
Abstract
Genetic factors contribute to the development of autism spectrum disorder (ASD), and although non-protein-coding regions of the genome are being increasingly implicated in ASD, the functional consequences of these variants remain largely uncharacterized. Induced pluripotent stem cells (iPSCs) enable the production of personalized neurons that are genetically matched to people with ASD and can therefore be used to directly test the effects of genomic variation on neuronal gene expression, synapse function, and connectivity. The combined use of human pluripotent stem cells with genome editing to introduce or correct specific variants has proved to be a powerful approach for exploring the functional consequences of ASD-associated variants in protein-coding genes and, more recently, long non-coding RNAs (lncRNAs). Here, we review recent studies that implicate lncRNAs, other non-coding mutations, and regulatory variants in ASD susceptibility. We also discuss experimental design considerations for using iPSCs and genome editing to study the role of the non-protein-coding genome in ASD.
Similar content being viewed by others


Overview
Autism spectrum disorder (ASD) is a neurodevelopmental disorder with complex genetic underpinnings, and our current understanding of specific genetic risk for ASD comes from the studies of rare mutations affecting DNA that encodes protein-coding exons and genes (for a comprehensive review of the neurobiology and genetics of ASD, see [1]). However, such protein-coding exons represent less than 2% of the human genome, and genome-wide association studies suggest that many ASD-associated variants map to intragenic and intronic regions, as well as non-protein-coding intervals [2]. The recent application of whole-genome sequencing (WGS), which captures the vast majority of chromosomal DNA, has led to the identification of increasing numbers of ASD-associated variants that affect RNA splicing [3,4,5,6], long non-coding RNAs (lncRNAs) [7], and transcriptional regulatory elements [4,5,6, 8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24] (Table 1). However, the functional consequences of non-coding variants are difficult to predict [25] and validate.
Although genetically modified rodents can be invaluable model systems to explore functions of ASD-associated protein-coding genes [26], human regulatory elements and non-coding RNAs are not always conserved in mice or rats. Notable interspecies differences have been identified across vertebrates for mechanisms governing the expression of conserved protein-coding genes [27]. Some human regulatory regions are newly evolved or undergo accelerated evolution [28, 29]. Furthermore, among the thousands of known human lncRNAs, nearly one third arose specifically in primate lineages [30]. Together, these observations suggest that human neurons are a more relevant model system for exploring, at least initially, the functions of ASD-associated non-coding variants.
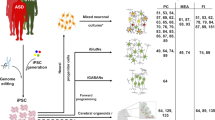
Induced pluripotent stem cells (iPSCs) can produce inexhaustible supplies of personalized neurons that are genetically matched to individuals with ASD or unaffected individuals [31]. CRISPR genome editing has also facilitated the generation of customized neurons with specific variants [32, 33]. iPSC-derived neurons have been used to model ASD, and these studies have consistently implicated altered synaptic function in the underlying pathophysiology of ASD, although the specific mechanisms of synaptic dysfunction vary between models [20, 32, 34,35,36,37,38,39,40,41,42,43,44] (Table 2). Compared to protein-coding genes, experimental perturbation of regulatory elements and non-coding RNAs are more technically difficult, and phenotypic effects may be challenging to detect or interpret [45,46,47]. We review recent insights into the role of non-coding and regulatory genetic variants in ASD, and we discuss future directions for using human iPSCs and genome editing to explore their functional consequences (Fig. 1).

Graphical summary of how hPSC-derived neurons can be used to model the functional consequences of ASD-associated non-coding variants. WGS will identify de novo variants in people with ASD (*), which will then be mapped to specific locations in the genome. Genomic loci are annotated as functional elements based on transcriptomics, chromatin state analyses, and computation predictions. ASD-associated variants can be modeled using personalized iPSCs from people with ASD or by genome editing to introduce or repair ASD-associated variants. hPSC-derived neurons are then made by directed differentiation (via a NPC stage) or direct conversion, and functional consequences of non-coding variants are determined by analyzing gene expression and connectivity
Gene regulatory factors in ASD
ASD is now increasingly considered a disorder of synaptic connectivity, and the growing list of ASD-relevant genes has largely converged on two biological processes: synaptic transmission and regulation of gene expression [1, 48, 49]. Known ASD genes involved in gene regulatory processes include transcription factors, RNA-binding proteins, and chromatin regulatory factors, many of which regulate expression of other autism risk genes, and additional regulators of synapse function [1]. ASD-associated chromatin regulators functionally converged upon methylation of H3K4 [50], which is important for the functional demarcation of promoters and enhancers [51]. Recurrent disruption of writers, erasers, and readers of H3K4 methylation in ASD [50] has led to speculation that ASD may be an “enhanceropathy” or a disorder caused by misregulated gene expression [52]. Neuronal gene expression must be finely tuned during development and in response to synaptic activity, so genetic variants that disrupt or alter regulatory elements could have a profound impact on the formation and refinement of synaptic networks [48, 53].
Non-coding regulatory variants in ASD
Non-coding elements were initially implicated in ASD by analyses of copy number variation (CNV), which identified 15 intergenic loci that were sites of recurrent genomic rearrangement found in ASD subjects [13]. Most of these variants disrupted putative regulatory elements or non-coding RNAs, and several mapped near protein-coding genes that were associated with ASD (CHD8, DIPK2A/C3orf58, and NRXN1) or neuronal function (ASTN2, EPH5A, SEMA3C, UNC5D). It was initially unknown whether non-coding variants would play a substantial role in ASD, but subsequent WGS studies suggest that approximately 5% of ASD cases may be accounted for by non-coding variants [54]. Also, the determination that non-coding variants largely converge on the same functional processes as ASD-associated protein-coding genes strongly supported the potential for non-coding variants to play causal roles in ASD development [4, 6]. Furthermore, the IQ of people with ASD inversely correlates with the burden of specific RNA regulatory variants, suggesting that non-coding variants may provide novel insights into the clinical heterogeneity of ASD [6]. Relative to exonic variants, it is difficult to predict the functional consequences of non-coding variants [25], although computational tools continue to evolve for prioritization of such variants [2, 4, 6, 12, 55,56,57]. Several approaches used epigenetic information (e.g., histone marks, chromatin accessibility, and transcription factor binding) from different cell types to computationally predict tissue-specific expression effects of non-coding variants [6, 56, 58]. Interestingly, when tested against known expression quantitative trait loci from the Genotype-Tissue Expression (GTEx) project, one such deep learning approach correctly predicted the directions of expression changes for hundreds of strong effect variants [58]. WGS studies implicated cis-regulatory elements like promoters, enhancers, and RNA regulatory sequences in ASD, and many of the genes that are regulated by these elements have been functionally or genetically linked to ASD (Table 1). Next, we highlight recent findings that illustrate the importance of non-coding variants in gene regulation and ASD.
Transcriptional regulatory elements in ASD
WGS studies have revealed extensive evidence for ASD-associated non-coding variants in transcriptional regulatory elements like promoters and enhancers, which are functionally annotated based on transcriptomics data and chromatin state analyses [59,60,61,62]. WGS studies reported that non-coding variants were enriched in conserved enhancers [4] that are accessible in the fetal brain [8] or predicted to regulate ASD genes [5, 8, 12]. WGS has also detected de novo ASD-associated variants in distal promoters (750–2000 bp upstream of transcription start sites), which had a significant association with transcription factor binding sites [9]. Rare recurrent variants were found disrupting predicted promoters for DLG2 and NR3C2, which have both been implicated in brain function or neurodevelopment [11].
Several ASD-associated variants of transcriptional regulatory elements have been shown to directly affect gene expression. For example, one ASD-associated single nucleotide variant mapped to a predicted transcription factor binding site and drove aberrant expression of a reporter gene in the developing mouse forebrain, where the reference sequence was not active [5]. Reporter assays were also used to test transcriptional regulatory properties of variants near known ASD risk genes. Specifically, when 59 prioritized variants from ASD probands were compared to control sequences from their unaffected siblings, 96% of these variants drove significant allele-specific changes in reporter gene expression [6]. Another study reported ASD-associated paternally inherited deletions mapping upstream of the LEO1 gene, which was previously implicated in ASD by exome sequencing [10]. Interestingly, fibroblast cell lines revealed elevated expression of two neighboring protein-coding genes (LEO1 and MAPK6) in carriers of the deletion who had ASD. The deletions encompass a predicted regulatory element that interacted with the promoters of both LEO1 and MAPK6 [10]. This regulatory element is thought to be important for ASD risk because the authors also reported a partially overlapping polymorphic deletion that preserved this element and was common in people who did not have ASD [10]. Together, these studies revealed that ASD-associated variants in non-coding regulatory elements can directly affect the expression of neurodevelopmental genes and ASD risk genes.
Post-transcriptional regulatory elements in ASD
Misregulation of RNA splicing has also been implicated in ASD (reviewed in [63]), although the underlying genetic mechanisms have not been extensively explored. Alternative splicing is the regulated inclusion or exclusion of specific exons during mRNA processing, which can have a profound impact on protein function. RNA-seq analyses of the brains from individuals with ASD revealed the downregulation of splicing proteins RBFOX1 [64] and nSR100 [65], which were associated with altered splicing of a subset of synaptic genes or genes with conserved microexons (~ 3–15 nucleotides), respectively. RNA-seq analyses also revealed ASD-associated changes in activity-dependent alternative splicing events and regional gene expression patterns in the cortex [23]. Activity-dependent alternative splicing of microexons in EIF4G1 and EIF4G3 is impaired in ASD, and deletion of the Eif4g1 microexon in mice led to prolonged neuronal activation, altered synaptic plasticity, and impaired social interactions [66]. These microexons encode prion-like domains, and their loss led to aberrant translation of proteins that control synaptic transmission and neuronal activity.
WGS studies have begun to uncover ASD-associated non-coding variants in splice signals and untranslated regions. For example, intronic single nucleotide variants that were predicted to alter splicing of transcripts from synaptic genes like GRIN1 [3], the ASD candidate gene PTEN [3], and the neurodevelopmental gene SMEK1 [6]. WGS studies also reported that ASD-associated de novo variants were enriched in splice sites or untranslated regions (UTRs) of mRNAs [4, 5], particularly UTRs of known ASD risk genes and neurodevelopmental genes [17]. The study of ASD-associated de novo variants in 3’ UTRs of brain-specific transcripts is of particular importance since neuronal mRNAs have the longest 3’ UTRs among all tissues, implying that these molecules are under strong post-transcriptional regulation [67]. Moreover, a few studies reported variants that disrupt untranslated exons of the ASD gene MBD5 [14,15,16] (Table 1).
UTRs often harbor binding sites for regulatory RNA-binding proteins and microRNAs (miRNAs) [68], and miRNAs have also been implicated in ASD. miRNAs are short (19–25 nucleotide), non-coding RNA molecules that bind to the UTRs of target mRNAs and affect mRNA stability or translation [69]. miRNAs have been implicated in ASD by genetic studies and animal models, although mechanisms remain largely unexplored. Heterozygous mutation of AGO1, which is critical for miRNA function, is associated with a syndromic neurodevelopmental disorder that includes ASD features [70]. Mice with targeted deletions of specific miRNAs or miRNA clusters exhibit pronounced changes in social behavior [71, 72]. The latest release of the miRBase database (v22) accounts for 2654 mature miRNAs identified in humans [73], the majority of which are expressed in the brain [74]. Dozens of microRNAs are consistently misregulated in ASD, and known ASD genes are enriched among targets of these miRNAs [75, 76]. For example, 28 miRNAs differentially expressed in the cerebellar cortexes of people with ASD, 7 of which target the SHANK3 mRNA [77]. Also, candidate genes for ASD and schizophrenia were enriched for two miRNA target sequences, although the associated miRNAs were not reported [78]. Despite these examples of ASD-associated miRNA misregulation, there are few reports describing causal links between mutations in the UTRs of ASD-risk genes that affect the protein outputs of their respective mRNAs.
Several WGS or whole-exome sequencing studies have reported genetic variants predicted to affect miRNAs or miRNA target sequences. One study specifically tested the hypothesis that ASD-associated synonymous variants in coding sequences may affect miRNA binding sites, although no significant enrichment for miRNA binding sites was detected [79]. The miR-133b/miR-206 cluster was implicated in ASD in a genome-wide association study (GWAS) discovery cohort, although no significant association was detected in a replication cohort [80]. WGS revealed sequence variants that affect the ability of miR-873-5p to bind and regulate ASD-associated target genes, including NRXN2 and CNTNAP2 [17]. Together, these findings suggest that ASD-associated variants in post-transcriptional regulatory elements may have the potential to affect splicing, stability, and translation of protein-coding transcripts.
Long non-coding RNAs in ASD
Most of ~ 16,000 lncRNAs encoded by the human genome [30, 81] have not yet been characterized, but 40% are expressed in the brain and some have been implicated in brain function [82]. For instance, lncRNAs influence neuronal versus glial fate in cortical progenitors [83], while others are upregulated in response to neuronal activity [84,85,86] and have roles in neuronal excitation and plasticity [87, 88]. When mouse fibroblasts were converted directly to neurons, ~ 60% of differentially expressed transcripts were non-coding RNAs [24], suggesting potential roles in neuronal development.
Data from genetic [18, 20,21,22, 24] and gene expression studies [23] have also implicated lncRNAs in ASD. The first lncRNA to be convincingly linked to ASD was PTCHD1-AS [21], which is frequently disrupted in people with ASD [89]. A recent study supported a role for PTCHD1-AS in ASD and also implicated the uncharacterized lncRNA AK127244 [18, 19]. Genetic and gene expression studies have also suggested potential roles for other lncRNAs in ASD, including MSNP1-AS [22], LINC00689, and LINC00693 [23], although the underlying mechanisms for their involvement with ASD remain largely unexplored and could be variable. A recent study reported several other developmentally regulated brain-expressed lncRNAs that are disrupted in ASD or intellectual disability, one of which (lnc-NR2F1) was shown to regulate autism risk genes and promote maturation of mouse stem cell-derived neurons [24]. These findings combined with our own recent work on PTCHD1-AS (described below) provide direct evidence that ASD-associated non-coding RNAs directly regulate neurodevelopmental processes relevant to ASD.
The aforementioned studies suggest that non-coding variants play important roles in the development of ASD, although the functions of these variants and the regulatory elements they disrupt remain largely unknown. Next, we discuss recent advances in cellular reprogramming and CRISPR technologies that are poised to greatly advance our understanding of ASD-associated non-coding variants.
Human pluripotent stem cell models of ASD
Human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) and human iPSCs, have the capacity to differentiate into unlimited supplies of brain cells and therefore have tremendous potential for modeling ASD [31]. To date, hPSC studies of ASD have focused largely on variants that disrupt protein-coding genes, which have revealed a wide range of synaptic phenotypes (Table 2). Synaptic phenotyping in iPSC models of ASD has focused primarily on excitatory synaptic function, which is impaired in some models and increased in others (Table 2). The majority of iPSC ASD models with synaptic phenotypes report decreased connectivity, and for some genes, the underlying mechanisms have been determined. Physiological decreases in synaptic function can result from fewer excitatory synapses (22q13.3+/−, SHANK3−/−), impairments in neurotransmitter release (STXBP1+/−, NRXN1+/−), or hypofunction of excitatory NMDA receptors (PTCHD1-AS) (Table 2). Conversely, some other genetic models of ASD show increased synaptic function via increased synapse numbers (NLGN4R704C, SHANK2+/−) or hyperfunction of NMDA receptors (EHMT1+/−) (Table 2). Together, these findings support the notion that increases or decreases in synaptic activity outside of the range that is typical of unaffected individuals may impair sensory processing and social interactions, thereby contributing to ASD development [90].
The protein-coding variants modeled to date are known or predicted to be of high penetrance, whereas the penetrance is unknown for most non-coding variants. Therefore, iPSC experiments modeling of non-coding variants must be carefully designed to minimize heterogeneity and experimental noise. Here, we discuss experimental design considerations for using iPSCs to model ASD, with a specific focus on challenges associated with modeling the consequences of non-coding variants.
Experimental controls in hPSC models of ASD
Published hPSC models of ASD have used two primary approaches: personalized iPSCs from donors with ASD or genome editing to introduce specific ASD-associated variants in reference lines. Modeling ASD with iPSCs typically uses a case-control model, where iPSC-derived neurons from people with ASD are compared to neurons from people who do not have ASD. These studies are often stratified by gene or shared neurodevelopmental phenotypes (Table 2), and controls are either unaffected family members [20, 41, 42] or unrelated people who are matched to the study subjects by age, sex, or both [20, 34, 35, 39, 40]. This approach is made challenging by extensive intra-individual variability and often requires extensive biological and technical controls/replicates to provide sufficient power to detect neuronal phenotypes [46]. Furthermore, iPSC reprogramming is associated with genomic instability and de novo genomic variants [91, 92], so multiple independent iPSC lines should be assessed for each donor. To overcome genetic and phenotypic heritability among individuals, several groups have increasingly employed “isogenic” genome editing approach for modeling ASD in hPSCs [20, 32, 36,37,38, 41, 43], which results in mutant and control cell lines with comparable genetic backgrounds.
Isogenic approaches have been extensively used for modeling ASD (Table 2) and are also very powerful when used together with case-control approaches [20, 41, 42]. Genome editing has been used to introduce mutations in several autism candidate genes, which has revealed a range of synaptic and gene expression phenotypes (Table 2). RNA-seq analyses in CRISPR-edited neurons with heterozygous CHD8 mutations revealed similar misregulated genes in monolayer neurons and organoids [93]. Interestingly, the top misregulated genes in CHD8+/− neurons included the lncRNA DLX6-AS1, which was also misregulated in organoids from people with idiopathic ASD and macrocephaly [94]. Genome editing approaches have also revealed a wide range of ASD-associated synaptic phenotypes in hESC-derived neurons with engineered variants in NRXN1, STXBP1, SHANK3, and NLGN4 (Table 2). The penetrance of ASD-associated non-coding variants is largely unknown, so introducing them by genome editing may not result in detectable phenotypes. Starting with iPSCs that are genetically matched to people with ASD improves construct validity and presumably increases the likelihood of detecting phenotypic differences. Upon identification of any ASD-associated phenotypes and correlated gene expression changes, genome editing can be used to repair the non-coding variant or introduce it into control iPSCs. We used this approach to determine that a nonsense mutation in the synaptic gene SHANK2 was both necessary and sufficient for overconnectivity that we observed in an iPSC model of ASD [41].
Another important consideration for modeling ASD-associated non-coding variants is the selection of controls. Although unaffected family members are often used as controls to partially account for genetic variability [20, 41, 42], some relatives have ASD-like features or carry other combinations of genetic variants that may cumulatively affect synapse function deleteriously [1]. Therefore, we recently reported a panel of iPSC lines from two males and two females who have no overt disease phenotypes and who were also shown by WGS to have minimal loads of genomic variants [95]. These iPSCs were reprogrammed using non-integrating Sendai virus vectors, which is less likely than retrovirus or mRNA reprogramming to result in de novo coding variants [91]. These iPSC lines (Personal Genome Project Canada participants) efficiently differentiate to neurons and other lineages and support genome editing to introduce specific variants. These iPSCs were also analyzed by WGS, which revealed surprising numbers of single nucleotide variants: each iPSC line had over 1000 de novo variants, and genome-edited iPSCs had hundreds of variants compared to the parental line [95]. Some of these de novo variants had the potential to affect disease-related cellular phenotypes, so we identified variant-preferred PGPC iPSC lines for specific applications like neuronal phenotyping. In addition to serving as controls for modeling ASD, we also foresee neurons from the PGPC iPSC lines being very useful for establishing the baseline range of “normal” synaptic functions in human iPSC-derived neurons.
Neuronal differentiation methods
Although a wide range of protocols has been developed for making specific neuronal and non-neuronal cell types from iPSCs [96], most iPSC approaches for modeling ASD have used neurons with a cortical forebrain identity [31]. Approaches for making iPSC-derived neurons rely primarily on either directed differentiation via a multipotent progenitor stage or direct conversion from pluripotent stem cells to post-mitotic neurons [96]. Both approaches have been used to model ASD (Table 2), although they each have relative advantages and disadvantages.
Phenotypic consequences of regulatory non-coding variants may be restricted to specific lineages or timepoints, and directed differentiation offers the advantage of interrogating multiple cell types within a single experiment. For example, directed differentiation to generate excitatory cortical neurons typically results in mixed cultures that also contain undifferentiated progenitors, inhibitory neurons, and astrocytes [97, 98]. Such mixed cell populations are observed when neurons are differentiated as adherent cultures or in suspension as three-dimensional organoids [99]. Neurons made by directed differentiation also mature asynchronously over the 6–14 weeks required for synapse development, so the resultant cultures contain neurons of varying ages. These mixed populations are advantageous for examining cell fate specification and migration [39] (particularly in organoids [99]) and for exploring interactions between different brain cell types [34, 40]. However, cell type composition in neuronal cultures made by direct differentiation varies between donors and batches, which introduces experimental noise and decreases statistical power to detect phenotypic differences [46, 100].
Direct conversion inherently overcomes heterogeneity by using specific transcription factors to swiftly generate pure populations of post-mitotic neurons [31, 96]. Ectopic expression of human or mouse neurogenin-2 (NGN2/Ngn2) in PSCs or neural progenitor cells results in homogeneous populations of excitatory cortical neurons that mature in only 3–4 weeks [101, 102]. Neurons made by direct conversion have been used to analyze synapse function and network connectivity in hPSC models of ASD (Table 2), and direct comparison with neurons from directed differentiation have revealed similar phenotypes [36]. Direct conversion can also generate homogenous cultures of inhibitory neurons [103] and astrocytes [104]. Due to their relative homogeneity, neurons made by direct conversion are particularly useful for gene expression analyses to detect regulatory consequences of non-coding variants. However, recent reports suggest that direct conversion may mask ASD-associated phenotypes that arise in the neuronal progenitor phase, such as epigenetic misregulation of synaptic gene expression [105].
As a first step in deciding which approach to use for modeling a potential regulatory variant, it is important to determine when and where the regulatory element is active. Publicly accessible transcriptome data from the developing and adult human brain and from differentiating iPSC-derived neurons [106,107,108,109,110,111,112] can be used to determine the cell type and developmental time point at which some regulatory elements are active. Transcription start site and chromatin state data can also be used to infer enhancer activity [60,61,62, 111,112,113], which can then be correlated with transcriptome data to identify potential target genes. If variants are predicted to have a function during early brain development or in cell-type specification, then directed differentiation would likely be the more suitable model. Conversely, if variants are predicted to function largely in post-mitotic neurons, then direct conversion may be preferable due to swifter maturation and homogeneous cultures. Upon determining the cell type for experimental analyses, it is of paramount importance that the resultant neurons be tested to ensure that the regulatory element of interest is active and that the cells are suitable for modeling ASD.
Identifying phenotypes in hPSC models of ASD
Gene expression analyses
We anticipate that non-coding variants will affect neuronal gene expression, but the design of expression analyses with iPSC-derived neurons can influence the interpretation of results. The heterogeneity that results from directed differentiation is undesirable for transcriptomics [20, 41] and other population-level analyses [98], although specific cell types can be enriched using genetic reporters [114] or cell surface markers [20, 41, 98]. If possible, transcriptomic analyses should be performed using isogenic controls to improve sensitivity to detect expression changes [46]. In our previous work modeling ASD with iPSCs, we found that isogenic pairs revealed robust expression changes [32, 41], whereas an analysis with a case-control design yielded no consistently misregulated genes [20]. It may be particularly difficult to detect expression changes in case-control studies because of the large sample size required to overcome inherent individual and technical variability in iPSC-derived neurons [46, 100]. However, computational advances may improve signal-to-noise for detecting expression changes in mixed populations of neurons, as was recently reported for modeling schizophrenia [115].
Another consideration when modeling transcriptional consequences of non-coding variants in ASD is the need for substantial sequencing depth in transcriptomic analyses. Detection of alternatively spliced exons and low abundance lncRNAs requires thorough read coverage. Indeed, we recently found that a read depth of 60 million paired-end reads per sample was necessary to detect the ASD-associated lncRNA PTCHD1-AS [20]. Furthermore, due to the potentially subtle or cis-acting effects of non-coding variants on the expression of target genes, it may also be necessary to perform allele-specific gene expression analyses, which also benefits from improved sequencing coverage [116].
Single-cell analyses: morphology and electrophysiology
iPSC models have confirmed the hypothesis that synaptic dysfunction underlies ASD (Table 2), but the mechanisms underlying synapse dysfunction vary considerably between models. The gold standard approach for assessing excitatory synaptic activity is patch-clamp electrophysiology to record miniature or spontaneous excitatory postsynaptic currents (mEPSCs or sEPSCs, respectively). The frequency of these excitatory synaptic events can be correlated with imaging data that quantify excitatory synapses and dendrite length [37, 38, 41]. Together, these metrics provide insight into the amount of synaptic connectivity and the potential mechanisms underlying any changes. iPSC-derived neurons have revealed a striking dichotomy in ASD-associated synaptic connectivity phenotypes, with different models displaying under- or over-connectivity that can arise by several distinct mechanisms (Table 2).
Some of the phenotypes observed in iPSC-derived neurons recapitulate those observed in mouse models, although contrasting phenotypes have also been reported from these two modeling approaches. Similar functional impairments in excitatory synapses and in hyperpolarization-activated cation (Ih) channels were observed in human and mouse neurons with SHANK3 mutations [38]. Human and mouse neurons deficient for EHMT1 also share decreased network burst frequency and increased NMDA receptor activity [44]. Conversely, heterozygous variants in STXBP1 [37], NRXN1 [36], and SHANK2 [41] are associated with ASD in people and affect connectivity in iPSC models, whereas heterozygous mutations have little or no phenotypic effect on synaptic function in mice. Future work will resolve whether phenotypic differences are species-specific or the result of differences in the cell types that were assessed.
iPSC-derived neurons are subject to both technical and biological variability that can introduce noise in assays of synaptic connectivity. We and others have used co-culture approaches to overcome this variability and record synaptic function in ASD neurons and control neurons within the same cultures [34, 41, 117]. We labeled mature excitatory neurons from controls and from people with ASD using two different fluorescent reporters [41]. These neurons were then sparsely seeded on a lawn of neurons (either from controls or people with ASD) and mouse astrocytes to provide a controlled synaptogenic environment. Simultaneous phenotyping of control neurons and ASD neurons in the same dish revealed increased connectivity in neurons with SHANK2 variants. This within-well normalization approach reduced experimental variability and improved sensitivity to detect phenotypic changes [41]. An extension of this approach compared isogenic neurons on the lawns of either mutant or control neurons to determine whether any functional impairments were due to pre- or post-synaptic dysfunction [117]. Within-well normalization combined with isogenic controls should reduce noise and improve consistency in human iPSC models of ASD-associated non-coding variants.
Another approach for combatting heterogeneity is to examine gene expression by single-cell transcriptomics. Single-cell RNA-seq has been used to identify misregulated cellular processes in iPSC models of Parkinson’s disease [118] and trisomy 21 [119]. This approach may also be combined with single-cell chromatin accessibility to detect coordinated changes in gene expression and enhancer usage [120]. Finally, single-cell transcriptomics can also be integrated with analyses of neuronal morphology and function using Patch-seq: this modification of patch-clamp electrophysiology uses the patching pipette to deliver a fluorescent dye to reveal neuronal morphology and then to acquire cytoplasmic RNA for single-cell RNA-seq following the completion of recordings [121].
Network activity
Assessing the function of neural networks can yield insight into how the underlying changes in gene expression, morphology, and synaptic transmission alter connectivity in models of ASD. Using microelectrode arrays (MEAs), neurons can be plated on a grid of microelectrodes to simultaneously record extracellular voltage changes within a synaptic network [122]. Multi-well MEAs can contain several hundred electrodes per plate and read at multiple timepoints to non-invasively acquire information on neural network development and function. Captured signals can be filtered to obtain higher frequency action potential spikes or lower frequency local field potential oscillations [123]. As neurons mature, synchronous network bursting patterns emerge, which can be used to assess differences in network dynamics and circuitry in control and ASD neural cultures. A recent study of 8-month-old cortical organoids even suggested that iPSC-derived neurons can mature to the point of displaying oscillatory network events similar to preterm human electroencephalograph recordings [124]. MEA datasets are also rich in positional information, which can provide insight into circuitry changes resulting from differences in neuron morphology or intrinsic function.
MEAs have recently been used to explore ASD-associated action potential firing and connectivity phenotypes (Table 2), which have revealed further evidence for both under- and over-connectivity in different models. Directed differentiation of mixed excitatory and inhibitory neuron cultures from people with idiopathic ASD exhibited reduced spiking activity and network bursting [39, 40]. Genome-edited excitatory neurons with mutations of several different ASD risk genes had decreased mean firing rate and network burst frequency [32]. Conversely, excitatory neurons from people with heterozygous deletion of CNTN5 and EHMT2 exhibited hyperactive networks [42]. iPSC-derived excitatory neurons from individuals with Kleefstra syndrome (who also had ASD diagnoses) showed network bursts with decreased frequency and altered kinetics, and these phenotypes could be rescued pharmacologically [44].
MEA phenotyping is attractive for modeling ASD because the simple non-invasive recordings facilitate higher throughput applications than imaging or patch-clamp electrophysiology. However, a careful experimental design will be necessary to overcome extensive technical and biological variability in baseline MEA metrics (i.e., mean firing rate, number of active electrodes). To overcome this variability, we recommend using isogenic controls when possible, performing ASD/control analyses on the same plate to account for batch effects, and establishing a schedule to ensure consistent latencies between media renewal and MEA recordings. Previous work in hPSC models of ASD has shown that some synaptic phenotypes can be rescued (Table 2). The medium-throughput nature of MEA phenotyping holds promise for building on these rescue approaches and establishing drug-screening platforms to find candidate compounds for correction of under- or over-connected neural networks.
Functional analyses of non-coding RNA in iPSC-derived neurons
Several recent studies have reported potential roles for miRNAs and lncRNAs in ASD-associated processes like neurodevelopment. Global expression analyses revealed that several miRNAs change in expression during differentiation of iPSC-derived neurons [125, 126]. Expression of miR-4449, miR-181a, and miR-1290 were altered in iPSC models of schizophrenia [127], fragile X syndrome [128], and ASD [129], respectively. miR-199 and miR-214 are upregulated in the neurodevelopmental disorder Rett syndrome, which is associated with impaired neurogenesis in human iPSCs and developing mice [130]. lncRNAs have also been implicated in ASD-associated processes like neurodevelopment and activity-dependent gene expression [53]. The lncRNAs TUNA [131] and lnc-NR2F1 [24] regulate gene expression and neuronal differentiation in mouse embryonic stem cells. LINC00473 is a primate-specific lncRNA that is robustly induced by synaptic excitation of human iPSC-derived neurons [85] and may regulate the activity-dependent transcription factor CREB [132]. NEAT1 is a highly abundant lncRNA that is downregulated in response to neuronal depolarization and interacts with epilepsy-associated potassium channels to regulate the excitability of human iPSC-derived neurons [88]. Together, these data suggest that non-coding RNAs contribute to a wide range of ASD-associated neuronal processes.
We recently reported a human iPSC approach for modeling ASD-associated non-coding variants focused on the lncRNA PTHCD1-AS [20]. We generated iPSCs from three unrelated males with ASD who had deletions that encompassed one or more exons of PTCHD1-AS, along with iPSCs from three unaffected individuals. These iPSCs were differentiated into forebrain neurons, and phenotypic analyses revealed pronounced deficits in excitatory synaptic function, including decreased frequency of mEPSCs and diminished amplitude of NMDA-evoked currents. We also used genome editing to replace a critical exon of PTCHD1-AS with a premature polyadenylation sequence, which recapitulated the mEPSC frequency impairment and confirmed the importance of PTCHD1-AS in excitatory synaptic function. Our work with PTCHD1-AS therefore provides proof of principle that ASD-associated non-coding variants can have pronounced phenotypic consequences in human iPSC-derived neurons.
Future directions
CRISPR-based tools for validating regulatory variants
Future analysis of non-coding variants in ASD will benefit from the concurrent application of CRISPR-based tools [133] for artificially manipulating genes and regulatory elements. For instance, variants that alter promoter activity can be independently modeled using CRISPR-interference (CRISPRi) and CRISPR-activation (CRISPRa) to deliver transcriptional repressors or activators to target promoters [133]. In a recent study [117], CRISPRi and CRISPRa were elegantly employed to model the functional consequences of 5 schizophrenia-associated common variants that were previously implicated in misregulation of neuronal genes. CRISPRi and CRISPRa are also potentially useful for functional analyses of lncRNAs, which often have cis functions at the endogenous site of lncRNA synthesis [82].
ASD-associated enhancers and splice sites can also be evaluated using CRISPR-based approaches. Enzymatically inactivated Cas9 can be fused to catalytic domains that add or remove histone modifications to directly manipulate enhancer function. For example, Cas9-mediated recruitment of catalytic domains of p300 and HDAC8 has been used to artificially activate or block dynamically regulated enhancers in mouse neurons [134]. CRISPR can also be used to deliver cytidine deaminase to target transcripts to force exon skipping or exon inclusion [135]. These approaches provide the opportunity to recapitulate expression changes caused by ASD-associated non-coding variants, which will independently verify their sufficiency to drive ASD-associated synaptic phenotypes.
Predicting lncRNA function: detecting cryptic coding capacity
Although lncRNAs are defined in part by their limited protein-coding potential, recent results have challenged the notion that all lncRNAs are devoid of translated open reading frames. Several approaches have been developed for characterizing the translational landscapes of human cells, resulting in the surprising discovery that some lncRNAs are associated with ribosomes and may therefore undergo translation [136]. Historically, open-reading frame prediction algorithms typically have a minimum threshold of 100 codons, leading some transcripts that encode small proteins or peptides to be classified as lncRNAs [137]. However, ribosome profile sequencing (Ribo-seq) has revealed that some of these lncRNAs have sequencing reads with 3 nucleotide periodicity, as is seen in normal ribosome movement on coding mRNAs [136]. Furthermore, Ribo-seq-enriched lncRNAs often encode conserved short open reading frames that are enriched in synonymous mutations [136, 137]. In a recent translational profile of human heart, 22% of expressed lncRNAs were translated into potential micropeptides [138]. Therefore, future studies of ASD-associated lncRNAs should first seek to rule out peptide/protein-coding potential before attempting to model any non-coding regulatory functions.
Conclusions
Continued WGS will invariably lead to increasing numbers of ASD-associated non-coding variants being discovered. iPSCs and genome editing provide exciting opportunities to model the consequences of these variants in human neurons and for correlating gene expression changes with functional differences in synaptic connectivity. Careful experimental design and use of well-selected experimental controls (including isogenic controls when possible) will reduce experimental noise and heterogeneity, leading to more sensitive analyses. Determination of the phenotypic consequences of non-coding variants will provide insights into both the neuronal dysfunction that underlies ASD and the mechanisms governing the regulation of human genetic information.
Availability of data and materials
Not applicable
References
Iakoucheva LM, Muotri AR, Sebat J. Getting to the cores of autism. Cell. 2019;178:1287–98.
Liu Y, Liang Y, Cicek AE, Li Z, Li J, Muhle RA, et al. A statistical framework for mapping risk genes from de novo mutations in whole-genome-sequencing studies. Am J Hum Genet. 2018;102:1031–47.
Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RKC, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347:1254806.
Yuen RKC, Merico D, Cao H, Pellecchia G, Alipanahi B, Thiruvahindrapuram B, et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom Med. 2016;1:160271–1602710.
Turner TN, Coe BP, Dickel DE, Hoekzema K, Nelson BJ, Zody MC, et al. Genomic patterns of de novo mutation in simplex autism. Cell. 2017;171:710–722.e12.
Zhou J, Park CY, Theesfeld CL, Wong AK, Yuan Y, Scheckel C, et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat Genet. 2019;51:973–80.
Yuen RK, Merico D, Bookman M, Howe J, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20:602–11.
Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, et al. Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am J Hum Genet. 2016;98:58–74.
An J-Y, Lin K, Zhu L, Werling DM, Dong S, Brand H, et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science. 2018;362:eaat6576.
Brandler WM, Antaki D, Gujral M, Kleiber ML, Whitney J, Maile MS, et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science. 2018;360:327–31.
Ruzzo EK, Pérez-Cano L, Jung J-Y, Wang L-K, Kashef-Haghighi D, Hartl C, et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 2019;178:850–866.e26.
He Z, Xu B, Buxbaum J, Ionita-Laza I. A genome-wide scan statistic framework for whole-genome sequence data analysis. Nat Commun. 2019;10:3018.
Walker S, Scherer SW. Identification of candidate intergenic risk loci in autism spectrum disorder. BMC Genomics. 2013;14:499.
Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BWM, Shen Y, Repnikova EA, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. 2011;89:551–63.
Hodge JC, Mitchell E, Pillalamarri V, Toler TL, Bartel F, Kearney HM, et al. Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol Psychiatry. 2014;19:368–79.
Woodbury-Smith M, Nicolson R, Zarrei M, Yuen RKC, Walker S, Howe J, et al. Variable phenotype expression in a family segregating microdeletions of the NRXN1 and MBD5 autism spectrum disorder susceptibility genes. NPJ Genom Med. 2017;2:17.
Williams SM, An JY, Edson J, Watts M, Murigneux V, Whitehouse AJO, et al. An integrative analysis of non-coding regulatory DNA variations associated with autism spectrum disorder. Mol Psychiatry. 2018;24:1707–19.
Zarrei M, Burton CL, Engchuan W, Young EJ, Higginbotham EJ, JR MD, et al. A large data resource of genomic copy number variation across neurodevelopmental disorders. NPJ Genom Med. 2019;4:1–13.
Duong LTT, Hoeffding LK, Petersen KB, Knudsen CD, Thygesen JH, Klitten LL, et al. Two rare deletions upstream of the NRXN1 gene (2p16.3) affecting the non-coding mRNA AK127244 segregate with diverse psychopathological phenotypes in a family. Eur J Med Genet. 2015;58:650–3.
Ross PJ, Zhang W-B, Mok RSF, Zaslavsky K, Deneault E, D’Abate L, et al. Synaptic dysfunction in human neurons with autism-associated deletions in PTCHD1-AS. Biol Psychiatr. 2019;87:139–49.
Noor A, Whibley A, Marshall CR, Gianakopoulos PJ, Piton A, Carson AR, et al. Disruption at the PTCHD1 Locus on Xp22.11 in autism spectrum disorder and intellectual disability. Sci Transl Med. 2010;2:49ra68.
Kerin T, Ramanathan A, Rivas K, Grepo N, Coetzee GA, Campbell DB. A noncoding RNA antisense to moesin at 5p14.1 in autism. Sci Transl Med. 2012;4:128ra40.
Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. 2016;540:423–7.
Ang CE, Ma Q, Wapinski OL, Fan S, Flynn RA, Lee QY, et al. The novel lncRNA lnc-NR2F1 is pro-neurogenic and mutated in human neurodevelopmental disorders. eLife. 8:–e41770.
Sanders SJ, Neale BM, Huang H, Werling DM, An J-Y, Dong S, et al. Whole genome sequencing in psychiatric disorders: the WGSPD consortium. Nat Neurosci. 2017;20:1661–8.
Kazdoba TM, Leach PT, Yang M, Silverman JL, Solomon M, Crawley JN. Translational mouse models of autism: advancing toward pharmacological therapeutics. Curr Top Behav Neurosci. 2016;28:1–52.
Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–40.
Marnetto D, Molineris I, Grassi E, Provero P. Genome-wide identification and characterization of fixed human-specific regulatory regions. Am J Hum Genet. 2014;95:39–48.
Doan RN, Bae B-I, Cubelos B, Chang C, Hossain AA, Al-Saad S, et al. Mutations in human accelerated regions disrupt cognition and social behavior. Cell. 2016;167:341–354.e12.
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89.
Kim D-S, Ross PJ, Zaslavsky K, Ellis J. Optimizing neuronal differentiation from induced pluripotent stem cells to model ASD. Front Cell Neurosci. 2014;8:109.
Deneault E, White SH, Rodrigues DC, Ross PJ, Faheem M, Zaslavsky K, et al. Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Rep. 2018;11:1211–25.
Tai DJC, Ragavendran A, Manavalan P, Stortchevoi A, Seabra CM, Erdin S, et al. Engineering microdeletions and microduplications by targeting segmental duplications with CRISPR. Nat Neurosci. 2016;19:517–22.
Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–71.
Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X, et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry. 2015;20:1350–65.
Pak C, Danko T, Zhang Y, Aoto J, Anderson G, Maxeiner S, et al. Human neuropsychiatric disease modeling using conditional deletion reveals synaptic transmission defects caused by heterozygous mutations in NRXN1. Cell Stem Cell. 2015;17:316–28.
Patzke C, Han Y, Covy J, Yi F, Maxeiner S, Wernig M, et al. Analysis of conditional heterozygous STXBP1 mutations in human neurons. J Clin Invest. 2015;125:3560–71.
Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science. 2016;352:aaf2669.
Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K, et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry. 2017;22:820–35.
Russo FB, Freitas BC, Pignatari GC, Fernandes IR, Sebat J, Muotri AR, et al. Modeling the interplay between neurons and astrocytes in autism using human induced pluripotent stem cells. Biol Psychiatry. 2018;83:569–78.
Zaslavsky K, Zhang W-B, McCready FP, Rodrigues DC, Deneault E, Loo C, et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat Neurosci. 2019;22:556–64.
Deneault E, Faheem M, White SH, Rodrigues DC, Sun S, Wei W, et al. CNTN5-/+ or EHMT2-/+ human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. Elife. 2019;8:e40092.
Marro SG, Chanda S, Yang N, Janas JA, Valperga G, Trotter J, et al. Neuroligin-4 regulates excitatory synaptic transmission in human neurons. Neuron. 2019;103:617–626.e6.
Frega M, Linda K, Keller JM, Gümüş-Akay G, Mossink B, van Rhijn J-R, et al. Neuronal network dysfunction in a model for Kleefstra syndrome mediated by enhanced NMDAR signaling. Nat Commun. 2019;10:4928.
Bassett AR, Akhtar A, Barlow DP, Bird AP, Brockdorff N, Duboule D, et al. Considerations when investigating lncRNA function in vivo. Elife. 2014;3:e03058.
Hoffman GE, Schrode N, Flaherty E, Brennand KJ. New considerations for hiPSC-based models of neuropsychiatric disorders. Mol Psychiatry. 2019;24:49–66.
Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci USA. 2014;111:6131–8.
Sullivan JM, De Rubeis S, Schaefer A. Convergence of spectrums: neuronal gene network states in autism spectrum disorder. Curr Opin Neurobiol. 2019;59:102–11.
Sanders SJ. First glimpses of the neurobiology of autism spectrum disorder. Curr Opin Genet Dev. 2015;33:80–92.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–15.
Shen E, Shulha H, Weng Z, Akbarian S. Regulation of histone H3K4 methylation in brain development and disease. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130514.
Vitriolo A, Gabriele M, Testa G. From enhanceropathies to the epigenetic manifold underlying human cognition. Hum Mol Genet. 2019;28:R226–34.
Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493:327–37.
Turner TN, Eichler EE. The role of de novo noncoding regulatory mutations in neurodevelopmental disorders. Trends Neurosci. 2019;42:115–27.
Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48:214–20.
Backenroth D, He Z, Kiryluk K, Boeva V, Pethukova L, Khurana E, et al. FUN-LDA: a latent dirichlet allocation model for predicting tissue-specific functional effects of noncoding variation: methods and applications. Am J Hum Genet. 2018;102:920–42.
Wells A, Heckerman D, Torkamani A, Yin L, Sebat J, Ren B, et al. Ranking of non-coding pathogenic variants and putative essential regions of the human genome. Nat Commun. 2019;10:5241.
Zhou J, Theesfeld CL, Yao K, Chen KM, Wong AK, Troyanskaya OG. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat Genet. 2018;50:1171–9.
FANTOM Consortium and the RIKEN PMI and CLST (DGT), Forrest ARR, Kawaji H, Rehli M, Baillie JK, de Hoon MJL, et al. A promoter-level mammalian expression atlas. Nature. 2014;507:462–470.
Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–61.
Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330.
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
Quesnel-Vallières M, Weatheritt RJ, Cordes SP, Blencowe BJ. Autism spectrum disorder: insights into convergent mechanisms from transcriptomics. Nat Rev Genet. 2019;20:51–63.
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–4.
Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159:1511–23.
Gonatopoulos-Pournatzis T, Niibori R, Salter EW, Weatheritt RJ, Tsang B, Farhangmehr S, et al. Autism-misregulated eIF4G microexons control synaptic translation and higher order cognitive functions. Molecular Cell. 2020;77:1176–92.
Miura P, Shenker S, Andreu-Agullo C, Westholm JO, Lai EC. Widespread and extensive lengthening of 3’ UTRs in the mammalian brain. Genome Res. 2013;23:812–25.
Rodrigues DC, Kim D-S, Yang G, Zaslavsky K, Ha KCH, Mok RSF, et al. MECP2 Is Post-transcriptionally regulated during human neurodevelopment by combinatorial action of RNA-binding proteins and miRNAs. Cell Rep. 2016;17:720–34.
Agarwal V, Bell GW, Nam J-W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005.
Sakaguchi A, Yamashita Y, Ishii T, Uehara T, Kosaki K, Takahashi T, et al. Further evidence of a causal association between AGO1, a critical regulator of microRNA formation, and intellectual disability/autism spectrum disorder. Eur J Med Genet. 2019;62:103537.
Cheng Y, Wang Z-M, Tan W, Wang X, Li Y, Bai B, et al. Partial loss of psychiatric risk gene Mir137 in mice causes repetitive behavior and impairs sociability and learning via increased Pde10a. Nat Neurosci. 2018;21:1689–703.
Lackinger M, Sungur AÖ, Daswani R, Soutschek M, Bicker S, Stemmler L, et al. A placental mammal-specific microRNA cluster acts as a natural brake for sociability in mice. EMBO Rep. 2019;20:e46429.
Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47:D155–62.
Fineberg SK, Kosik KS, Davidson BL. MicroRNAs potentiate neural development. Neuron. 2009;64:303–9.
Hicks SD, Middleton FA. A comparative review of microRNA expression patterns in autism spectrum disorder. Front Psychiatry. 2016;7:176.
Poelmans G, Franke B, Pauls DL, Glennon JC, Buitelaar JK. AKAPs integrate genetic findings for autism spectrum disorders. Transl Psychiatry. 2013;3:e270.
Abu-Elneel K, Liu T, Gazzaniga FS, Nishimura Y, Wall DP, Geschwind DH, et al. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics. 2008;9:153–61.
Li J, Cai T, Jiang Y, Chen H, He X, Chen C, et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol Psychiatry. 2016;21:290–7.
Takata A, Ionita-Laza I, Gogos JA, Xu B, Karayiorgou M. De novo synonymous mutations in regulatory elements contribute to the genetic etiology of autism and schizophrenia. Neuron. 2016;89:940–7.
Toma C, Torrico B, Hervás A, Salgado M, Rueda I, Valdés-Mas R, et al. Common and rare variants of microRNA genes in autism spectrum disorders. World J Biol Psychiatry. 2015;16:376–86.
Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–73.
Briggs JA, Wolvetang EJ, Mattick JS, Rinn JL, Barry G. Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron. 2015;88:861–77.
Mercer TR, Qureshi IA, Gokhan S, Dinger ME, Li G, Mattick JS, et al. Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci. 2010;11:14.
Lipovich L, Dachet F, Cai J, Bagla S, Balan K, Jia H, et al. Activity-dependent human brain coding/noncoding gene regulatory networks. Genetics. 2012;192:1133–48.
Pruunsild P, Bengtson CP, Bading H. Networks of cultured iPSC-derived neurons reveal the human synaptic activity-regulated adaptive gene program. Cell Rep. 2017;18:122–35.
Bitar M, Kuiper S, O’Brien E, Barry G. Using human iPSC-derived neurons to uncover activity-dependent non-coding RNAs. Genes (Basel). 2017;8:401.
Raveendra BL, Swarnkar S, Avchalumov Y, Liu X-A, Grinman E, Badal K, et al. Long noncoding RNA GM12371 acts as a transcriptional regulator of synapse function. Proc Natl Acad Sci USA. 2018;115:E10197–205.
Barry G, Briggs JA, Hwang DW, Nayler SP, Fortuna PRJ, Jonkhout N, et al. The long non-coding RNA NEAT1 is responsive to neuronal activity and is associated with hyperexcitability states. Sci Rep. 2017;7:40127.
Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev. 2012;22:229–37.
Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012;4:a009886.
Bhutani K, Nazor KL, Williams R, Tran H, Dai H, Džakula Ž, et al. Whole-genome mutational burden analysis of three pluripotency induction methods. Nat Commun. 2016;7:10536.
Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Närvä E, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62.
Wang P, Mokhtari R, Pedrosa E, Kirschenbaum M, Bayrak C, Zheng D, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol Autism. 2017;8:11.
Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell. 2015;162:375–90.
Hildebrandt MR, Reuter MS, Wei W, Tayebi N, Liu J, Sharmin S, et al. Precision health resource of control iPSC lines for versatile multilineage differentiation. Stem Cell Rep. 2019;13:1126–41.
Mertens J, Marchetto MC, Bardy C, Gage FH. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat Rev Neurosci. 2016;17:424–37.
Djuric U, Cheung AYL, Zhang W, Mok RS, Lai W, Piekna A, et al. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol Dis. 2015;76:37–45.
Djuric U, Rodrigues DC, Batruch I, Ellis J, Shannon P, Diamandis P. Spatiotemporal proteomic profiling of human cerebral development. Mol Cell Proteomics. 2017;16:1548–62.
Adams JW, Cugola FR, Muotri AR. Brain organoids as tools for modeling human neurodevelopmental disorders. Physiology (Bethesda). 2019;34:365–75.
Schwartzentruber J, Foskolou S, Kilpinen H, Rodrigues J, Alasoo K, Knights AJ, et al. Molecular and functional variation in iPSC-derived sensory neurons. Nat Genet. 2018;50:54–61.
Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–98.
Ho S-M, Hartley BJ, Tcw J, Beaumont M, Stafford K, Slesinger PA, et al. Rapid Ngn2-induction of excitatory neurons from hiPSC-derived neural progenitor cells. Methods. 2016;101:113–24.
Yang N, Chanda S, Marro S, Ng Y-H, Janas JA, Haag D, et al. Generation of pure GABAergic neurons by transcription factor programming. Nat Methods. 2017;14:621–8.
Canals I, Ginisty A, Quist E, Timmerman R, Fritze J, Miskinyte G, et al. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat Methods. 2018;15:693–6.
Schafer ST, Paquola ACM, Stern S, Gosselin D, Ku M, Pena M, et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat Neurosci. 2019;22:243–55.
Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206.
Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, et al. Conserved cell types with divergent features in human versus mouse cortex. Nature. 2019;573:61–8.
Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362:eaat8464.
Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018;362:eaat8127.
van de Leemput J, Boles NC, Kiehl TR, Corneo B, Lederman P, Menon V, et al. CORTECON: a temporal transcriptome analysis of in vitro human cerebral cortex development from human embryonic stem cells. Neuron. 2014;83:51–68.
Amiri A, Coppola G, Scuderi S, Wu F, Roychowdhury T, Liu F, et al. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science. 2018;362:eaat6720.
Song M, Yang X, Ren X, Maliskova L, Li B, Jones IR, et al. Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat Genet. 2019;51:1252–62.
Forrest MP, Zhang H, Moy W, McGowan H, Leites C, Dionisio LE, et al. Open chromatin profiling in hipsc-derived neurons prioritizes functional noncoding psychiatric risk variants and highlights neurodevelopmental loci. Cell Stem Cell. 2017;21:305–318.e8.
Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell. 2014;14:781–95.
Hoffman GE, Hartley BJ, Flaherty E, Ladran I, Gochman P, Ruderfer DM, et al. Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nat Commun. 2017;8:2225.
Castel SE, Levy-Moonshine A, Mohammadi P, Banks E, Lappalainen T. Tools and best practices for data processing in allelic expression analysis. Genome Biol. 2015;16:195.
Schrode N, Ho S-M, Yamamuro K, Dobbyn A, Huckins L, Matos MR, et al. Synergistic effects of common schizophrenia risk variants. Nat Genet. 2019;51:1475–85.
Lang C, Campbell KR, Ryan BJ, Carling P, Attar M, Vowles J, et al. Single-cell sequencing of iPSC-dopamine neurons reconstructs disease progression and identifies HDAC4 as a regulator of Parkinson cell phenotypes. Cell Stem Cell. 2019;24:93–106.e6.
Czermiński JT, Lawrence JB. Silencing trisomy 21 with XIST in neural stem cells promotes neuronal differentiation. Dev Cell. 2020;52:294–308.e3.
Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361:1380–5.
Bardy C, van den Hurk M, Kakaradov B, Erwin JA, Jaeger BN, Hernandez RV, et al. Predicting the functional states of human iPSC-derived neurons with single-cell RNA-seq and electrophysiology. Mol Psychiatry. 2016;21:1573–88.
Obien MEJ, Deligkaris K, Bullmann T, Bakkum DJ, Frey U. Revealing neuronal function through microelectrode array recordings. Front Neurosci. 2014;8:423.
Spira ME, Hai A. Multi-electrode array technologies for neuroscience and cardiology. Nat Nanotechnol. 2013;8:83–94.
Trujillo CA, Gao R, Negraes PD, Gu J, Buchanan J, Preissl S, et al. Complex oscillatory waves emerging from cortical organoids model early human brain network development. Cell Stem Cell. 2019;25:558–569.e7.
Tu J, Cao D, Li L, Cheung H, Chan W. MicroRNA profiling during directed differentiation of cortical interneurons from human-induced pluripotent stem cells. FEBS Open Bio. 2018;8:502–12.
Zhao D, Lin M, Chen J, Pedrosa E, Hrabovsky A, Fourcade HM, et al. MicroRNA profiling of neurons generated using induced pluripotent stem cells derived from patients with schizophrenia and schizoaffective disorder, and 22q11.2 Del. PLoS One. 2015;10:e0132387.
Maekawa M, Yamada K, Toyoshima M, Ohnishi T, Iwayama Y, Shimamoto C, et al. Utility of scalp hair follicles as a novel source of biomarker genes for psychiatric illnesses. Biological Psychiatry. 2015;78:116–25.
Achuta VS, Möykkynen T, Peteri U-K, Turconi G, Rivera C, Keinänen K, et al. Functional changes of AMPA responses in human induced pluripotent stem cell-derived neural progenitors in fragile X syndrome. Sci Signal. 2018;11:eaan8784.
Moore D, Meays BM, Madduri LSV, Shahjin F, Chand S, Niu M, et al. Downregulation of an evolutionary young miR-1290 in an iPSC-derived neural stem cell model of autism spectrum disorder. Stem Cells Int. 2019;2019:8710180.
Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry. 2018;23:1051–65.
Lin N, Chang K-Y, Li Z, Gates K, Rana ZA, Dang J, et al. An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol Cell. 2014;53:1005–19.
Chen Z, Li J-L, Lin S, Cao C, Gimbrone NT, Yang R, et al. cAMP/CREB-regulated LINC00473 marks LKB1-inactivated lung cancer and mediates tumor growth. J Clin Invest. 2016;126:2267–79.
Engreitz J, Abudayyeh O, Gootenberg J, Zhang F. CRISPR tools for systematic studies of RNA regulation. Cold Spring Harb Perspect Biol. 2019;11:a035386.
Chen L-F, Lin YT, Gallegos DA, Hazlett MF, Gómez-Schiavon M, Yang MG, et al. Enhancer histone acetylation modulates transcriptional bursting dynamics of neuronal activity-inducible genes. Cell Rep. 2019;26:1174–1188.e5.
Yuan J, Ma Y, Huang T, Chen Y, Peng Y, Li B, et al. Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol Cell. 2018;72:380–394.e7.
Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJS, Jackson SE, et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014;8:1365–79.
Makarewich CA, Olson EN. Mining for micropeptides. Trends Cell Biol. 2017;27:685–96.
van Heesch S, Witte F, Schneider-Lunitz V, Schulz JF, Adami E, Faber AB, et al. The translational landscape of the human heart. Cell. 2019;178:242–260.e29.
Funding
Research in the Ross lab is supported by the National Science and Engineering Research Council of Canada (RGPIN-2017-05469 to PJR). Research on non-coding elements and autism in the Ellis lab is supported by the Canadian Institutes of Health Research (PJT-148746 to JE), the Simons Foundation (SFARI 514918 to JE), and the University of Toronto McLaughlin Centre (MC-2014-06 to JE). SWS is the GlaxoSmithKline-CIHR Endowed Chair in Genome Sciences at the Hospital for Sick Children.
Author information
Authors and Affiliations
Contributions
Conceptualization: PJR and JE. Writing—original draft: PJR, RSFM, BSS, DR, MM, SWS, and JE. Writing—review and editing: PJR, RSFM, BSS, DR, SWS, and JE. Visualization: PJR. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
SWS is on the Scientific Advisory Committees of Population Bio and Deep Genomics, and the intellectual property from aspects of his research held at the Hospital for Sick Children is licensed to Athena Diagnostics, Lineagen, and co-held with Population Bio. The other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ross, P.J., Mok, R.S.F., Smith, B.S. et al. Modeling neuronal consequences of autism-associated gene regulatory variants with human induced pluripotent stem cells. Molecular Autism 11, 33 (2020). https://doi.org/10.1186/s13229-020-00333-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-020-00333-6