
Abstract
Efforts to identify the causes of autism spectrum disorders have highlighted the importance of both genetics and environment, but the lack of human models for many of these disorders limits researchers’ attempts to understand the mechanisms of disease and to develop new treatments. Induced pluripotent stem cells offer the opportunity to study specific genetic and environmental risk factors, but the heterogeneity of donor genetics may obscure important findings. Diseases associated with unusually high rates of autism, such as SCN2A syndromes, provide an opportunity to study specific mutations with high effect sizes in a human genetic context and may reveal biological insights applicable to more common forms of autism. Loss-of-function mutations in the SCN2A gene, which encodes the voltage-gated sodium channel NaV1.2, are associated with autism rates up to 50%. Here, we review the findings from experimental models of SCN2A syndromes, including mouse and human cell studies, highlighting the potential role for patient-derived induced pluripotent stem cell technology to identify the molecular and cellular substrates of autism.
Similar content being viewed by others


Background
Our understanding of autism spectrum disorders (ASD) has increased exponentially over the last 20 years. We now know that though a clear diagnosis may not be made until age two or later, developmental dysfunction may begin far earlier [1]. Researchers have identified hundreds of risk genes for autism, and increasing evidence indicates that parental age, prenatal exposure to certain medications, and even pollution also affect the likelihood an individual will develop the disorder [2,3,4,5,6]. Only a small proportion of autism cases are clearly linked to a single genetic mutation; the vast majority of cases are complicated, likely the result of a combination of multiple genetic and environmental factors [7].
The complex etiology of autism presents a significant challenge to researchers and clinicians alike: there are hundreds of possible relevant variables to control for and consider, while pharmaceutical treatments are difficult to develop in the absence of genetic or molecular target(s). Recent research suggests that neurodevelopmental abnormalities associated with ASD begin in utero [1]. Furthermore, earlier treatment is correlated with greater independence later in life [8], suggesting that some aspects of dysfunction can be ameliorated by appropriate treatment during key windows or sensitive periods in brain development. It is thus increasingly apparent that autism is a disorder of early brain development, and as such, our scientific efforts need to likewise be directed toward development. While a comprehensive review of ASD is beyond the scope of this review, we refer readers to several excellent recent reviews for further detail on ASD genetics, etiology, and phenotype [2, 8,9,10].
Animal models have yielded great strides toward our understanding of ASD, both mechanistically and behaviorally. However, as we have learned from decades of translational research, drugs that appear promising in animal models often fail in clinical trials due to adverse effects or lack of efficacy in humans. Human subject studies, and in particular functional MRI (fMRI) experiments, have shed light on the differences between brain function in individuals with typical cognition versus those with autism. However, human studies are limited by a variety of factors, including the ethics and challenges of working with small children or infants, high subject-to-subject variability, and the inability to obtain live primary tissue for in vitro experiments.
Human embryonic stem cell (hESC) research first opened the door to in vitro experiments with human neurons as researchers developed techniques to differentiate hESCs into neurons. In 2007, the ability to transform differentiated human cells such as fibroblasts into induced pluripotent stem cells (iPSCs) changed the playing field [11,12,13]. For the first time, scientists were able to generate different cell types from an individual who had (or had not) been diagnosed with a particular condition of interest. Stem cells produced from ASD subjects already contain at least one of the genetic cocktails needed to generate autism phenotypes, and thus the cell types produced through subsequent differentiation of these stem cells should likewise be more representative of neurons from an individual with ASD and therefore may facilitate the study of autism on a molecular and cellular level.
Despite the advances provided by iPSCs, significant caveats remain. For one, multiple iPSC clones derived from the same subject show significant clone-to-clone variability [14,15,16]. Additionally, as with fMRI and other human subject studies, there is variability between study subjects. A further complication is found in the variety of factors which contribute to the development of autism: subjects diagnosed with ASD likely converge on a similar phenotype through different paths. Thus, even with the use of human-derived iPSC lines, high variability between subjects and samples might generate enough experimental “noise” that critical phenotypes may be missed. To maximize the effectiveness of iPSCs for studying autism-linked genes, it may be necessary to focus on less genetically complex forms of ASD to reduce variability.
Although it is difficult to minimize variability between iPSC clones, a meaningful step toward enhancing experimental signal can be achieved by narrowing the scope of the disorder in question from the very broad umbrella diagnosis of ASD to a single model with a simple, clearly identified etiology. A small subset of autism cases are monogenic in origin and can be directly tied to a single causal mutation [5], such as loss-of-function mutations in SCN2A which cause SCN2A syndromes [17,18,19]. While such cases are often on the severe end of the phenotypic spectrum, the classical characteristics of autism such as repetitive behaviors and interests, difficulty with social cues, and rarely initiating social interaction, are present and consistent with more common forms of ASD. Stem cell models of monogenic forms of ASD may allow researchers, in combination with careful isogenic controls, to subtract out a significant amount of experimental noise caused by genetic and environmental variability. Monogenic ASD iPSC lines can then be used for making isogenic comparison lines using CRISPR or TALENs, which allow the experimenter to control for genetic background. While this does not eliminate all variability, it does allow stronger inferences to be made about the specific effect of a mutation in a single gene, especially in situations where there are multiple donors from which multiple isogenic lines can be created.
The eventual long-term goal of using iPSC models for SCN2A-related diseases is to expand the technique from modeling monogenic diseases to modeling more complex polygenic diseases. However, as highlighted by Zhu et al. in their thoughtful review of the subject [20], cell culture phenotypes are often much more subtle than clinical phenotypes, which can make them difficult to detect and quantify in vitro. In this case, the use of monogenic models is helpful, and potentially even critical, as a first step toward developing in vitro assays which detect cellular and molecular phenotypes that can be correlated with clinical phenotypes. Once this initial foundation has been laid, more variables can be introduced to the experimental system, such as disease models with multiple genetic or environmental factors. As Zhu et al. suggest, the original monogenic models can then serve as a form of positive control against which more complex models can be compared. iPSC models have already been successfully used to identify cellular and molecular dysfunction that occurs in other forms of monogenic ASD-related disorders, including fragile X syndrome [21,22,23,24], Timothy syndrome [25,26,27,28], Rett syndrome [29,30,31,32], tuberous sclerosis [33,34,35,36], and neurofibromatosis type 1 [37,38,39,40]. As more monogenic models are added to the list, in vitro phenotypes can be compared and contrasted between the models to better understand the phenotypes from autism models with a more complex etiology.
To date, the Simons Foundation for Autism Research has listed 174 genes as “high confidence” for association with autism spectrum disorders [41]. SCN2A, which encodes the voltage-gated sodium channel NaV1.2, is of particular interest as an in vitro model for several reasons, including the following: (1) It is among the most common monogenic sources of autism-causing mutations [19]; (2) Gain-of-function and loss-of-function mutations have both been identified in humans but with markedly different phenotypes (epilepsy vs autism/intellectual disability, respectively) [17], allowing for a compare-and-contrast opportunity between variants; (3) Several mouse models already exist which may complement iPSC models [42,43,44,45,46]; (4) Sodium channels are “druggable” targets [47,48,49,50]; and (5) Elucidation of the role of SCN2A in neural development and function may also contribute to the development of therapies to treat SCN2A-related diseases such as epilepsy, which is often intractable [51].
SCN2A and NaV1.2
SCN2A encodes the voltage-gated sodium channel NaV1.2, which contains 27 exons encoded by 2005 amino acids [Fig. 1]. Sodium channels are large (~ 250 kD) membrane proteins composed of 24 transmembrane (TM) segments that are organized into four homologous domains (DI-DIV). Each domain contains 6 TM segments where the first four TM segments (S1–S4) make a voltage sensor, and the second two segments (S5 and S6) create a central ion pore. The pore domain houses the “selectivity filter,” an unusual protein fold which helps to select for sodium over other monovalent cations, as well as the “gates” formed by the S6 bundle-crossing which control the transmembrane flow of sodium ions. The status of the central gate is tightly coupled to voltage-sensor domains which display sub-millisecond activation in response to depolarizing stimulus. The resulting inward flow of cationic sodium ions down their electro-chemical gradient into the neuron—roughly one million ions per second for each channel—produces a rapid depolarization of the membrane, seen as the upstroke of the action potential. After opening (within milliseconds), sodium channels spontaneously inactivate (cease to conduct) to curb the inward flow of more sodium ions and to allow for membrane repolarization. The overall gating scheme and protein architecture is conserved for all nine mammalian sodium channel isoforms [52].

Structure, architectural elements, and sodium channel (NaV) topology. a Side view of the voltage-gated sodium channel pore forming (alpha) subunit. Each domain is color depicted. Extracellular IG-fold domain of the beta subunit is shown. A sodium ion and tetrodotoxin (TTX), both in orange, are shown outside of the external vestibule. GX-936, a potent and isoform-selective acylsulfonamide, is shown near its DIV voltage-sensor domain (VSD) binding site. The DEKA box represents the selectivity filter comprised of Asp, Glu, Lys, and Ala sidechains. Traditional sodium channel blockers are shown at the intracellular entrance to the of pore domain. b Top view of (a) depicting the four-fold symmetry and central permeation pathway for by the Pore Module (PM) of each domain and voltage-sensing domain (VSD). Note the domain swapped architecture between each PD and VSD, a feature which underlies allosteric coupling in NaV gating. c Classic topology plot showing the same features
NaV1.2 is predominantly expressed in the brain, specifically in cortical pyramidal neurons and cerebellar granule neurons [53,54,55,56]. There are two known developmentally relevant splice isoforms, “neonatal” (5N) and “adult” (5A), distinguished by which of two mutually exclusive variants of exon 5 they contain. As can be inferred by their names, the neonatal variant is the dominant isoform found in cortical neurons during early development, and this is gradually exchanged for the adult variant after birth [57]. In contrast, in cerebellum, the adult isoform is approximately twice as abundant as the neonatal isoform at birth, and the proportion of 5A only slightly increases during development [57]. Studies have shown that the 5N variant, which contains an asparagine (N) at residue 209, is less excitable than its adult counterpart, which contains an aspartic acid (D) at residue 209 [44]. Mice engineered to express the 5A variant throughout development display an increased likelihood of developing seizures that does not abate after development, suggesting permanent developmental effects occur as a result of the specific temporal expression patterns of the two isoforms [44].
During early development, NaV1.2 is localized along the axon length in cortical pyramidal neurons [58]. As development continues, NaV1.2 is gradually replaced by NaV1.6 along the axon, and NaV1.2 expression becomes localized mainly to the axon initial segment [59, 60]. Recent work by Spratt et al. suggests that NaV1.2 enhances backpropagation of action potentials in cortical pyramidal neurons, and thus NaV1.2 may play a role in synaptic plasticity and learning [61]. In contrast, in cerebellum, NaV1.2 expression persists at a much higher level in adulthood [57]. Combined with the different expression patterns of the 5N/5A isoforms in cerebellum, these data suggest the channel may play different roles in the different regions of the brain.
The complex nature of NaV1.2 expression, both in the ratio of 5N/5A isoforms and its replacement with NaV1.6 in certain brain regions, suggests there is a fine-tuned balance of these channels during brain development, and any disturbance of this regulation might result in some form of developmental defect. It is little wonder, then, that mutations in SCN2A lead to intellectual disability, epilepsy, and autism.
SCN2A syndrome(s)
In general, mutations that alter neuronal sodium channel structure, function, or expression lead to epilepsy and neurological disorders, but most SCN2A mutations that cause autism result from mutations that produce premature stop codons (a.k.a. nonsense mutations), which cause termination of protein translation. This effectively results in NaV1.2 haploinsufficiency (homozygous loss of NaV1.2 appears incompatible with life) [45]. As of 2018, 276 patients were identified with SCN2A-mediated disorders, approximately 50% of whom displayed autism [19]. However, the current rates of SCN2A mutations in both ASD and other neurodevelopmental disorder populations are likely underestimated, and the incidence is expected to increase significantly as genetic testing for these patients becomes more widespread. Indeed, Sanders et al. estimate that disease-causing SCN2A mutations may occur as frequently as 7.5 per 100,000 births in the general population [19]. Of the 50% of SCN2A syndrome patients diagnosed with ASD, their symptoms are similar to those seen in more common forms of ASD, such as reduced social interaction and repetitive behaviors. Some patients also exhibit additional symptoms such as intellectual disability (ID) and developmental delay, seizures, and symptoms of cerebellar dysfunction such as abnormal gait, clumsiness, and hypotonia [19].
Of the SCN2A mutations linked to autism, intellectual disability, and/or epilepsy, many of these variants appear to be de novo, rather than inherited. In 2014, Baasch et al. performed exome sequencing of a patient with multiple symptoms including seizures, severe intellectual disability, structural abnormalities in the brain, as well as abnormal brain activity via EEG [62]. They discovered a de novo missense mutation in SCN2A that had not been previously reported. Based on their conclusions and literature review, they suggest that de novo mutations of SCN2A are linked to more severe phenotypes. Likewise, Nakamura et al. also suggest that inherited SCN2A mutations result in less severe phenotypes than de novo SCN2A mutations [63]. This also suggests that apparent rates of SCN2A mutations may increase as new deleterious variants are identified.
It has been proposed that within SCN2A mutations, those that cause a gain of function and increased neural excitability cause epilepsy, whereas mutations that cause loss of function, either dampening channel function or leading to haploinsufficiency, cause autism [17]. However, Wolff et al. identified several loss-of-function mutations that cause later-onset epilepsy, suggesting that the relationship between channel function and phenotype may be more complicated [51]. Emerging evidence suggests that SCN2A may also contribute to schizophrenia and intellectual disability in the absence of autism [64, 65], although these associations require further investigation [66]. Nevertheless, the variety of phenotypes associated with different types of SCN2A mutations may provide an opportunity to more fully investigate the relationship between channel function and phenotype.
SCN2A studies in humans
Histological postmortem examination of NaV1.2 in hippocampus and temporal lobe shows that expression begins in these brain regions at 19 weeks of gestation, peaks at 7–9 months of age, is sustained throughout childhood, and gradually decreases in adulthood [56]. Although EEG has been used in SCN2A epilepsy patients to characterize neural dysfunction during seizures [63, 67], it has not been used to measure brain activity in nonepileptic states. MRI studies have revealed that many SCN2A syndrome patients display structural abnormalities in the brain, including (from most to least common) cerebral atrophy, thin corpus callosum, delayed myelination, and cerebellar atrophy [63].
In one study assessing the effects of different SCN2A variants in schizophrenia, postmortem examination of SCN2A expression in the prefrontal cortex revealed that lower levels of SCN2A expression were associated with poorer cognitive performance [64]. Subsequent fMRI studies showed that the SCN2A SNP rs10174400 is a significant predictor of cognitive ability not only in schizophrenia patients but in healthy adults as well [64, 68]. Combined, these results suggest that common variation in SCN2A plays a role in cognitive ability, which may explain why mutations in SCN2A often cause intellectual disability.
Although it is true that many of the characteristics of SCN2A patients are not representative of more common (and less severe) forms of autism, the distinctive nature of the phenotypes observed with SCN2A syndrome facilitates our ability to connect phenotypes observed in experimental models to those observed clinically in patients. These distinct phenotypes can then be compared and contrasted with those observed in other monogenic ASD models (and eventually, to complex polygenic models) to determine which characteristics are unique to SCN2A syndrome and which are more broadly relevant to ASD in general. As more monogenic ASD models are compared and contrasted, it will be easier to identify those characteristics common to autism specifically versus those belonging to each unique syndrome. The broader developmental dysfunction phenotypes observed in SCN2A syndromes may also serve as a model to aid our understanding of developmental dysfunction in other neurodevelopmental disorders with which it is genetically linked, such as intellectual disability and schizophrenia.
Mouse models of SCN2A syndrome
Scn2aQ54
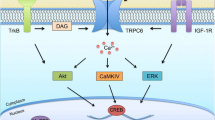
The longest standing mouse model used to investigate the role of SCN2A in vivo is the Scn2aQ54 mouse, which expresses a gain-of-function SCN2A mutant, GAL879-881QQQ, at a low level over a WT Scn2a+/+ background. Scn2aQ54 mice exhibit epilepsy, persistent sodium current, spontaneous action potential firing, and repetitive behaviors [69]. To date, these mice have primarily served as a model for epilepsy, and there are no published studies investigating ASD endophenotypes in Scn2aQ54 mice. Multiple genetic modifiers that influence the severity of Scn2aQ54 epilepsy have since been discovered, including Kcnq2 [70], Cacna1g [71], and Hlf [72]. Interestingly, the severity of the Scn2aQ54 phenotypes produced are highly dependent upon mouse strain: Scn2aQ54 mice on the SJL/J background exhibit more frequent and more severe seizures than Scn2aQ54 mice on a C57BL/6J background [73, 74]. Further investigation into the differences between the two models has identified calcium/calmodulin protein kinase II (CaMKII) as a modulator of the Scn2aQ54 phenotype [74], suggesting the CaMKII pathway may serve as a therapeutic target for SCN2A-associated epilepsy.
Haploinsufficient mouse models
Homozygous knockout of SCN2A is perinatally lethal [45], prohibiting the use of complete, unconditional gene knockout for all but in vitro assays. Nevertheless, Scn2a haploinsufficient mouse models (Scn2a+/−) show significant phenotypes linked to human disease and have been used to further elucidate the role of NaV1.2 in both normal neurodevelopment and disease. Scn2a+/− mice display mild absence-like seizures, delayed spatial learning, increased contextual fear learning, impaired fear extinction, and hyperactivity [42, 46, 75, 76], all of which may be related to ASD symptoms in humans. Cortical neurons from these mice display impaired excitability and impaired excitatory synapse function [61]. Additionally, Scn2a+/− mice display reduced anxiety in novel environments [42, 43] but show an increased aversion to light [42], suggesting the model may recapitulate some of the sensory sensitivity seen in ASD.
Other potential endophenotypes of ASD, such as reduced sociability, impaired communication, and repetitive behaviors, are not consistently observed in Scn2a+/− mice. Léna and Mantegazza found that Scn2a+/− mice displayed repetitive behaviors and reduced vocalization and communication as juveniles, but that these phenotypes disappeared in adulthood [43]. In contrast, Tatsukawa et al. did not observe repetitive behaviors or communication deficits in juvenile or adult Scn2a+/− mice [42]. Therefore, it is unclear whether simple loss of one copy of SCN2A is sufficient to generate the social and behavioral phenotypes of autism spectrum disorders. More work is needed, possibly with specific genetic models of ASD-linked SCN2A mutations, to fully address specific differences in genotype-phenotype relationships.
Non-mammalian SCN2A models
An SCN2A homolog occurs in both zebrafish (scn1Lab) and Drosophila (paralytic). In both organisms, the homolog is not specific to SCN2A but instead bears significant homology to three human sodium channel counterparts: SCN1A, SCN2A, and SCN3A. Knockout models have been created for both organisms (reviewed in [77]), but to date, these have generally been used as models for SCN1A but not SCN2A or SCN3A.
Mutations in the SCN1A gene, which encodes the voltage-gated sodium channel NaV1.1, lead to Dravet syndrome, which is characterized by temperature-sensitive epilepsy. Both zebrafish and Drosophila models for Dravet syndrome display seizure phenotypes which respond to drugs used to treat Dravet epilepsy, suggesting the models may be effective for drug screening [78].
Interestingly, based on MUSCLE homology analysis [79], zebrafish scn1Lab is slightly more homologous to human SCN2A than SCN1A, with 81% and 77% identities, respectively. The high sequence homology to SCN2A suggests that these zebrafish models may be useful tools for drug screening in the context of SCN2A-related disorders, as well. The Drosophila paralytic gene is much less homologous to human sodium channels, with 57% identity to SCN2A and 47.5% identity to SCN1A. However, Sun et al. [80] and Schutte et al. [81] were able to recapitulate temperature-sensitive seizure phenotypes in Drosophila using knock-in mutations homologous to Dravet-causing mutations in human SCN1A. These models were subsequently used to further elucidate neural dysfunction in Dravet syndrome. In summary, while zebrafish and Drosophila may not serve as an ideal behavioral model for SCN2A-related disorders, they may prove useful for drug screening and as a tool to identify genetic interactions.
Cell-based SCN2A models
Human stem cell models of SCN2A mutations
To date, one patient-derived SCN2A syndrome iPSC line has been published, though no data have yet been published using it as an experimental model [82]. However, a number of investigators have used CRISPR to genetically modify the SCN2A gene in stem cells derived from control subjects, which have revealed phenotypes that mirror those observed in mouse models of SCN2A syndrome mentioned above. In 2018, Deneault et al. used CRISPR to create a homozygous knockout of SCN2A in a human iPSC line generated from the unaffected father of a child with ASD [83]. These stem cells were subsequently differentiated into excitatory neurons through ectopic expression of NEUROG2 and assessed for neural activity. The authors found that, compared to neurons generated from the control SCN2A+/+ line, SCN2A−/− neurons showed a significant reduction in spontaneous excitatory postsynaptic current, mean firing rate, and network burst frequency. Based on both patch-clamp and multi-electrode array analyses, Deneault et al. concluded that knockout of SCN2A led to an overall decrease in neuronal activity at both the single cell and population levels [83].
In 2019, Lu et al. used CRISPR to create an SCN2A haploinsufficient (SCN2A+/−) hESC line [84]. Using overexpression of NEUROG1 and NEUROG2, the authors differentiated the cell line into a network of both glutamatergic excitatory and GABAergic inhibitory neurons. Similarly to what Deneault et al. reported with SCN2A−/− neurons [83], Lu et al. found that SCN2A+/− neurons also displayed a significant reduction in spontaneous neural activity. However, SCN2A haploinsufficiency did not appear to disrupt inhibitory synaptic transmission in GABAergic cells. Lu et al. thus concluded that haploinsufficiency of SCN2A is sufficient to cause an overall decrease in neural network activity.
These two studies demonstrate that both hESCs and iPSCs can be used to create in vitro models to study SCN2A activity in neurons on a human genetic background. Furthermore, the consistency in results between both studies—from two different groups using completely different cell lines—supports the use of induced neurons from stem cells as a viable model system. What is unknown from these models is how disease-causing mutations from actual SCN2A syndrome patients affect the channel’s electrophysiological properties, and whether the mutations observed in patients are associated with disruptions of other intracellular signaling pathways or channel functions.
The future of SCN2A iPSCs
While stem cell models are already proving valuable, the ability to direct differentiation into specific, desired cell types is still relatively new. Many types of neurons have yet to be reliably produced in vitro, and existing differentiation protocols often yield heterogeneous populations of neurons. As proper neuronal development and function often depends on the interplay between different cell types, future efforts should focus on creating more physiologically relevant in vitro neural environments. Specifically, since NaV1.2 is primarily expressed in glutamatergic neurons of the cerebral and cerebellar cortex, and these neurons develop in tandem with GABAergic neurons and glia, it would be ideal to utilize model systems in which these neurons differentiate together in the ratios normally observed in the embryonic brain, and if possible, in their normal three-dimensional physical relationships.
In addition to the difficulty in generating specific cell types, in vitro neural differentiation often results in functionally immature neurons. Generally speaking, iPSC-derived neurons have depolarized resting membrane potentials, high input resistance, low membrane capacitance, and are often unable to fire repetitive, narrow-spike-width action potentials [85,86,87,88,89,90], all of which suggest that they may not express the full complement of ion channels found in mature neurons despite having typical neuronal morphology. For this reason, induced neurons may be a good tool to investigate neurodevelopment in vitro, but they are currently a poor model for mature neural networks. It may be that physiologically relevant heterogeneous populations of neurons are required to achieve neural maturity from stem cells. It is also probable that three-dimensional structure affects development, and thus more elaborate and multi-dimensional culture methods will likely need to be utilized.
Organoids
As discussed above, neurodevelopment occurs in a tightly temporally and spatially regulated manner, and aberrant NaV1.2 expression (either high or low) is likely to disrupt this process in various brain regions. While this process can be studied in non-human animal models, these do not recapitulate the human genetic context that likely contributes to SCN2A syndrome. To study the role of NaV1.2 in human neurodevelopment, organoid models seem particularly promising. Organoids are three-dimensional systems in which iPSCs partially regulate their own differentiation into a specific tissue type, resulting in a model tissue that closely recapitulates the normal process of development. The majority of brain organoid protocols generate tissues with gene expression patterns most similar to the second trimester of in utero development or younger [91,92,93,94]. Recent technical improvements in organoid technology and prolonged periods in culture may enable scientists to model later stages of fetal development [95]. Organoids have been used to model both idiopathic autism [96] and autism related to mutations in the voltage-gated calcium channel CaV1.2, a.k.a. Timothy syndrome [25], and studies suggest that these models will be useful for studying both neurodevelopmental and genetic factors that contribute to ASD [97]. Indeed, cortical organoids that were developed using iPSCs derived from patients with CDK5RAP2-associated microcephaly accurately recapitulated human phenotypes that were not apparent in animal models likely due to differences in neurodevelopment, highlighting the relevance of organoid technology [98]. In the case of SCN2A syndrome, organoids may help reveal both cell-autonomous and non-cell-autonomous effects of NaV1.2 function. For example, although NaV1.2 is mostly expressed in glutamatergic neurons, we do not know whether loss of NaV1.2 in glutamatergic neurons might have downstream effects on the development or function of GABAergic neurons or glia, which tend to develop later. In the context of organoids, it is possible to detect how abnormalities in one cell type affect another cell type in a human developmental context.
Neural Differentiation
In addition to organoids, iPSCs can be differentiated in 2D cultures, allowing for experiments that may be challenging in 3D organoids, such as patch-clamping of individual neurons. Although neurons can be isolated and cultured from animal models, human iPSC-derived neurons have been shown to successfully recapitulate human phenotypes that did not manifest in the animal model, such as with 16p11.2del models [99]. The available data suggest that the gene product of SCN2A, NaV1.2, is primarily expressed in glutamatergic neurons of the cerebral and cerebellar cortex. Fortunately, forebrain cortical glutamatergic neurons are one cell type for which the field has developed robust and reproducible iPSC differentiation protocols. Overexpression of NEUROG2 can drive consistent and rapid maturation of this neuronal type [100], which may help reveal how NaV1.2 is localized and functions in human neurons. It is important to note, however, that NEUROG2 overexpression protocols may bypass developmental stages of interest and thus are likely best suited to studies of ion channel localization and function rather than neurodevelopment. The role of NaV1.2 during early neurodevelopment may be better recapitulated using differentiation protocols that rely on small molecules and morphogens, such as dual-SMAD inhibition and dorsalization via small molecules [101, 102].
The other brain region noted for persistently high levels of NaV1.2 expression is the cerebellar cortex, specifically, cerebellar granule neurons. As described above, these cells express NaV1.2 throughout development and maintain high expression of this channel throughout life, rather than replacing it with NaV1.6 (as occurs in the cerebral cortex). There are a small number of published protocols for generating cerebellar tissue types from iPSCs [103,104,105], and given the increasing interest in using iPSCs to model human neurodevelopment, more differentiation protocols are likely to follow. These will open the door for scientists to interrogate the unusual NaV1.2 expression pattern observed in the cerebellum and investigate its contribution to SCN2A syndromes.
One particular strength of human iPSC models is that they retain the genetic code of the patients from whom they are derived. Unlike postmortem tissue, iPSC models effectively turn back the clock on a variety of potentially confounding lifetime exposures, such as medication, comorbid diseases, age, and substance use. This yields a powerful tool for determining which genetic pathways may be altered by a specific mutation or set of common variants. In the case of SCN2A syndromes, iPSCs from patients who have known deleterious SCN2A mutations could help identify which pathways are perturbed during specific developmental stages in specific brain regions, potentially revealing new druggable therapeutic targets and advancing our understanding of this complex syndrome.
Xenograft studies
Although many human neuronal culture systems are limited in their ability to generate mature neurons with normal electrophysiological properties, xenograft systems have not only met with success in this area, but they have also shown that many of the signaling cues for development and migration of neurons are common between mice and humans. Multiple groups have shown that human iPSC-derived cells introduced into the mouse brain migrate to their proper location and develop mature characteristics of their specified cell type [91, 106, 107]. Although xenograft systems do not mimic a fully human context, they are uniquely well suited for studies of how iPSC-derived neurons integrate into a functional neuronal circuit in a brain, which no current iPSC-only model can (yet) do. As with the organoid studies mentioned above, xenograft studies could allow for more precise analyses of how SCN2A mutations affect the assembly and function of neural circuits that rely on SCN2A-expressing cells in the cerebral and cerebellar cortices.
Conclusions
Autism spectrum disorders are a complex group of neuropsychiatric disorders with genetic and environmental causes. A lack of robust cellular models for ASD remains a bottleneck to progress for researchers and clinicians compelled to expand the mechanistic understanding of ASD, as well as those working to develop new therapeutic interventions for ASD management. Monogenic causes of ASD, while rare, hold promise as cellular testbeds for drug development and biological insights alike. While SCN2A may not be alone in this potential, the development and critical assessment of SCN2A channelopathies in the context of experimental neuronal systems will be an essential step for expanding the palette of experimental systems needed to achieve a better molecular understanding of ASD. SCN2A and other as-yet undefined genes may play a role in redefining our understanding of neurodevelopment and ASD.
Availability of data and materials
Not applicable
Abbreviations
- 5A :
-
Adult splice isoform of NaV1.2
- 5N:
-
Neonatal splice isoform of NaV1.2
- ASD:
-
Autism spectrum disorder
- CACNA1G:
-
Calcium voltage-gated channel subunit alpha1 G
- CaMKII:
-
Calcium/calmodulin dependent protein kinase II
- EEG:
-
Electroencephalogram
- fMRI:
-
Functional magnetic resonance imaging
- hESCs:
-
Human embryonic stem cells
- HLF:
-
HLF transcription factor, PAR bZIP family member
- ID:
-
Intellectual disability
- iPSCs:
-
Induced pluripotent stem cells
- KCNQ2:
-
Potassium voltage-gated channel subfamily Q member 2
- NEUROG1:
-
Neurogenin 1
- NEUROG2:
-
Neurogenin 2
- SCN1A:
-
Sodium voltage-gated channel alpha subunit 1
- SCN1LAB:
-
Sodium channel, voltage-gated type 1 like, alpha b
- SCN2A:
-
Sodium voltage-gated channel alpha subunit 2
- SCN3A:
-
Sodium voltage-gated channel alpha subunit 3
References
Bonnet-Brilhault F, Rajerison TA, Paillet C, Guimard-Brunault M, Saby A, Ponson L, et al. Autism is a prenatal disorder: evidence from late gestation brain overgrowth. Autism Res. 2018;11(12):1635–42.
Vorstman JAS, Parr JR, Moreno-De-Luca D, Anney RJL, Nurnberger JI Jr, Hallmayer JF. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet. 2017;18(6):362–76.
Siu MT, Weksberg R. Epigenetics of autism spectrum disorder. Adv Exp Med Biol. 2017;978:63–90.
Modabbernia A, Velthorst E, Reichenberg A. Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol Autism. 2017;8:13.
de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nat Med. 2016;22(4):345–61.
De Rubeis S, Buxbaum JD. Genetics and genomics of autism spectrum disorder: embracing complexity. Hum Mol Genet. 2015;24(R1):R24–31.
Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46(8):881–5.
Lai MC, Lombardo MV, Baron-Cohen S. Autism Lancet. 2014;383(9920):896–910.
Lord C, Elsabbagh M, Baird G, Veenstra-Vanderweele J. Autism spectrum disorder. Lancet. 2018;392(10146):508–20.
Muhle RA, Reed HE, Stratigos KA, Veenstra-VanderWeele J. The emerging clinical neuroscience of autism spectrum disorder: a review. JAMA Psychiatry. 2018;75(5):514–23.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–20.
Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–7.
Germain PL, Testa G. Taming human genetic variability: transcriptomic meta-analysis guides the experimental design and interpretation of iPSC-based disease modeling. Stem Cell Reports. 2017;8(6):1784–96.
Thatava T, Kudva YC, Edukulla R, Squillace K, De Lamo JG, Khan YK, et al. Intrapatient variations in type 1 diabetes-specific iPS cell differentiation into insulin-producing cells. Mol Ther. 2013;21(1):228–39.
Kyttala A, Moraghebi R, Valensisi C, Kettunen J, Andrus C, Pasumarthy KK, et al. Genetic variability overrides the impact of parental cell type and determines iPSC differentiation potential. Stem Cell Reports. 2016;6(2):200–12.
Ben-Shalom R, Keeshen CM, Berrios KN, An JY, Sanders SJ, Bender KJ. Opposing effects on NaV1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol Psychiatry. 2017;82(3):224–32.
Tavassoli T, Kolevzon A, Wang AT, Curchack-Lichtin J, Halpern D, Schwartz L, et al. De novo SCN2A splice site mutation in a boy with autism spectrum disorder. BMC Med Genet. 2014;15:35.
Sanders SJ, Campbell AJ, Cottrell JR, Moller RS, Wagner FF, Auldridge AL, et al. Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci. 2018;41(7):442–56.
Zhu H, Lensch MW, Cahan P, Daley GQ. Investigating monogenic and complex diseases with pluripotent stem cells. Nat Rev Genet. 2011;12(4):266–75.
Achuta VS, Grym H, Putkonen N, Louhivuori V, Karkkainen V, Koistinaho J, et al. Metabotropic glutamate receptor 5 responses dictate differentiation of neural progenitors to NMDA-responsive cells in fragile X syndrome. Dev Neurobiol. 2017;77(4):438–53.
Boland MJ, Nazor KL, Tran HT, Szucs A, Lynch CL, Paredes R, et al. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome. Brain. 2017;140(3):582–98.
Gerhardt J. Epigenetic modifications in human fragile X pluripotent stem cells; implications in fragile X syndrome modeling. Brain Res. 1656;2017:55–62.
Zhou Y, Kumari D, Sciascia N, Usdin K. CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol Autism. 2016;7:42.
Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545(7652):54–9.
Krey JF, Pasca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16(2):201–9.
Panagiotakos G, Haveles C, Arjun A, Petrova R, Rana A, Portmann T, et al. Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy syndrome. Elife. 2019;8.
Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17(12):1657–62.
Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, et al. Isolation of MECP2-null Rett syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. 2011;20(11):2103–15.
Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143(4):527–39.
Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry. 2018;23(4):1051–65.
Tang X, Kim J, Zhou L, Wengert E, Zhang L, Wu Z, et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc Natl Acad Sci U S A. 2016;113(3):751–6.
Blair JD, Hockemeyer D, Bateup HS. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat Med. 2018;24(10):1568–78.
Ebrahimi-Fakhari D, Saffari A, Wahlster L, Di Nardo A, Turner D, Lewis TL Jr, et al. Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. 2016;17(4):1053–70.
Nadadhur AG, Alsaqati M, Gasparotto L, Cornelissen-Steijger P, van Hugte E, Dooves S, et al. Neuron-glia interactions increase neuronal phenotypes in tuberous sclerosis complex patient iPSC-derived models. Stem Cell Reports. 2019;12(1):42–56.
Zucco AJ, Pozzo VD, Afinogenova A, Hart RP, Devinsky O, D'Arcangelo G. Neural progenitors derived from tuberous sclerosis complex patients exhibit attenuated PI3K/AKT signaling and delayed neuronal differentiation. Mol Cell Neurosci. 2018;92:149–63.
Anastasaki C, Gutmann DH. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum Mol Genet. 2014;23(25):6712–21.
Anastasaki C, Woo AS, Messiaen LM, Gutmann DH. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum Mol Genet. 2015;24(12):3518–28.
Carrio M, Mazuelas H, Richaud-Patin Y, Gel B, Terribas E, Rosas I, et al. Reprogramming captures the genetic and tumorigenic properties of neurofibromatosis type 1 plexiform neurofibromas. Stem Cell Reports. 2019;12(2):411–26.
Wegscheid ML, Anastasaki C, Gutmann DH. Human stem cell modeling in neurofibromatosis type 1 (NF1). Exp Neurol. 2018;299(Pt B):270–80.
SFARI Gene 3.0. SFARI.org. 2019. https://gene.sfari.org/database/gene-scoring. Accessed 12 Dec 2019.
Tatsukawa T, Raveau M, Ogiwara I, Hattori S, Miyamoto H, Mazaki E, et al. Scn2a haploinsufficient mice display a spectrum of phenotypes affecting anxiety, sociability, memory flexibility and ampakine CX516 rescues their hyperactivity. Mol Autism. 2019;10:15.
Lena I, Mantegazza M. NaV1.2 haploinsufficiency in Scn2a knock-out mice causes an autistic-like phenotype attenuated with age. Sci Rep. 2019;9(1):12886.
Gazina EV, Leaw BT, Richards KL, Wimmer VC, Kim TH, Aumann TD, et al. ‘Neonatal’ Nav1.2 reduces neuronal excitability and affects seizure susceptibility and behaviour. Hum Mol Genet. 2015;24(5):1457–68.
Planells-Cases R, Caprini M, Zhang J, Rockenstein EM, Rivera RR, Murre C, et al. Neuronal death and perinatal lethality in voltage-gated sodium channel alpha (II)-deficient mice. Biophys J. 2000;78(6):2878–91.
Middleton SJ, Kneller EM, Chen S, Ogiwara I, Montal M, Yamakawa K, et al. Altered hippocampal replay is associated with memory impairment in mice heterozygous for the Scn2a gene. Nat Neurosci. 2018;21(7):996–1003.
Kwong K, Carr MJ. Voltage-gated sodium channels. Curr Opin Pharmacol. 2015;22:131–9.
Catterall WA. Forty years of sodium channels: structure, function, pharmacology, and epilepsy. Neurochem Res. 2017;42(9):2495–504.
de Lera RM, Kraus RL. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J Med Chem. 2015;58(18):7093–118.
Ahern CA, Payandeh J, Bosmans F, Chanda B. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J Gen Physiol. 2016;147(1):1–24.
Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140(5):1316–36.
Falkson G, Gelman RS, Dreicer R, Tormey DC, Alberts AS, Coccia-Portugal MA, et al. Myelodysplastic syndrome and acute nonlymphocytic leukemia secondary to mitolactol treatment in patients with breast cancer. J Clin Oncol. 1989;7(9):1252–9.
Yamagata T, Ogiwara I, Mazaki E, Yanagawa Y, Yamakawa K. Nav1.2 is expressed in caudal ganglionic eminence-derived disinhibitory interneurons: mutually exclusive distributions of Nav1.1 and Nav1.2. Biochem Biophys Res Commun. 2017;491(4):1070–6.
Martinez-Hernandez J, Ballesteros-Merino C, Fernandez-Alacid L, Nicolau JC, Aguado C, Lujan R. Polarised localisation of the voltage-gated sodium channel Na(v)1.2 in cerebellar granule cells. Cerebellum. 2013;12(1):16–26.
Schaller KL, Caldwell JH. Expression and distribution of voltage-gated sodium channels in the cerebellum. Cerebellum. 2003;2(1):2–9.
Wang W, Takashima S, Segawa Y, Itoh M, Shi X, Hwang SK, et al. The developmental changes of Na(v)1.1 and Na(v)1.2 expression in the human hippocampus and temporal lobe. Brain Res. 2011;1389:61–70.
Gazina EV, Richards KL, Mokhtar MB, Thomas EA, Reid CA, Petrou S. Differential expression of exon 5 splice variants of sodium channel alpha subunit mRNAs in the developing mouse brain. Neuroscience. 2010;166(1):195–200.
Garrido JJ, Fernandes F, Giraud P, Mouret I, Pasqualini E, Fache MP, et al. Identification of an axonal determinant in the C-terminus of the sodium channel Na(v)1.2. EMBO J. 2001;20(21):5950–61.
Kole MH, Stuart GJ. Signal processing in the axon initial segment. Neuron. 2012;73(2):235–47.
Bender KJ, Trussell LO. The physiology of the axon initial segment. Annu Rev Neurosci. 2012;35:249–65.
Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ Jr, Clarkson RL, Sanders SJ, et al. The Autism-associated gene Scn2a contributes to dendritic excitability and synaptic function in the prefrontal cortex. Neuron. 2019;103(4):673–85 e5.
Baasch AL, Huning I, Gilissen C, Klepper J, Veltman JA, Gillessen-Kaesbach G, et al. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia. 2014;55(4):e25–9.
Nakamura K, Kato M, Osaka H, Yamashita S, Nakagawa E, Haginoya K, et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 2013;81(11):992–8.
Dickinson D, Straub RE, Trampush JW, Gao Y, Feng N, Xie B, et al. Differential effects of common variants in SCN2A on general cognitive ability, brain physiology, and messenger RNA expression in schizophrenia cases and control individuals. JAMA Psychiatry. 2014;71(6):647–56.
Li J, Cai T, Jiang Y, Chen H, He X, Chen C, et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol Psychiatry. 2016;21(2):290–7.
Rees E, Kirov G, Walters JT, Richards AL, Howrigan D, Kavanagh DH, et al. Analysis of exome sequence in 604 trios for recessive genotypes in schizophrenia. Transl Psychiatry. 2015;5:e607.
Kong Y, Yan K, Hu L, Wang M, Dong X, Lu Y, et al. Association between SCN1A and SCN2A mutations and clinical/EEG features in Chinese patients from epilepsy or severe seizures. Clin Chim Acta. 2018;483:14–9.
Scult MA, Trampush JW, Zheng F, Conley ED, Lencz T, Malhotra AK, et al. A common polymorphism in SCN2A predicts general cognitive ability through effects on PFC physiology. J Cogn Neurosci. 2015;27(9):1766–74.
Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, et al. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience. 2001;102(2):307–17.
Kearney JA, Yang Y, Beyer B, Bergren SK, Claes L, Dejonghe P, et al. Severe epilepsy resulting from genetic interaction between Scn2a and Kcnq2. Hum Mol Genet. 2006;15(6):1043–8.
Calhoun JD, Hawkins NA, Zachwieja NJ, Kearney JA. Cacna1g is a genetic modifier of epilepsy caused by mutation of voltage-gated sodium channel Scn2a. Epilepsia. 2016;57(6):e103–7.
Hawkins NA, Kearney JA. Hlf is a genetic modifier of epilepsy caused by voltage-gated sodium channel mutations. Epilepsy Res. 2016;119:20–3.
Bergren SK, Chen S, Galecki A, Kearney JA. Genetic modifiers affecting severity of epilepsy caused by mutation of sodium channel Scn2a. Mamm Genome. 2005;16(9):683–90.
Thompson CH, Hawkins NA, Kearney JA, George AL Jr. CaMKII modulates sodium current in neurons from epileptic Scn2a mutant mice. Proc Natl Acad Sci U S A. 2017;114(7):1696–701.
Miyamoto H, Tatsukawa T, Shimohata A, Yamagata T, Suzuki T, Amano K, et al. Impaired cortico-striatal excitatory transmission triggers epilepsy. Nat Commun. 2019;10(1):1917.
Ogiwara I, Miyamoto H, Tatsukawa T, Yamagata T, Nakayama T, Atapour N, et al. Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun Biol. 2018;1.
Schutte SS, Schutte RJ, Barragan EV, O'Dowd DK. Model systems for studying cellular mechanisms of SCN1A-related epilepsy. J Neurophysiol. 2016;115(4):1755–66.
Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun. 2013;4:2410.
Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47(W1):W636–W41.
Sun L, Gilligan J, Staber C, Schutte RJ, Nguyen V, O'Dowd DK, et al. A knock-in model of human epilepsy in Drosophila reveals a novel cellular mechanism associated with heat-induced seizure. J Neurosci. 2012;32(41):14145–55.
Schutte RJ, Schutte SS, Algara J, Barragan EV, Gilligan J, Staber C, et al. Knock-in model of Dravet syndrome reveals a constitutive and conditional reduction in sodium current. J Neurophysiol. 2014;112(4):903–12.
Sampaio GLA, Martins GLS, Paredes BD, Nonaka CKV, da Silva KN, Rossi EA, et al. Generation of an induced pluripotent stem cell line from a patient with autism spectrum disorder and SCN2A haploinsufficiency. Stem Cell Res. 2019;39:101488.
Deneault E, White SH, Rodrigues DC, Ross PJ, Faheem M, Zaslavsky K, et al. Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Reports. 2018;11(5):1211–25.
Lu C, Shi X, Allen A, Baez-Nieto D, Nikish A, Sanjana NE, et al. Overexpression of NEUROG2 and NEUROG1 in human embryonic stem cells produces a network of excitatory and inhibitory neurons. FASEB J. 2019;33(4):5287–99.
Pre D, Nestor MW, Sproul AA, Jacob S, Koppensteiner P, Chinchalongporn V, et al. A time course analysis of the electrophysiological properties of neurons differentiated from human induced pluripotent stem cells (iPSCs). PLoS One. 2014;9(7):e103418.
Livesey MR, Magnani D, Hardingham GE, Chandran S, Wyllie DJ. Functional properties of in vitro excitatory cortical neurons derived from human pluripotent stem cells. J Physiol. 2016;594(22):6573–82.
Belinsky GS, Rich MT, Sirois CL, Short SM, Pedrosa E, Lachman HM, et al. Patch-clamp recordings and calcium imaging followed by single-cell PCR reveal the developmental profile of 13 genes in iPSC-derived human neurons. Stem Cell Res. 2014;12(1):101–18.
Lam RS, Topfer FM, Wood PG, Busskamp V, Bamberg E. Functional maturation of human stem cell-derived neurons in long-term cultures. PLoS One. 2017;12(1):e0169506.
Nicholas CR, Chen J, Tang Y, Southwell DG, Chalmers N, Vogt D, et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell. 2013;12(5):573–86.
Bardy C, van den Hurk M, Kakaradov B, Erwin JA, Jaeger BN, Hernandez RV, et al. Predicting the functional states of human iPSC-derived neurons with single-cell RNA-seq and electrophysiology. Mol Psychiatry. 2016;21(11):1573–88.
Zhou FW, Fortin JM, Chen HX, Martinez-Diaz H, Chang LJ, Reynolds BA, et al. Functional integration of human neural precursor cells in mouse cortex. PLoS One. 2015;10(3):e0120281.
Kelava I, Lancaster MA. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Dev Biol. 2016;420(2):199–209.
Yoon SJ, Elahi LS, Pasca AM, Marton RM, Gordon A, Revah O, et al. Reliability of human cortical organoid generation. Nat Methods. 2019;16(1):75–8.
Lancaster MA, Corsini NS, Wolfinger S, Gustafson EH, Phillips AW, Burkard TR, et al. Guided self-organization and cortical plate formation in human brain organoids. Nat Biotechnol. 2017;35(7):659–66.
Trujillo CA, Gao R, Negraes PD, Gu J, Buchanan J, Preissl S, et al. Complex oscillatory waves emerging from cortical organoids model early human brain network development. Cell Stem Cell. 2019;25(4):558–69 e7.
Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell. 2015;162(2):375–90.
Amiri A, Coppola G, Scuderi S, Wu F, Roychowdhury T, Liu F, et al. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science. 2018;362(6420).
Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–9.
Deshpande A, Yadav S, Dao DQ, Wu ZY, Hokanson KC, Cahill MK, et al. Cellular phenotypes in human iPSC-derived neurons from a genetic model of autism spectrum disorder. Cell Rep. 2017;21(10):2678–87.
Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78(5):785–98.
Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc. 2012;7(10):1836–46.
Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci. 2012;15(3):477–86 S1.
Holmes DB, Heine VM. Streamlined 3D cerebellar differentiation protocol with optional 2D modification. J Vis Exp. 2017;130.
Muguruma K. Self-organized cerebellar tissue from human pluripotent stem cells and disease modeling with patient-derived iPSCs. Cerebellum. 2018;17(1):37–41.
Erceg S, Lukovic D, Moreno-Manzano V, Stojkovic M, Bhattacharya SS. Derivation of cerebellar neurons from human pluripotent stem cells. Curr Protoc Stem Cell Biol. 2012; Chapter 1:Unit 1H 5.
Grealish S, Diguet E, Kirkeby A, Mattsson B, Heuer A, Bramoulle Y, et al. Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell. 2014;15(5):653–65.
Grealish S, Heuer A, Cardoso T, Kirkeby A, Jonsson M, Johansson J, et al. Monosynaptic tracing using modified rabies virus reveals early and extensive circuit integration of human embryonic stem cell-derived neurons. Stem Cell Reports. 2015;4(6):975–83.
Acknowledgements
The authors would like to thank Leah Schust from the FamilieSCN2A Foundation for serving as an enthusiastic and valuable resource regarding SCN2A syndromes and for providing a comprehensive summary of currently identified SCN2A mutations.
Funding
Funding for the authors was provided by: NIH 5KL2TR002536, the Simons Foundation for Autism Research Initiative, and the Carver Charitable Trust.
Author information
Authors and Affiliations
Contributions
All authors contributed to the literature review for and writing of the manuscript. CAA and TMG created the figure. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kruth, K.A., Grisolano, T.M., Ahern, C.A. et al. SCN2A channelopathies in the autism spectrum of neuropsychiatric disorders: a role for pluripotent stem cells?. Molecular Autism 11, 23 (2020). https://doi.org/10.1186/s13229-020-00330-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-020-00330-9