
Abstract
Background
Both de novo variants and recessive inherited variants were associated with autism spectrum disorder (ASD). This study aimed to use exome data to prioritize recessive inherited genes (RIGs) with biallelically inherited variants in autosomes or X-linked inherited variants in males and investigate the functional relationships between RIGs and genes with de novo variants (DNGs).
Methods
We used a bioinformatics pipeline to analyze whole-exome sequencing data from 1799 ASD quads (containing one proband, one unaffected sibling, and their parents) from the Simons Simplex Collection and prioritize candidate RIGs with rare biallelically inherited variants in autosomes or X-linked inherited variants in males. The relationships between RIGs and DNGs were characterized based on different genetic perspectives, including genetic variants, functional networks, and brain expression patterns.
Results
Among the biallelically or hemizygous constrained genes that were expressed in the brain, ASD probands carried significantly more biallelically inherited protein-truncating variants (PTVs) in autosomes (p = 0.038) and X-linked inherited PTVs in males (p = 0.026) than those in unaffected siblings. We prioritized eight autosomal, and 13 X-linked candidate RIGs, including 11 genes already associated with neurodevelopmental disorders. In total, we detected biallelically inherited variants or X-linked inherited variants of these 21 candidate RIGs in 26 (1.4%) of 1799 probands. We then integrated previously reported known or candidate genes in ASD, ultimately obtaining 70 RIGs and 87 DNGs for analysis. We found that RIGs were less likely to carry multiple recessive inherited variants than DNGs were to carry multiple de novo variants. Additionally, RIGs and DNGs were significantly co-expressed and interacted with each other, forming a network enriched in known functional ASD clusters, although RIGs were less likely to be enriched in these functional clusters compared with DNGs. Furthermore, although RIGs and DNGs presented comparable expression patterns in the human brain, RIGs were less likely to be associated with prenatal brain regions, the middle cortical layers, and excitatory neurons than DNGs.
Limitations
The RIGs analyzed in this study require functional validation, and the results should be replicated in more patients with ASD.
Conclusions
ASD RIGs were functionally associated with DNGs; however, they exhibited higher heterogeneity than DNGs.
Similar content being viewed by others

Background
Autism spectrum disorder (ASD) is an early-onset neurodevelopmental disorder with a global prevalence of 1% and is clinically diagnosed based on social impairment, repetitive behaviors and restricted interests [1]. In addition to these core symptoms, other variable traits in patients with ASD include neuropsychiatric comorbidities such as intellectual disability (ID) and developmental delay [2], leading to marked clinical heterogeneity. ASD is highly heritable, indicating that genetic factors play vital roles in its pathogenesis [3]. Microarray technology [4,5,6,7,8], whole-exome sequencing (WES) [9,10,11,12,13,14,15,16,17,18,19], and whole-genome sequencing [20,21,22,23,24,25,26,27,28] have been widely used in genetic studies of large ASD cohorts. Numerous de novo variants (DNVs), particularly protein-truncating variants (PTVs), have been detected, and several exome-wide significant genes with DNVs (DNGs) have been elucidated, contributing to a better understanding of the genetic causes of ASD. In our previous studies, we focused on DNVs to explore the genetic architecture and genotype–phenotype correlations in Chinese ASD patients [29,30,31], demonstrating that analyzing DNG-expression patterns and functional networks could provide clues to elucidate the ASD etiology and subtypes [32,33,34].
Although DNVs are clearly important in ASD, relatively few exome studies have explored rare inherited variants in ASD. Krumm et al. [35] reported significant maternal transmission bias of inherited truncating variants in male ASD cases. Additionally, studies on both consanguineous and nonconsanguineous families identified several recessive inherited genes (RIGs) with rare biallelically inherited variants in autosomes or X-linked inherited variants in males that are associated with ASD [36,37,38]. Specifically, Chahrour et al. [36] performed WES for 16 nonconsanguineous families and identified four RIGs (UBE3B, CLTCL1, NCKAP5L, and ZNF18). Yu et al. [37] performed WES for multiple consanguineous families and identified six RIGs (AMT, PEX7, SYNE1, VPS13B, PAH, and POMGNT1). Lim et al. [38] identified genes with recessive inherited PTVs using WES data from 933 ASD cases and 869 controls, estimating that complete knockouts of autosomal and X-linked genes accounted for 3% of all ASD cases and 2% of male ASD cases, respectively. Recently, Doan et al. [39] reanalyzed WES data of individuals with ASD from the Autism Sequencing Consortium (ASC), demonstrating that biallelically autosomal PTVs and deleterious missense (Dmis) variants were present in 5% of all ASD cases and 10% of female ASD cases. Moreover, they identified 41 autosomal ASD-associated RIGs [39]; however, recessive X-linked genes were not included in their analysis.
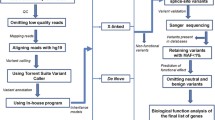
To provide resources for the study of ASD genetics, the Simons Simplex Collection (SSC) [40] established a permanent repository of quad samples, each comprising of one ASD proband, one unaffected sibling, and their unaffected parents. To better understand the contributions of RIGs to ASD, we performed genetic burden analysis on biallelically and X-linked inherited coding variants in 1799 quads from the SSC, prioritized a total of 21 candidate RIGs, and investigated the functional relationships between RIGs and DNGs based on functional clusters and expression patterns in the human brain (Fig. 1).

Study workflow. The study included six phases: (1) genetic data collection, (2) identification and annotation of inherited variants, (3) burden analysis, (4) prioritization of ASD-associated RIGs, (5) RIG/DNG functional network analysis, and (6) RIG/DNG brain-expression analysis. ASD, autism spectrum disorder; Dmis, deleterious missense variants; DNGs, genes with de novo variants; PTVs, protein truncating variants; RIGs, recessive inherited genes
Materials and methods
Annotation and identification of inherited variants
Genetic data in VCF format (SSC_WES_3), and the corresponding phenotypic information were downloaded from the SFARI Base (https://www.sfari.org/resource/sfari-base/). Only coding variants with sequencing depth > 20, genotype quality > 50, and call rate > 0.9 were retained for further analysis. Quad samples containing a proband, an unaffected sibling, and their unaffected parents were analyzed. Informed consent was provided in the original study [15]. Comprehensive annotation of all variants was performed by ANNOVAR [41] and VarCards [42], as previously described [29, 32, 33]. Annotation included gene regions, amino acid alterations, mutation effects, GenBank mRNA accession numbers, and minor allele frequencies (MAFs) in the Genome Aggregation Database (gnomAD database, https://gnomad.broadinstitute.org/) [43]. All coding variants were classified into five functional categories: (1) PTVs, including alternative splice variants (defined as variants that occurred within ± 2 bp of an exon–intron boundary), stop-gain and stop-loss single-nucleotide variants, and frameshift indels; (2) Dmis variants with ReVe scores > 0.7 [44]; (3) tolerated missense variants with ReVe scores ≤ 0.7; (4) synonymous variants; and (5) non-frameshift indels.
For the 1,799 quads, we focused on biallelically inherited variants in autosomes with MAF ≤ 1% and X-linked inherited variants in males with MAF ≤ 0.1% based on the SSC cohort and the gnomAD database. For all probands and siblings, we counted the number of samples carrying inherited variants and classified them in the following six groups: (1) biallelically inherited PTVs, including protein-truncating homozygous or compound heterozygous variants in autosomes (genes carrying one PTV and one Dmis in trans were also defined as biallelic PTVs); (2) biallelically inherited Dmis variants, including homozygous or compound heterozygous Dmis variants in autosomes; (3) biallelically inherited synonymous variants in autosomes; (4) X-linked inherited PTVs in males; (5) X-linked inherited Dmis variants in males, and (6) X-linked inherited synonymous variants in males. For each family, biallelically inherited variants in autosomes that were shared between the probands and siblings were removed, and only proband- or sibling-specific biallelically inherited variants were counted. Additionally, the X-linked inherited variants that were shared between male probands and male siblings were excluded. Fisher's exact test was used to calculate the statistical significance of differences in mutational burden between probands with ASD and unaffected siblings. Moreover, we limited the mutational burden analysis to brain-expressed genes and biallelically or hemizygous constrained genes. The brain-expressed genes were defined as genes with an average expression value ≥ 1 read per kilobase per million map reads [RPKM] in human fetal brain samples based on the BrainSpan database (https://www.brainspan.org/) [45]. The biallelically or hemizygous constrained genes were defined as genes carrying no more than five homozygous or hemizygous Dmis variants and no more than two homozygous or hemizygous PTVs in the gnomAD database.
Prioritization of ASD-associated RIGs
Because biallelically inherited and X-linked inherited PTVs significantly differed between the probands and unaffected siblings, we focused on these PTVs to further prioritize candidate genes and filtered them, as the following standard: (1) PTVs recorded as benign variants in the ClinVar database; (2) X-linked inherited PTVs that were present in > 10 males in the gnomAD database; (3) genes in autosomes harboring biallelically inherited PTVs in the unaffected siblings or genes in the X chromosome harboring X-linked inherited PTVs in the unaffected male siblings; and (4) genes in the X chromosome with a probability of loss-of-function intolerance (pLI) < 0.5, as sourced from the gnomAD database. The phenotypes and inheritance patterns of the prioritized RIGs were curated using the Online Mendelian Inheritance in Man (OMIM: https://omim.org) database [46]. We applied the guidelines of the American College of Medical Genetics and Genomics [47] to interpret the clinical pathogenicity of inherited variants of known disease-associated genes, which were classified into pathogenic/likely pathogenic and variants of uncertain significance (VUS). Inherited variants of newly reported candidate genes were all interpreted as VUS. Additionally, the RIGs prioritized in this study were integrated with 49 RIGs from the ASC cohort [39] and other studies [36, 37] for further analysis.
A total of 87 DNGs with a false discovery rate (FDR) < 0.1 were sourced from our recently developed Gene4Denovo database (https://genemed.tech/gene4denovo) [48]. As described in our previous studies [29, 32], we used the RNA sequencing (RNA-seq) data of 524 human brain samples from 16 brain regions across different developmental stages from the BrainSpan database and protein–protein interaction (PPI) data from the STRING database (https://string-db.org/) [49] to evaluate functional relationships between ASD-associated RIGs and DNGs. Pearson’s correlation coefficients (r) between gene pairs were determined based on their expression in the human brain samples. When |r|≥ 0.8, genes were regarded as co-expressed, and if two proteins had a PPI confidence score ≥ 400, they were regarded to interact. A permutation test, as described in our previous studies [29, 32], was performed to evaluate the relationships between ASD-associated RIGs and DNGs while considering gene-level background DNV rates and gene lengths.
Functional clusters of ASD-associated RIGs and DNGs
Co-expression and PPI data were used to construct a functional network of RIGs and DNGs. We paired any two ASD-associated RIGs or DNGs if they were co-expressed or found to interact at the protein level and combined these relationships to form an interconnected network, which was visualized in Cytoscape (v.3.6: https://cytoscape.org/). Gene ontology (GO) enrichment analysis of RIGs and DNGs was performed by using Metascape (https://metascape.org/) with default parameters. Similar GO terms were merged, and only the most significant GO term in each cluster was shown. Additionally, we sourced fragile X mental retardation protein (FMRP) targets [50] and essential genes [51], as described in a previous study [18].
Brain expression patterns of RIGs and DNGs
RNA-seq data from the BrainSpan database were used to determine the spatiotemporal expression patterns of ASD-associated RIGs and DNGs, as described in previous studies [29, 32]. Hybrid-weighted gene co-expression network analysis (WGCNA) [52] was conducted to cluster ASD-associated RIGs and DNGs into different co-expression modules using standard protocols and at a power of four. Additionally, we sourced transcriptome data from different layers of the developing neocortex (n = 526) [53] and characterized the laminar expression patterns of ASD-associated RIGs and DNGs by using WGCNA at a power of three. Furthermore, we downloaded a single-cell RNA-seq dataset from 15,928 nuclei in the human middle temporal gyrus from the Allen Brain Map database (https://portal.brain-map.org/atlases-and-data/rnaseq). These transcriptional profiles showed the RNA levels of all genes in 45 types of inhibitory neurons and 24 types of excitatory neurons. For each gene in each nucleus, the RPKM value (i.e., mRNA expression) was calculated based on the counts per million value downloaded from the Allen Brain Map database. Subsequently, the average expression of each gene in each neuronal type was quantified. Differences in ASD gene expression in inhibitory and excitatory neurons were evaluated by the Wilcoxon-rank sum test.
Results
Excess biallelically inherited and X-linked inherited PTVs in patients with ASD
In this study, we analyzed biallelically inherited variants in autosomes and X-linked inherited variants in samples from males in 1799 quad families, including 1,799 probands (1571 males and 228 females), 1799 unaffected siblings (847 males and 952 females), and their unaffected parents. Because autosomal biallelically inherited variants shared between probands and their unaffected siblings are less likely to contribute to ASD etiology, they were removed from burden analysis, thus only proband- and sibling-specific inherited variants were included (Table 1). Moreover, X-linked inherited variants shared between male probands and unaffected males were also removed. As a result, 25 of 1799 probands and 19 of 1,799 siblings harbored biallelically inherited PTVs with no significant difference (p = 0.45, odds ratio (OR) = 1.32). Furthermore, there was no significant difference between the numbers of probands and siblings carrying biallelically inherited Dmis variants in the 1799 quads (n = 32 vs. 30, p = 0.89, OR = 1.07). However, significantly more male probands (55 of 1571) carried X-linked inherited PTVs than male siblings (15 of 847) (p = 0.015, OR = 2.01). Additionally, there was no significant difference in X-linked inherited Dmis variants between male probands and male siblings (n = 125 vs. 55, p = 0.22, OR = 1.24). Following negative control comparisons, we observed no significant enrichment in biallelically inherited synonymous variants in probands compared to that in siblings (n = 65 vs. 61, p = 0.79, OR = 1.07) or in X-linked inherited synonymous variants in male probands compared to male siblings (n = 392 vs. 202, p = 0.55, OR = 1.06; Table 1).
We then assessed inherited PTVs and Dmis variants in biallelically or hemizygous constrained genes (according to the gnomAD database) that were expressed in the brain (according to the BrainSpan database), and observed that probands harbored significantly more biallelically inherited PTVs in autosomes (n = 10 vs 2, p = 0.038, OR = 5.02) and X-linked inherited PTVs in males (n = 23 vs. 4, p = 0.026, OR = 3.13) relative to their siblings (Table 1). However, we did not observe significant differences in biallelically inherited Dmis variants (n = 19 vs. 15, p = 0.61, OR = 1.27) or X-linked inherited Dmis variants (n = 73 vs. 29, p = 0.17, OR = 1.37) between probands and siblings. These results suggest that biallelically and X-linked inherited PTVs contribute more significantly to ASD than do Dmis variants (Table 1).
RIGs present higher genetic heterogeneity than DNGs in ASD
Based on the above analysis, we detected biallelically and X-linked inherited PTVs and Dmis variants in 33 genes in ASD probands. After filtering (Table S1), 21 ASD-associated RIGs were prioritized, including eight autosomal genes, and 13 X-linked genes (Fig. 1 and Table 2). Of these, 11 genes (AFF2, ATAD3A, CCDC22, CUL4B, HDAC8, MED12, RBMX, RFT1, UBE2A, USP9X, and VPS13B) have known links to neurodevelopmental disorders, two (ANO5 and CYBB) are involved in other genetic disorders, and associations between the remaining eight genes and genetic disorders are uncertain according to the OMIM database. We noted that 14 probands with biallelically or X-linked inherited variants in the RIGs, including five patients with biallelic variants in autosomal genes and nine male patients with X-linked variants, had an intelligence quotient (IQ) ≤ 90. Four of the identified genes (CUL4B, HDAC8, MED12, and USP9X) have been previously reported to harbor de novo PTVs and Dmis variants in male cases with neurodevelopmental disorders (Table S2), providing more genetic evidence of their pathogenicity.
Proband 12,651.p1, with an IQ of 34, carried pathogenic/likely pathogenic compound heterozygous variants (c.8185G > A, p.G2729R; c.2889G > A, p.W963X) in VPS13B, and was previously confirmed to have Cohen syndrome [37]. Proband 12,440.p1 with an IQ of 58, carried a likely pathogenic X-linked hemizygous splicing variant in UBE2A (c.329A > T, p.Q110L, at the end of exon 5), a known causative gene of ID. Among the eight newly reported genes, two are involved in the nervous system. First, we identified an X-linked inherited variant in GRIPAP1 at the end of exon 20 (c.1830G > A, p.A610A), which might alter RNA splicing. GRIPAP1 is specifically expressed in the nervous system and encodes a neuron-specific guanine nucleotide exchange factor for the Ras family of small G proteins [54]. Moreover, GRIPAP1 might regulate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor location and synaptic signal transmission in the brain [54]. Besides, a recent study identified an X-linked variant of apolipoprotein (APOO: c.350 T > c, p.I117T) among members of a family with mitochondrial disease, and most affected individuals exhibited developmental delay, cognitive impairment, and autistic features [55]. Here, we identified a potential inherited splicing variant of APOO that was not present in the gnomAD database.
The 21 RIGs harbored biallelically or X-linked inherited PTVs or Dmis variants in 26 of 1,799 (1.4%) patients in the SSC cohort (Table 2). X-linked inherited variants in three genes (IL13RA1, MED12, and CCDC22) were detected in two unrelated probands, and X-linked inherited variants in USP9X were detected in three unrelated probands. Additionally, 41 autosomal ASD-associated RIGs identified in a previous study based on the ASC cohort [39] were present in 44 of 2343 (1.88%) patients; these RIGs included three genes (CDHR3, FEV, and PAH) with variants in two unrelated patients and the others only in one patient (Table S3). No overlap was observed between the 41 autosomal RIGs from the ASC cohort and eight autosomal RIGs from the SSC cohort, suggesting high genetic heterogeneity among ASD-associated RIGs. Based on the genetic data regarding probands in the SSC cohort, we found that four of the 21 RIGs harbored multiple recessive inherited variants, whereas 36 of 87 DNGs with FDR < 0.1 from the Gene4Denovo database (Table S4) harbored multiple DNVs with slight differences (Fisher’s exact test: p = 0.078, OR = 0.34), indicating a higher genetic heterogeneity in RIGs than DNGs in ASD.
RIGs have higher functional heterogeneity than DNGs in ASD
After combining the 21 RIGs prioritized in this study with 41 from the ASC cohort [39] and 10 from two other previous studies [36, 37], 70 RIGs were included for further analysis after removing redundancy (Table S3). We investigated associations between RIGs and DNGs using transcription data from the BrainSpan database and PPI data from the STRING database. RIGs were co-expressed with 16 DNGs (p = 0.045) and formed 30 connections (p = 0.043), with the frequency of co-expression and connections significantly higher than randomly expected (Figure S1a). Additionally, we observed that ASD-associated RIGs interacted with 14 DNGs (p = 0.042) and 23 connections (p = 0.015) at the protein level based on PPI data, which was also significantly higher than those randomly expected (Figure S1b), again suggesting that ASD-associated RIGs were functionally related to DNGs.
We then connected any RIG or DNG that was co-expressed in the human brain at the transcript level or via interaction at the protein level to develop a functional network, that comprised 58 DNGs and 37 RIGs (Fig. 2a). This network was enriched in genes involved in histone modification (GO: 0016570, p = 1.07 10–9), proteolysis involved in cellular protein catabolic process (GO: 0051603, p = 2.76 10–4), neuron death (GO: 0070997, p = 2.02 10–5), cell morphogenesis involved in differentiation (GO: 0000904, p = 1.96 10–6) and chemical synaptic transmission (GO: 0007268, p = 4.30 10–8) (Fig. 2b and Table S5). Additionally, we observed significant enrichment of FMRP targets (p = 2.96 10–11) and essential genes (p = 2.69 10–9) in this functional network. We found that DNGs were more likely to be associated with chemical synaptic transmission and essential genes (p < 0.05) than RIGs (Fig. 2b). Moreover, functional analysis of all 157 genes (70 RIGs and 87 DNGs) indicated that DNGs were more likely to be associated with chemical synaptic transmission and cell morphogenesis involved in differentiation, FMRP targets, and essential genes than RIGs (all p < 0.05) (Fig. 2b). The same analysis was performed between the 70 RIGs and 102 ASD-associated DNGs recently identified by Satterstrom et al. [56] with similar results observed (Figure S2). These findings indicated that RIGs were less likely to be enriched in functional clusters involved in ASD etiology and might present higher functional heterogeneity than DNGs.

Functional network of ASD-associated RIGs and DNGs. a Network of ASD-associated RIGs and DNGs based on PPI and co-expression in the human brain. b Comparison of the numbers of RIGs and DNGs in the functional clusters. There were 85 genes in the network and 157 genes (RIGs and DNGs) in total. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. ASD, autism spectrum disorder; RIGs, recessive inherited genes; DNGs, genes with de novo variants; PPI, protein–protein interaction
RIGs have higher heterogeneity than DNGs in brain expression patterns
To explore the spatiotemporal expression patterns of RIGs and DNGs in the human brain, we performed WGCNA of all samples at different developmental stages from the BrainSpan database to identify co-expression modules. We identified two independent modules with different spatiotemporal expression patterns, comprising a total of 43 (61.43%) of 70 RIGs and 67 (77.01%) of the 87 DNGs (Fig. 3a and Table S6). Genes in module 1 (M1: n = 70) were highly expressed during the prenatal stages, but gradually decreased toward the end of the prenatal stage and remained relatively stable after birth. In contrast, genes in module 2 (M2: n = 40) exhibited the opposite spatiotemporal expression pattern. Although RIGs presented a spatiotemporal expression pattern similar to that of DNGs, as described in our previous study [29] and others studies [57, 58], we found that DNGs were more likely to be associated with M1 than RIGs (n = 47 vs. 23, Fisher’s test, p = 0.022, OR = 2.19) (Table S7).

Expression patterns of ASD-associated RIGs and DNGs in the human brain. a Heat map of the normalized expression levels of two spatiotemporal co-expression modules (M1 and M2), corresponding to 17 developmental stages and 16 sub-regions in the human brain. pcw, post-conceptual weeks; yrs, years. CBC, cerebellum cortex; MFC, anterior cingulate cortex; OFC, orbital frontal cortex; DFC, dorsolateral prefrontal cortex; VFC, ventrolateral prefrontal cortex; M1C, primary motor cortex; S1C, primary somatosensory cortex; IPC, posteroinferior parietal cortex; A1C, primary auditory cortex; STC, posterior superior cortex; ITC, inferolateral temporal cortex; VIC, primary visual cortex; HIP, hippocampus; AMY, amygdaloid complex; STR, striatum; MD, mediodorsal nucleus of thalamus; CBC, cerebellar cortex. b Heat map of the normalized expression levels of all RIGs and DNGs in different cortical layers. SG, subpial granular zone; MZ, marginal zone; CPo, outer cortical plate; CPi, inner cortical plate; SP, subplate zone; IZ, intermediate zone; SZo, outer subventricular zone; SZi, inner subventricular zone; VZ, ventricular zone. (C) Heat map of the normalized expression levels of RIGs and DNGs in 45 inhibitory and 24 excitatory neuronal types. P values were calculated by Wilcoxon rank sum test. Expression level is presented according to the log2 value of the average expression of each gene
Because the expression of RIGs and DNGs showed significant fluctuation in the human brain during the prenatal period, we characterized prenatal neocortical-expression patterns using previously reported laser-microdissection data from prenatal neocortical samples [53]. Interestingly, we identified three co-expression modules (Ma, Mb, and Mc) with distinct laminar expression patterns, and comprising 44 (62.86%) RIGs and 61 (70.11%) DNGs (Fig. 3b and Table S6). The expression of genes within Ma (n = 52), the biggest module, was higher in the middle layers (from the marginal zone to subplate zone) and lower in the deep cortical layers and the outer subventricular zone to the ventricular zone. In contrast, gene expression in Mb (n = 25) displayed the opposite trends. However, genes within Mc (n = 31) did not display any laminar expression features. Although the three modules contained both RIGs and DNGs, we found that DNGs were significantly more likely to be associated with Ma than RIGs (n = 35 vs. 17, p = 0.041, OR = 2.09), whereas RIGs were more likely to be associated with Mb than DNGs (n = 10 vs. 15, p = 0.12, OR = 0.48).
Complex neuronal diversity and connectivity are vital to human brain function [59]. Single-nucleus RNA-seq analysis of the human brain allows the characterization of the inhibitory and excitatory neuronal expression patterns of ASD-associated RIGs. Therefore, we compared the expression of RIGs and DNGs in 45 types of inhibitory (GABAergic) neurons and 24 types of excitatory (glutamatergic) neurons. In total, 53 genes (nRIG = 18 and nDNG = 35) showed higher expression in excitatory neurons than in inhibitory neurons, whereas 27 genes (nRIG = 13 and nDNG = 14) showed higher expression in inhibitory neurons than in excitatory neurons, with 77 genes exhibiting no significant difference between neuron classes (Fig. 3c and Table S7). Although 50% of genes were broadly expressed in both inhibitory and excitatory neurons, more genes containing RIGs and DNGs were preferentially expressed in excitatory neurons, thereby highlighting their essential roles in the cortical circuit. Notably, although the difference was not significant, DNGs were more likely to be associated with excitatory neurons than RIGs (p = 0.063, OR = 1.94). Taken together, these results revealed that RIGs were less likely to present distinct expression patterns in the human brain, suggesting higher transcriptomic heterogeneity than DNGs.
To validate whether the observations between RIGs and DNGs were ASD specific, we extracted ID-associated RIGs (n = 120) from the OMIM database and DNGs (n = 82) from the Gene4Denovo database. ID-associated DNGs tended to be associated with the module that was highly expressed during the prenatal stages (M1: OR = 1.64, p = 0.088) (Table S7), and were more likely to be associated with excitatory neurons than RIGs (OR = 2.06, p = 0.040) (Table S7). We then compared the expression patterns of congenital heart disease (CHD) associated RIGs (n = 88) and DNGs (n = 78) from a previous study [60] and the Gene4Denovo database, respectively, and did not observe a significant enrichment of DNGs relative to RIGs in M1 (OR = 1.22, p = 0.53) or in the middle cortical layers (Ma: OR = 1.04, p = 1.0). Additionally, compared with RIGs, DNGs associated with CHD were not more enriched in excitatory neurons (OR = 0.98, p = 1.0) (Table S7). These observations suggest that the differences in expression patterns between DNGs and RIGs are specific to neurodevelopmental disorders.
Discussion
ASD is highly heritable, and DNVs are estimated to contribute to the disease in up to 30% of cases; however, the genetic defects in most ASD cases remain unclear [61]. Few studies have attempted to evaluate the effects of inherited PTVs and prioritize ASD-associated RIGs [36,37,38,39]. In 1799 quads from the SSC cohort, we observed that ASD probands were more likely to carry biallelically and X-linked inherited PTVs in brain-expressed and biallelically or hemizygous constrained genes than unaffected siblings, consistent with a previous study [38]. Given that a trend toward the enrichment of recessive inherited Dmis variants has been observed in ASD [39], we believe that a fraction of these variants might be involved in disease etiology. However, larger sample sizes and functional studies will be required to estimate the contributions of these variants to ASD.
We prioritized 21 RIGs with biallelically or X-linked inherited PTVs and Dmis variants found in ASD probands only, including 13 genes associated with known genetic disorders. We hypothesized that these ASD-associated RIGs contribute to a broad range of neurological phenotypes, with different penetrance in each phenotype. Furthermore, we reported eight genes yet to be linked to any genetic disorders, with possible associations with ASD. The biallelically or X-linked inherited variants of the 21 RIGs were present in 1.4% of ASD cases in the SSC cohort, which was close to estimations (1.8–3%) in previous large cohort studies [38, 39]. We encourage further studies to validate our candidate genes, detect more inherited variants in neurodevelopmental disorders, and characterize genotype–phenotype correlations, which would contribute to ASD subtype definitions.
Although a previous study detected four RIGs in ASD [36], none of them harbored biallelically or X-linked inherited variants in the ASC cohort or in this study. Yu et al. [37] detected six RIGs, one of which (PAH) was found to harbor different biallelically inherited variants in the ASC cohort (Table S3). Similarly, Doan et al. [39] reported 41 RIGs in 2343 probands, and identified only three genes with variants in more than one proband. In the present study, we identified 21 RIGs from 1799 quads and found four X-linked genes harboring inherited variants in multiple probands. Most RIGs harbored biallelically or X-linked inherited variants in only one proband, in both the SSC and ASC cohorts, with few RIGs overlapping between cohorts; this highlighted the high genetic heterogeneity of ASD-associated RIGs and the need for larger sample sizes in future studies. However, we cannot fully exclude the influence of technical differences between different studies.
This study highlights the genetic similarities and differences between ASD-associated RIGs and DNGs from different perspectives. First, although both DNGs and RIGs displayed genetic heterogeneity, RIGs were less likely to harbor multiple biallelically or X-linked inherited variants than DNGs were to carry multiple DNVs, suggesting a higher genetic heterogeneity in RIGs. Second, RIGs and DNGs were functionally interconnected, forming a functional network of known pathways involved in ASD [13]. However, some functional clusters showed a preference for DNGs, whereas no functional cluster showed a preference for RIGs, further suggesting higher functional heterogeneity in RIGs. Third, RIGs were significantly co-expressed with DNGs at the mRNA level in the human brain and shared spatiotemporal expression patterns, neocortex laminar expression patterns, and excitatory neuronal expression patterns with DNGs [29, 62]. However, RIGs were less likely to be associated with prenatal brain regions, middle cortical layers, and excitatory neurons, which are associated with ASD and other neuropsychiatric disorders [29, 34, 62,63,64], again suggesting the higher transcriptomic heterogeneity of RIGs. Furthermore, expression patterns of RIGs and DNGs were similar in ASD and ID cases but differed from those in CHD cases, suggesting that these expression patterns might be specific to neurodevelopmental disorders.
Limitations
We acknowledge several limitations in this study. First, because we only observed significant differences in biallelically and X-linked inherited PTVs between probands and siblings rather than Dmis variants, we prioritized candidate genes with PTVs and might have missed promising candidate genes harboring Dmis variants. Second, the 21 candidate genes and biallelically or X-linked inherited variants should be confirmed in additional studies and require functional validation. Third, because the samples were derived from the SSC cohort, we were unable to re-examine patients with biallelically or X-linked inherited variants to further characterize their detailed clinical phenotypes.
Conclusions
In summary, our study showed that biallelically or X-linked inherited variants contribute to ASD but only occur in only a small fraction of ASD cases. Moreover, the analyses of functional clusters and expression patterns suggested that ASD-associated RIGs were functionally correlated with DNGs; however, they presented higher genetic and functional heterogeneity, providing strong evidence for the need for further studies regarding the molecular etiology of ASD.
Availability of data and materials
The data used in this study are available from the corresponding author upon reasonable request.
Abbreviations
- ASC:
-
Autism sequencing consortium
- ASD:
-
Autism spectrum disorder
- CHD:
-
Congenital heart disease
- Dmis:
-
Deleterious missense
- DNGs:
-
Genes with de novo variants
- DNV:
-
De novo variant
- FDR:
-
False discovery rate
- FMRP:
-
Fragile X mental retardation protein
- gnomAD:
-
Genome aggregation database
- GO:
-
Gene ontology
- ID:
-
Intellectual disability
- IQ:
-
Intelligence quotients
- MAF:
-
Minor allele frequency
- OMIM:
-
Online Mendelian Inheritance in Man
- OR:
-
Odds ratio
- PPI:
-
Protein–protein interaction
- PTVs:
-
Protein truncating variants
- RIGs:
-
Recessive inherited genes
- RNA-seq:
-
RNA sequencing
- RPKM:
-
Reads per kilobase per million
- SSC:
-
Simons simplex collection
- VUS:
-
Variants of uncertain significance
- WES:
-
Whole-exome sequencing
- WGCNA:
-
Weighted gene co-expression network analysis
References
Elsabbagh M, Divan G, Koh YJ, Kim YS, Kauchali S, Marcin C, et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012;5:160–79.
Lord C, Elsabbagh M, Baird G, Veenstra-Vanderweele J. Autism spectrum disorder. Lancet. 2018;392:508–20.
Tick B, Bolton P, Happe F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry. 2016;57:585–95.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–9.
Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu X-Q, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–28.
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–73.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72.
O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–9.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99.
Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–5.
O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–50.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–15.
Dong S, Walker MF, Carriero NJ, DiCola M, Willsey AJ, Ye AY, et al. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. 2014;9:16–23.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21.
Hashimoto R, Nakazawa T, Tsurusaki Y, Yasuda Y, Nagayasu K, Matsumura K, et al. Whole-exome sequencing and neurite outgrowth analysis in autism spectrum disorder. J Hum Genet. 2016;61:199–206.
Lim ET, Uddin M, De Rubeis S, Chan Y, Kamumbu AS, Zhang X, et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Neurosci. 2017;20:1217–24.
Takata A, Miyake N, Tsurusaki Y, Fukai R, Miyatake S, Koshimizu E, et al. Integrative analyses of De Novo mutations provide deeper biological insights into autism spectrum disorder. Cell Rep. 2018;22:734–47.
Feliciano P, Zhou X, Astrovskaya I, Turner TN, Wang T, Brueggeman L, et al. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom Med. 2019;4:19.
Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–5.
Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–42.
Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21:185–91.
Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, et al. Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am J Hum Genet. 2016;98:58–74.
Yuen R, Merico D, Cao H, Pellecchia G, Alipanahi B, Thiruvahindrapuram B, et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom Med. 2016;1:160271–1602710.
Yuen RKC, Merico D, Bookman M, Howe JL, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20:602–11.
Guo H, Duyzend MH, Coe BP, Baker C, Hoekzema K, Gerdts J, et al. Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Genet Med. 2019;21:1611–20.
Ruzzo EK, Perez-Cano L, Jung JY, Wang LK, Kashef-Haghighi D, Hartl C, et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 2019;178(850–866):e826.
Zhou J, Park CY, Theesfeld CL, Wong AK, Yuan Y, Scheckel C, et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat Genet. 2019;51:973–80.
Li J, Wang L, Guo H, Shi L, Zhang K, Tang M, et al. Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol Psychiatry. 2017;22:1282–90.
Guo H, Wang T, Wu H, Long M, Coe BP, Li H, et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol Autism. 2018;9:64.
Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. 2016;7:13316.
Li J, Cai T, Jiang Y, Chen H, He X, Chen C, et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol Psychiatry. 2016;21:290–7.
Li J, Wang L, Yu P, Shi L, Zhang K, Sun ZS, et al. Vitamin D-related genes are subjected to significant de novo mutation burdens in autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet. 2017;174:568–77.
Li J, Hu S, Zhang K, Shi L, Zhang Y, Zhao T, et al. A comparative study of the genetic components of three subcategories of autism spectrum disorder. Mol Psychiatry. 2018.
Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015.
Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, Hill RS, et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8:e1002635.
Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–73.
Lim ET, Raychaudhuri S, Sanders SJ, Stevens C, Sabo A, MacArthur DG, et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013;77:235–42.
Doan RN, Lim ET, De Rubeis S, Betancur C, Cutler DJ, Chiocchetti AG, et al. Recessive gene disruptions in autism spectrum disorder. Nat Genet. 2019;51:1092–8.
Fischbach GD, Lord C. The simons simplex collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–5.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Li J, Shi L, Zhang K, Zhang Y, Hu S, Zhao T, et al. VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018;46:D1039–48.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Li J, Zhao T, Zhang Y, Zhang K, Shi L, Chen Y, et al. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018;46:7793–804.
Sunkin SM, Ng L, Lau C, Dolbeare T, Gilbert TL, Thompson CL, et al. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 2013;41:D996–1008.
Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–D798.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Zhao G, Li K, Li B, Wang Z, Fang Z, Wang X, et al. Gene4Denovo: an integrated database and analytic platform for de novo mutations in humans. Nucleic Acids Res. 2019.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–8.
Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–61.
Georgi B, Voight BF, Bucan M. From mouse to human: evolutionary genomics analysis of human orthologs of essential genes. PLoS Genet. 2013;9:e1003484.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559.
Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206.
Ye B, Liao D, Zhang X, Zhang P, Dong H, Huganir RL. GRASP-1: a neuronal RasGEF associated with the AMPA receptor/GRIP complex. Neuron. 2000;26:603–17.
Beninca C, Zanette V, Brischigliaro M, Johnson M, Reyes A, Valle DAD, et al. Mutation in the MICOS subunit gene APOO (MIC26) associated with an X-linked recessive mitochondrial myopathy, lactic acidosis, cognitive impairment and autistic features. J Med Genet. 2020.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-Scale Exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180(568–584):e523.
Ben-David E, Shifman S. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol Psychiatry. 2013;18:1054–6.
Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–21.
Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352:1586–90.
Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat Genet. 2017;49:1593–601.
Vorstman JAS, Parr JR, Moreno-De-Luca D, Anney RJL, Nurnberger JI Jr, Hallmayer JF. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet. 2017;18:362–76.
Coe BP, Stessman HAF, Sulovari A, Geisheker MR, Bakken TE, Lake AM, et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet. 2019;51:106–16.
Nguyen HT, Bryois J, Kim A, Dobbyn A, Huckins LM, Munoz-Manchado AB, et al. Integrated Bayesian analysis of rare exonic variants to identify risk genes for schizophrenia and neurodevelopmental disorders. Genome Med. 2017;9:114.
Shohat S, Ben-David E, Shifman S. Varying intolerance of gene pathways to mutational classes explain genetic convergence across neuropsychiatric disorders. Cell Rep. 2017;18:2217–27.
Acknowledgements
We thank the Simons Simplex Collection for their excellent work in the collection and management of patient data, and the patients and their families for their participation. We acknowledge SFARI Base for providing access to phenotypic data.
Funding
This work was supported by the National Natural Science Foundation of China (81730036 and 81525007 to K Xia; 31601027 to ZS Sun; 8200053490 to L Wang; and 81801133 and 32070591 to JC Li), the Key R&D Program of Hunan Province (2016JC2055 to K Xia), Science and Technology Innovation Leader of Hunan Province (2016RS2001 to K Xia), Science and Technology Major Project of Hunan Provincial Science and Technology Department (2018SK1030 to K Xia), Guangdong Key Project in Development of new tools for diagnosis and treatment of Autism (2018B030335001 to ZS Sun), Young Elite Scientist Sponsorship Program by CAST (2018QNRC001 to JC Li) and the Innovation-Driven Project of Central South University (20180033040004 to JC Li).
Author information
Authors and Affiliations
Contributions
XK, SZ, and LJ designed the study. WL and LJ wrote the manuscript. WL, ZY, LK, WZ, and WX collected and organized the data and performed bioinformatics analysis. All authors contributed to the preparation of the manuscript and read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent from all subjects was obtained by SFARI. All procedures performed in this study were in accordance with the ethical standards of the Institutional Review Board of the School of Life Sciences at Central South University, Changsha, Hunan, China.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Figure S1. Permutation test of connections between RIGs and DNGs. Figure S2. The functional network of 70 RIGs in this study and 102 DNGs from Satterstrom et al. Table S1. Summary of genes excluded when prioritizing candidate genes.Table S2. De novo variants in genes with X-linked inherited variants based on the Gene4Denovo database. Table S3. All RIGs in ASD included in this study. Table S4. ASD-associated DNGs were sourced from the Gene4denovo database. Table S5. Functional enrichment of ASD-associated RIGs and DNGs. Table S6. Expression patterns of 70 RIGs and 87 DNGs in ASD. Table S7. Comparison of expression patterns between DNGs and RIGs in ASD, ID and CHD.
Web Resources
Allen Brian Map, https://portal.brain-map.org/atlases-and-data/rnaseq
BrainSpan, https://www.brainspan.org
Cytoscape, https://cytoscape.org/
Gene4Denovo, http://genemed.tech/gene4denovo
Metascape, https://metascape.org/
OMIM, https://omim.org
SFARI Base, https://www.sfari.org/resource/sfari-base/
STRING, https://string-db.org〹
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, L., Zhang, Y., Li, K. et al. Functional relationships between recessive inherited genes and genes with de novo variants in autism spectrum disorder. Molecular Autism 11, 75 (2020). https://doi.org/10.1186/s13229-020-00382-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-020-00382-x