oat cell/small cell neuroendocrine, and neuroblastoma). Other soft tissue sarcomas and rhabdomyosarcoma should also be considered in the differential diagnosis. Lymphoblastic lymphomas closely resemble PNET because they have sheets of small cells with a lack of glandular or squamous differentiation.
Immunohistochemistry is critical in the diagnosis. Small cell neuroendocrine carcinomas are usually positive for chromogranin, synaptophysin, and neuron-specific enolase, and overlap highly with PNET. The vast majority of EFTs express high levels of a cell surface glycoprotein CD99 or MIC2 surface antigen that is encoded by the CD99 (MIC2X) gene [7, 8]. The finding of membrane-localized MIC2 expression in a small blue cell malignancy is a sensitive diagnostic marker for the EFTs. MIC2 lacks specificity because other tumors, like rhabdomyosarcoma, can be MIC2 positive. However, small cell carcinomas are negative for MIC2. Benbrahim et al. reported a case of cervical PNET that was initially misdiagnosed as lymphoma [9]. Lymphoid markers, leucocyte common antigen, CD20, and CD3 can be utilized to differentiate lymphoma from PNET [9].
Molecular genetic characterization of chromosomal anomalies specific to EFT has led to increased detection. The characteristic signature translocation involving the EWS gene at 22q12.2 and various erythroblast transcription specific-family genes, like FLI (friend leukemia virus integration 1) at 11q24.1-q24.3, is seen in 85–90 % of cases [10, 11]. ESWR encodes a multifunctional protein that regulates multiple cellular processes. FLI1 encodes the FLI1 protein, which controls cellular development, proliferation, and carcinogenesis [12]. The EWSR1–ERG translocation [t (21; 22) (q22; q12)] is present in 5–10 % of EFTs, while other translocations are less common [13].
Most of the cases reported were diagnosed based on histopathology and immunohistochemistry (Table 1). Cytogenetic analysis [14] was done in one case. Fluorescent in situ hybridization [14] was used in two cases including our case, while reverse transcriptase polymerase chain reaction [15, 16] was used in two cases.
Currently there is no uniformity of treatment, owing to the rarity of this neoplasm. Snijders-Keilholz et al. recommended a multidisciplinary approach similar to that used in osseous PNETs with induction chemotherapy, surgery, adjuvant chemotherapy, and radiation [17]. When surgery is feasible, wide excision performed at a sarcoma center is preferable.
Most patients (Table 1) with early stage disease underwent total abdominal hysterectomy and bilateral salpingo-oophorectomy. Pelvic lymph node dissection was performed in 11 out of 19 cases. Of these 11 cases, eight patients are alive without recurrence, the outcome of two cases is unknown, and one patient died from metastatic disease 4 years later. Of the eight cases that did not use lymph node dissection, two tumors were inoperable owing to stage IIIB [5] and IV [16] disease. The stage of one patient is unknown. The remaining five cases were early stage (three stage I B2, two stage IIB). Of these five cases, one patient did not receive any chemotherapy and died 4 years later. Our patient (stage IIB) died 10 months later despite chemotherapy and radiation. The other three cases received chemotherapy and are alive without relapse. The contribution of Pelvic lymph node dissection (PLND) to overall survival, especially in patients who had chemoradiation, is unclear.
Chemotherapy was used in 16 of the 19 cases (Table 1). Two patients who were metastatic at diagnosis received palliative chemotherapy. Although adjuvant chemotherapy was used in the earlier reported cases, a combined regimen of neoadjuvant and adjuvant chemotherapy has been used frequently in recent years, with favorable results. The chemotherapy regimens and schedules used varied considerably. The use of ifosfamide and etoposide alternating with vincristine, doxorubicin (Adriamycin) and cyclophosphamide, which is the regimen of choice for Ewing’s sarcoma of the bone, has increased in recent years with good outcomes. The overall survival appears to have dramatically improved with the use of chemotherapy.
Eight of 19 patients (Table 1) received radiation therapy. The intent of radiation was palliative in one case, definitive in three, and adjuvant in four. The definitive chemoradiation given to our patient was part of the small cell carcinoma regimen owing to an initial small cell diagnosis, but she subsequently developed pelvic recurrence 9 months after diagnosis. Another case death from pelvic recurrence 9 months after diagnosis was reported by Xiao et al. [18], but the authors did not report if radiation was given. Radiation doses ranged from 40 to 55 Gy with the fractionation schedule of 180–200 cGy over 4–5 weeks. Overall, we conclude that adjuvant radiation may have a role in preventing local recurrence and should be considered when appropriate.
The follow-up of these cases ranged between 5 months and 8 years, with 15 of the 19 cases being alive and recurrence-free at the time of follow-up. Two patients died of metastatic disease 12 days and 4.2 years after presentation, respectively. Our patient developed metastatic disease while on adjuvant chemotherapy and died 10 months after diagnosis. The outcome of a case reported by Goda et al. [19] is unknown.
The best outcomes were noted in patients who underwent tri-modality therapy with surgery, chemotherapy, and radiation; although owing to the paucity of cases the best approach is still unknown.
Similar to skeletal Ewing’s sarcoma, the most unfavorable prognostic factor is the presence of distant metastasis, with stage IV disease being universally fatal. A recent review by Baldini et al. [20] suggested that age may be a prognostic factor in survival and elderly patients do poorly.
Conclusion
It is important to identify these rare cases of Ewing’s sarcoma to provide early and appropriate treatment. With prompt diagnosis and aggressive multimodality treatment, PNET of the cervix appears to be a potentially curable disease. Once diagnosis is made, referral to a tertiary care center with dedicated multidisciplinary tumor boards and special expertise in sarcoma management is recommended. Hopefully, identification of more cases in the future may help establish a meaningful pattern of behavior and guide clinical management of this rare entity.
Consent
Written informed consent was obtained from the patient’s next of kin for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Jedicka P. Ewing sarcoma, an enigmatic malignancy of likely progenitor cell origin, driven by transcription factor oncogenic fusions. Int J Clin Exp Pathol. 2010;3(4):338–47.
Horn LC, Fischer U, Bilek, K. Primitive neuroectodermal tumor of the cervix uteri. A case report. Gen Diagn Pathol. 1996;142:227–30.
Tsao A, Roth L, Sandler A, Hurteau JA. Cervical primitive neuroectodermal tumor. Gynecol Oncol. 2001;83:138–42.
Arora N, Kalra A, Kausar H, Ghosh TK, Majumdar A. Primitive neuroectodermal tumour of uterine cervix - a diagnostic and therapeutic dilemma. Gynecol Case Rep. 2012;32(7):711–3.
Li B, Ouyang L, et al. Primary primitive neuroectodermal tumor of the cervix. Onco Targets Ther. 2013;6:707–11.
Malpica A, Moran C. Primitive neuroectodermal tumor of the cervix: a clinicopathologic and immunohistochemical study of two cases. Ann Diagn Pathol. 2002;6:281–7.
Khoury JD. Ewing sarcoma family of tumors. Adv Anat Pathol. 2005;12:212–20.
De Alava E, Gerald WL. Molecular biology of Ewing’s sarcoma/primitive neuroectodermal tumor family. J Clin Oncol. 2000;18:204–13.
Benbrahim Z, Haie-Meder C, Duvillard P, El Mesbahi O, Cesne AL, Pautier P. Primitive neuroectodermal tumor of the cervix uteri: a case report and review of literature. Int J Hematol Oncol Stem Cell Res. 2012;6:4.
Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359(6391):162.
Zucman J, Delattre O, Desmaze C, Plougastel B, Joubert I, Melot T, et al. Cloning and characterization of the Ewing’s sarcoma and peripheral neuroepithelioma t(11;22) translocation breakpoints. Genes Chromosomes Cancer. 1992;5(4):271.
Hromas R, Klemsz M. The ETS oncogene family in development, proliferation and neoplasia. Int J Hematol. 1994;59(4):257.
Obata K, Hiraga H, Nojima T, Yoshida MC, Abe S. Molecular characterization of the genomic breakpoint junction in a t(11;22) translocation in Ewing sarcoma. Genes Chromosomes Cancer. 1999;25(1):6.
Pauwels P, Ambros P, Hattinger C, Lammens M, Dalcin P, Ribot J, et al. Peripheral primitive neuroectodermal tumour of the cervix. Virchows Arch. 2000;436:68–73.
Cenacchi G, Pasquinelli G, Montanaro L, Cerasolis S, Vici M, Bisceglia M, et al. Primary endocervical extraosseous Ewing’s Sarcoma/PNET. Gynecol Pathol. 1998;17:83–8.
Masoura S, Kourtis A, Kalogiannidis I, Kotoula V, Anagnostou E, Angelidou S, et al. Primary primitive neuroectodermal tumor of the cervix confirmed with molecular analysis in a 23 year old woman: a case report. Pathol Res Pract. 2012;208:245–9.
Snijders-Keilholz A, Ewing P. Primitive neruoectodermal tumor of the cervi uteri: a case report changing concepts in therapy. Gynecol Oncol. 2005;98:516–9.
Xiao C, Zhao J, Guo P, Wang D, Zhao D, Ren T, et al. Clinical analysis of primary neuroectodermal tumors in the female genital tract. Gynecol Cancer. 2014;24:404–9.
Goda JS, Nirah B, Mayur K, Pramod P, Vdayan K, Udayan K. Primitive neuroectodermal tumour of the cervix: a rare entity. Internet J Radiol. 2007; 6.doi:10.5580/269d.
Baldini EH, Demetri GD, Fletcher CD, Foran J, Marcus KC, Singer S. Adults with Ewing’s sarcoma/primitive neuroectodermal tumor: adverse effect of older age and primary extraosseous disease on outcome. Ann Surg. 1999;230(1):79–86.
Russin V, Valente P, Hanjani P. Psammoma bodies in neuroendocrine carcinoma of the uterine cervix. Acta Cytol. 1987;31:791–5.
Sato S, Yajima A, Kimura N, Namiki T, Furuhashi N, Sakuma H. Peripheral neuroepithelioma (peripheral primitive neuroectodermal tumor) of the uterine cervix. Tohoku J Exp. 1996;180:187–95.
Farzaneh F, Rezvani H, Boroujeni PT, Rahimi F. Primitive neuroectodermal tumor of the cervix: a case report. Case Rep. 2011;5(5):489.
Khosla D, Rai B, Firuza P, Sreedharanunni S, Dey P, Sharma S. Primitive neuroecodermal tumor of the uterine cervix diagnosed during pregnancy: a rare case with review of literature. J Obstet Gynaecol Res. 2013;40:878–82.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NM gathered and interpreted data regarding the disease. JG helped analyze data. MZ helped obtain histological images and with interpretation. JS helped analyze images and contributed to the paper. MY was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mashriqi, N., Gujjarlapudi, J.k., Sidhu, J. et al. Ewing’s sarcoma of the cervix, a diagnostic dilemma: a case report and review of the literature. J Med Case Reports 9, 255 (2015). https://doi.org/10.1186/s13256-015-0733-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-015-0733-2