
Abstract
Background
Genotype and environment can interact during development to produce novel adaptive traits that support life in extreme conditions. The development of the annual killifish Austrofundulus limnaeus is unique among vertebrates because the embryos have distinct cell movements that separate epiboly from axis formation during early development, can enter into a state of metabolic dormancy known as diapause and can survive extreme environmental conditions. The ability to enter into diapause can be maternally programmed, with young females producing embryos that do not enter into diapause. Alternately, embryos can be programmed to “escape” from diapause and develop directly by both maternal factors and embryonic incubation conditions. Thus, maternally packaged gene products are hypothesized to regulate developmental trajectory and perhaps the other unique developmental characters in this species.
Results
Using high-throughput RNA sequencing, we generated transcriptomic profiles of mRNAs, long non-coding RNAs and small non-coding RNAs (sncRNAs) in 1–2 cell stage embryos of A. limnaeus. Transcriptomic analyses suggest maternal programming of embryos through alternatively spliced mRNAs and antisense sncRNAs. Comparison of these results to those of comparable studies on zebrafish and other fishes reveals a surprisingly high abundance of transcripts involved in the cellular response to stress and a relatively lower expression of genes required for rapid transition through the cell cycle.
Conclusions
Maternal programming of developmental trajectory is unlikely accomplished by differential expression of diapause-specific genes. Rather, evidence suggests a role for trajectory-specific splice variants of genes expressed in both phenotypes. In addition, based on comparative studies with zebrafish, the A. limnaeus 1–2 cell stage transcriptome is unique in ways that are consistent with their unique life history. These results not only impact our understanding of the genetic mechanisms that regulate entrance into diapause, but also provide insight into the epigenetic regulation of gene expression during development.
Similar content being viewed by others


Background
Development in the annual killifish Austrofundulus limnaeus is unique for four major reasons. First, the embryos develop slowly for their size and can enter into a state of developmental and metabolic arrest termed diapause at three distinct developmental stages [1, 2]. Second, during early development the cell movements associated with epiboly are separated temporally and spatially from gastrulation and formation of the embryonic axis [3, 4]. Third, embryonic development is plastic and embryos can develop along at least two alternative pathways based on an interaction of maternal programming and incubation environment [5]. Finally, embryos of A. limnaeus can tolerate and survive extreme environmental stresses, such as long-term anoxia and dehydration [6]. Despite these unique characters, the development of A. limnaeus is quintessentially vertebrate and appears to utilize the same conserved genetic networks that govern development of the typical vertebrate body plan [4]. The mix of unique and apparently conserved developmental characteristics of this species makes it an excellent model for examining the evolutionary and mechanistic adaptations of novelty in vertebrate development.
In all vertebrates, the activation of the embryonic genome is delayed for several to many cell divisions following fertilization [7, 8]. During this time, cellular processes are directed by maternal products (RNA transcripts, proteins, ribosomes and hormones) packaged in the egg during oogenesis [9–12]. In the zebrafish Danio rerio, and most other organisms, maternally derived mRNA transcripts direct early gene expression in the zygote through cleavage and blastulation, but are targeted for degradation coincident with activation of embryonic genome transcription [11, 13–16]. This process has been termed the maternal-to-zygotic transition and coincides with what is known as the mid-blastula transition in some vertebrates [15]. While many studies have reported on the existence of the maternal-to-zygotic transition during vertebrate development, very few have actually profiled the contents of newly fertilized eggs to identify maternally contributed gene products [13, 17].
Maternally packaged RNAs underlie cellular programming in vertebrate embryos that ensures proper early development such as cleavage, formation of the blastula and gastrulation [7, 17, 18]. Patterns of early cleavage determine distribution of yolk resources, maternally derived factors, and establish the morphogenetic fields that define the vertebrate body plan. Thus, maternally packaged RNAs and their regulation, modification and stability are especially important during the earliest phases of embryonic development. Understanding the key transcripts that must be packaged into an oocyte, and the mechanisms that determine their stability, translatability and thus their ultimate expression and action could help explain a diversity of developmental phenomena.
There are two major avenues for altered expression of the maternal transcriptome during early development: alternative packaging of transcripts and modifications of stability or translatability to existing transcripts. For example, alternative mRNA splicing has been found in many vertebrate systems as part of cellular responses to environmental stimuli [19, 20]. Alternatively, small non-coding RNAs (sncRNAs) have been implicated in the regulation of gene expression by altering mRNA stability or translation in a variety of contexts including embryonic development [21–26]. The role of sncRNAs in the highly conserved processes of early vertebrate development has received far less attention than other potential mechanisms of regulation. The ability of a single sncRNA to target multiple transcripts and effect global alterations in gene expression makes this an attractive mechanism for control of gene networks in the absence of an active genome. Many recent studies suggest a critical role of regulatory RNAs in early vertebrate development [27].
Annual killifish (Aplocheiloidei) are represented by hundreds of species of small tropical and subtropical fishes in Africa and South America [28]. The annual killifish, A. limnaeus, is native to ephemeral ponds on the coast of Venezuela [29–31]. These ponds are short-lived (several weeks to several months), typically small (3–200 m2), and offer a harsh developmental environment with large fluctuations in key environmental parameters such as temperature, oxygen partial pressure, pH and water availability [6, 32]. These fish grow rapidly to sexual maturity and spawn continuously during their typically short (a few months) adult life, leaving their embryos to survive the dry season. Importantly, an expanding set of genomic tools, including a draft genome assembly [33], are now available for A. limnaeus, making it possible to explore genetic and epigenetic mechanisms during development.
Annual killifish embryos are a unique system for examining developmental physiology because they are capable of entering an endogenously cued metabolic dormancy termed diapause as an adaptive phenotype to survive the seasonal drying of their pond habitats [34, 35]. Diapause can occur at three distinct developmental stages, diapause I, II, III [2]. There are unique physiological traits associated with each stage of diapause; however, diapause II embryos show the greatest degree of tolerance to environmental stresses such as desiccation and anoxia [6, 36]. Entrance into diapause II (from here forward referred to as diapause) is one of two possible trajectories during the embryonic development of annual killifish [34, 35]. While a large proportion of embryos enter diapause as their normal mode of development, others are capable of “escaping” diapause and instead develop continuously until the pre-hatching stage [2]. Early embryos on either trajectory are indistinguishable; however, during somitogenesis the trajectories diverge in both morphological and physiological characters such that the timing of developmental events is unique for each trajectory [5]. The mechanisms that regulate these trajectories are currently unknown, but recent studies suggest possible maternal provisioning during oogenesis or another form of epigenetic programming [5, 37]. Typically, younger females produce almost exclusively escape embryos and older females produce almost exclusively diapausing embryos [5]. Yet, there is a great deal of interindividual variation and some females will consistently produce escape or diapausing embryos independent of age. Interestingly, embryonic incubation temperature can override maternal influences and lead to embryos that develop exclusively along the escape and diapause trajectories. Incubation at 20 °C results in 100% diapausing embryos, while incubation at 30 °C results in 100% escape embryos [5]. Thus, both the pre-fertilization and post-fertilization environment can affect developmental trajectory. Given the harsh conditions in which these embryos exist and the short duration of pond inundation, the developmental trajectory of an embryo will likely have a profound effect on its survival and on the survival of the local population. The mechanisms that mediate this type of critical genome–environment interaction are unknown but critically important for understanding the basic mechanisms of development in the natural world.
The events that occur in early development are thought to be some of the most conserved processes in biology. For example, there is striking conservation of function in the genes that regulate blastulation and gastrulation in all animals [38, 39]. A great deal of work has gone into characterizing these shared molecular pathways, while relatively few studies have focused on gene expression changes that may underlie plasticity during vertebrate development [40]. In fact, the vast majority of gene expression studies on developing vertebrates have focused on systems that exhibit little to no intraspecific plasticity in development [41–43]. Here, we report on the transcriptome of newly fertilized eggs of the annual killifish A. limnaeus collected from females that are known to produce 100% escape and 100% diapausing embryos. This paper describes for the first time the maternally derived transcriptome of early embryos of A. limnaeus, explores the possibility of maternal control of entrance into diapause through differential packaging of RNA and uses a comparative approach to identify aspects of the transcriptome that may explain some of the unique attributes of development in this species compared to more typical teleosts. Evidence is presented that suggests splice variants of genes common to both trajectories and differential packaging of sncRNAs are both possible routes for maternal control of developmental trajectory. Further, comparative analysis of A. limnaeus to zebrafish suggests a unique maternally packaged transcriptome in A. limnaeus that is consistent in many ways with the unique developmental patterns observed in annual killifishes.
Results
Maternally derived poly-A RNA transcriptome
Details regarding poly-A RNA transcriptome library sequencing and bioinformatics can be found in Additional file 1. RNA-seq methods detected 12,329 transcripts in the 1–2 cell stage transcriptome of A. limnaeus with a mean expression of 2 or greater fragments per kilobase of transcript per million mapped reads (FPKM), representing about 60% of all sequences in the libraries (Additional file 2). The 20 most abundant transcripts included nuclear- and mitochondrially encoded protein-coding, ribosomal RNA and long non-coding RNA genes (Table 1). Gene ontology (GO) analysis revealed enrichment for highly expressed genes (500 most abundant transcripts; >180 FPKM) in categories that include RNA-binding proteins, cytoskeletal proteins as well as redox reaction enzymes and pathways enriched for ATP synthesis and G-protein-coupled signaling (Additional file 3).
Differential expression of poly-A RNA
Gene-level analysis determined that none of the 12,329 genes were differentially packaged in diapause- and escape-destined embryos (t test, FDR > 0.10). However, 57 genes showed differential exon usage between diapause- and escape-destined embryos (Fig. 1a). GO analysis suggests that the differentially expressed exons are enriched in a number of pathways including glycolysis and insulin signaling (Fig. 1b). These exons reside within genes with a variety of functions including: heat-shock protein (hsp), hspa14; pre-mRNA splicing, snrnp200; cytoskeletal proteins, vinculin; transmembrane ion transport, sideroflexin; cell adhesion, svep1; and mTOR signaling of cell growth and proliferation, ribosome protein s6 kinase. The 10 genes with the most significant differentially expressed exons (based on FDR adjusted P values) are presented in Fig. 2, and the entire list of 57 genes can be found in Additional file 4.

Alternative splicing of poly-A RNA in embryos of A. limnaeus that will develop along two alternative developmental trajectories. a Differential exon usage in mRNA gene transcripts that are packaged into diapause- and escape-destined 1–2 cell stage embryos of A. limnaeus. Of the 57 exons that were significantly different between trajectories (red symbols, FDR < 0.1, t test) 49 are upregulated in diapause-bound embryos, while only 8 are upregulated in escape-bound embryos. b GO term analysis for transcript variants between diapause- and escape-bound embryos of A. limnaeus (P < 0.05) suggests enrichment for exons expressed in genes for a variety of molecular and metabolic pathways including glycolysis and the insulin/IGF signaling pathway

Top 10 genes with developmental trajectory-specific splice variants based on statistical significance. Each biological replicate is graphed separately in the exon usage graphs with orange lines indicating escape-bound embryos and blue lines indicating diapause-bound embryos. The x-axis on the plots indicates the exon number and the mapping location of the exon on the appropriate contig from the A. limnaeus genome file. Note that the y-axis is a log scale which tends to mask the differential expression of the exons, and thus we have provided a bar graph on a linear scale to better illustrate the mean (±SD) levels of expression for the differentially expressed exon within each gene. Blue bars indicate diapause-bound embryos, while orange bars represent escape-bound embryos
Comparative transcriptomics
Comparison of the transcriptome of 1–2 cell stage embryos from A. limnaeus to the same stage embryos of D. rerio (as reported by Harvey et al. [17]) revealed thousands of species-specific transcripts that likely identify major differences in developmental programs between annual killifish and zebrafish (Fig. 3). Of the 9018 transcripts present in the 1–2 cell stage transcriptome of D. rerio (≥2 FPKM, protein-coding only [17]) less than 10% (841) were identified as orthologous to the transcripts expressed in A. limnaeus (Fig. 3). Comparatively, 89% (10,997) and 91% (8174) of the expressed transcripts (≥2 FPKM, protein-coding only) were unique to A. limnaeus and D. rerio, respectively. The group of 841 genes that were expressed in both species shared similar patterns of abundance. However, there were significant differences in expression of these transcripts between the two species, with the most differentially expressed transcripts coming from mitochondrially encoded genes, claudin and ubiquitin, among others (Table 2). Transcripts encoded in the mitochondrial genome are twofold to fourfold higher in most cases for D. rerio compared to A. limnaeus (Table 3). While there are only 3 comparable datasets available in the literature for 1–2 cell stage embryos (Hippoglossus hippoglossus [44], D. rerio [17] and data presented here for A. limnaeus), it appears that each species has a unique expression pattern for the 20 most abundant maternally packaged transcripts (Fig. 4a). When comparing the 100 most abundantly expressed transcripts in the D. rerio and A. limnaeus 1–2 cell stage transcriptomes (Fig. 4b–d), the A. limnaeus transcriptome is enriched in GO terms for ion binding and transport as well as cytoskeletal structure and function (Fig. 4d). Alternatively, the A. limnaeus transcriptome is underrepresented compared to D. rerio in transcripts with GO terms for metabolic processes (Fig. 4d, Additional file 5).

Comparative analysis of poly-A transcriptomes in 1–2 cell stage embryos of D. rerio and A. limnaeus. a The 20 most abundant transcripts and their FPKM values that are unique to the transcriptome of either A. limnaeus or D. rerio. b Venn diagram depicting the number of shared (orthologous) and non-shared transcripts in 1–2 cell stage embryos of A. limnaeus (turquoise) and D. rerio (gray). Frequency histograms show the distribution of expression values of shared (orthologous) and non-shared genes and indicate similar patterns of transcript abundance between the two species

The relationship of the maternally packaged transcriptome of A. limnaeus to other teleosts. a Venn diagram showing shared numbers of the 20 most abundant transcripts in 3 species of fish with 1–2 cell stage Illumina-sequenced transcriptomes: A. limnaeus (this study), D. rerio [17] and H. hippoglossus [44]. b–d Gene ontology analysis comparing the top 100 transcripts in the 1–2 cell stage transcriptomes of A. limnaeus and D. rerio. Pie charts represent the quantities of GO categories in (b) D. rerio and c A. limnaeus for biological process classification (top) and protein class (bottom). d Go terms enriched in the A. limnaeus compared to D. rerio (P < 0.05) transcriptome. For more details see the text and Additional file 5
Small RNA transcriptome
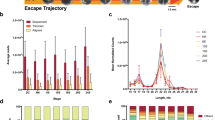
Details of the small RNA library sequencing and bioinformatics results can be found in Additional file 1. The A. limnaeus 1–2 cell stage sncRNA transcriptome is characterized by 3379 sncRNA transcripts expressed at a level of ≥2 normalized counts per million reads (Additional file 6). To better examine the diversity of sequence reads we grouped only identical sequences, not making any assumptions about mature sequence length, clustering or shared gene origin. At fertilization, embryos possess a wide diversity of sncRNA sequences. The majority of the sequence reads were short in length (<20 nt), while longer sequences (>25 nt) were less abundant (Fig. 5a). However, analysis of unique sncRNA sequence abundance as a function of sequence length revealed two dominating size classes; the highest abundance size class was 16 nt, while the second most abundant was 26 nt (Fig. 5b).

Maternally packaged sncRNA transcriptome of A. limnaeus. Frequency distribution of a normalized sequence reads and b unique sequences as a function of sequence length in the sncRNA libraries (n = 12). Each library is a different color. c There is a high diversity of sncRNA sequences with lengths between 15 and 23 nucleotides that are unknown (blue line) compared to those that could be annotated (red line) by sequence similarity to those cataloged in RNA databases
Between 57 and 73% of sncRNA sequence reads in each sample library could be annotated to known RNAs based on sequence alignment with miRBase Release 21 and Rfam version 12.1 [45, 46]. All sncRNAs were also annotated based on sequence alignment to a database of piwiRNA (piRNA) sequences in RNAdp v. 2.0 [47]. Annotations of miRNA sequences were similar in Rfam and miRBase, and the following summary of annotations is based on the Rfam results only. Of the 3379 unique sncRNA sequences, the majority had lengths below 20 nt and were not annotatable, while only 21% (722) annotated to known RNAs (Fig. 5c). The sncRNA sequences with the highest abundance annotated as antisense RNAs (55% of total reads, Fig. 6a) with the remainder including fragments of ribosomal RNAs (<1% of total reads) and small nucleolar RNAs (<1% of total reads; Fig. 6b, c). Surprisingly, miRNA annotations comprised <1% of the sncRNA transcriptome in 1–2 cell stage embryos (Fig. 6a–c). Of the 22 unique mature sequence variants that annotated as miRNAs, consensus sequences were generated for mir-181 and mir-10. High confidence precursor sequences, based on sequence and secondary structure modeling, were prepared for submission to miRBase for Alim-mir-10 and Alim-mir-181 (Fig. 6d).

Rfam database annotation of the maternally packaged sncRNA transcriptome of A. limnaeus. a The most abundant sncRNAs are 16–17 nucleotides in length and annotate as antisense RNAs, while the second most abundant group are sequences that are 26 nucleotides in length that annotate as ribosomal RNA (see panel c for a color key to annotation category). b The highest diversity of unique sncRNA sequences is in the 15 and 17 nucleotide length categories. Note the enrichment of miRNA sequences in the 20–22 nucleotide range as expected, even though miRNAs are not a dominant part of the sncRNA transcriptome. c Some annotation categories have distinct size ranges, while others span the entire range of sizes explored in this study. d Putative micro-RNA precursor structures and consensus mature sequences (highlighted in yellow) annotated as mir-181a (Alim-mir-181a1-3) and mir-10b (Alim-mir-10b1-4)
The 3379 unique sncRNA sequences mapped to approximately 33,000 locations in the genome, with 61% mapping to intergenic regions and 39% within exons (Additional file 6). The remaining alignments were positioned within introns (8%) or as a combination of categories (1%). Most sequences (2796) mapped to the genome in multiple locations (2–100). Only 583 mapped to exactly one position, with 51% of these in intergenic regions, 21% in introns, 27% in exons. Interestingly, 41% aligned in an antisense orientation, and 4 sequences mapped to the mitochondrial genome (3 of 4 to the ND3 gene).
The two most abundant sncRNA sequences had average counts per million reads of 168,769 and 79,972 and annotated as antisense RNAs in Rfam, while the third-most abundant (43,648 counts) annotated as rRNA based on genome alignments (Table 4, Additional file 7). The top twenty most abundantly expressed sncRNAs are dominated by sequences unknown to Rfam. However, they appear to be unique and non-repetitive sequences as determined by a low number of alignments to the genome (1–2 alignments each with perfect match of leftmost 15 bases). However, loosening the parameters just slightly increases the genomic alignments to over 100 locations (Additional file 8).
Differential expression of sncRNAs
Only four sncRNA sequences (≥2 normalized counts) were differentially expressed between embryos on the escape and diapause trajectories (Table 5). These four sequences do not match any known RNA sequences in Rfam or miRBase. The most differentially expressed gene sequence, ACAACGTGTGATACA, aligned once to the genome in an intronic region of a gene predicted to be zinc finger protein 646-like (LOC106517361; Additional file 8). The other differentially expressed sequences had two alignment positions in the genome (using strict parameters aligning the left-most 15 nt with 0 mismatches). Exploring the role of these sncRNAs is critical to understanding their possible involvement in post-transcriptional modification of other components of the 1–2 cell stage transcriptome.
Discussion
This study demonstrates for the first time a profile of maternally packaged RNAs for A. limnaeus. Due to the critical nature of early developmental events across multiple timescales (individual life span to evolutionary time) it is highly likely that each of the transcripts identified in this study plays an essential role in supporting early development and diapause in this species. This work serves as a foundation of information from which to build working hypotheses concerning the maternal control of the unique developmental attributes of annual killifishes.
General patterns in the poly-A RNA transcriptome
Perhaps not surprisingly, the protein-coding transcriptome of 1–2 cell stage embryos from A. limnaeus shares many similarities with fertilized eggs from other teleosts when analyzed on a presence–absence basis [17, 43, 44, 48, 49] and GO analysis indicates similar function between the transcriptomes of A. limnaeus and D. rerio. Early vertebrate development is thought to be highly constrained and conserved, and thus, it is reasonable to hypothesize that similar gene expression patterns would be required in all species. However, for the 3 species for which data are directly comparable (same developmental stage and sequencing methodology) each species appears to have a unique pattern of highly abundant transcripts with only 2 of the top 20 transcripts shared across all three species. In a direct comparison of A. limnaeus and D. rerio, each species expresses an order of magnitude more transcripts that are unique, compared to those that are shared. This species-specific expression in the maternally derived transcriptome likely reflects the different developmental strategies and environments of each species.
Stress tolerance
Early development, specifically the cleavage stages, tends to be sensitive to environmental stress. It is certainly the case in A. limnaeus that stress tolerance is lowest during the first 4 days of development [6, 50, 51]. It has been proposed that overexpression of stress tolerance genes may have a negative effect on development in some species. For example, heat shock that induces HSP expression in zebrafish embryos leads to a high proportion of abnormal embryos [52]. Also, overexpression of the molecular chaperone HSP 70 in Drosophila larvae increased thermal tolerance, but also caused a notable decrease in developmental rate and had other negative effects [53, 54]. In contrast, reduction of two small molecular weight chaperones, p26 and artemin, appears to slow development in diapause-bound embryos of Artemia [55, 56]. In other studies, inhibition of HSP 90 led to teratogenic effects due to an unmasking of cryptic variation in Drosophila and zebrafish [57–59]. Thus, buffering the effects of environmental stresses is likely an essential component of development, but may affect other processes such as developmental rate or phenotypic expression. The transcriptome of A. limnaeus has a number of characters that suggest an increased ability to mitigate the potentially negative impacts of environmental stress. A better understanding of these characters may shed light on how embryos can evolve to tolerate high levels of environmental stress while preserving essential processes required for normal development.
The small molecular chaperone, hsp 27 (hspb1), is one of the top 20 expressed transcripts (>3000 FPKM) in A. limnaeus and is represented at a substantially higher level than in zebrafish (2.1 FPKM; Harvey et al. [17]) and was not detected in Atlantic halibut [44]. In another report, in situ hybridization did not detect hspb1 in zebrafish embryos until the gastrula stage [60]. In a study of Fundulus heteroclitus, Tingaud-Sequiera et al. [42] identified the transcriptome of pooled embryonic stages (including the 2–4 cell stage) during exposure to air and addressed the expression of larger inducible heat-shock molecular chaperones such as 40-, 70- and 90-kDa classes, but made no mention of any small heat-shock proteins. In zebrafish, HSP 27 possesses the ability to prevent protein precipitation and is induced under heat stress in post-blastula stage embryos [61, 62]. Further, small hsps have been shown to play a critical role in the survival of heat stress in adult desert fishes [63, 64] and are highly differentially expressed in response to fluctuating daily temperatures in adult A. limnaeus [65]. Thus, the high abundance of hspb1 in the A. limnaeus 1–2 cell stage transcriptome appears to be unique and may contribute to their ability to survive stresses imposed by their harsh developmental environment.
Protection from reactive oxygen species is essential for normal cell function and is thought to be critical in mediating survival of oxygen deprivation [66, 67]. Embryos of A. limnaeus are the most anoxia-tolerant vertebrates [6, 36, 51] and can develop normally even under extreme hypoxia [68]. Thus, it may not be surprising that maternal provisioning of antioxidant systems would be elevated in this species. Some of the most highly expressed unique sequences in the A. limnaeus transcriptome are involved with glutathione metabolism, a key mechanism to deal with oxidative stress [69]. In addition, transcripts for the superoxide dismutase genes (sod1 and sod2) are packaged at a much higher level in embryos of A. limnaeus (863 and 46 FPKM) compared to D. rerio (87 and 19 FPKM). Maternal packaging of sod1 is thought to protect embryos from oxidative damage during development, when metabolic demands are high [70]. However, the low metabolic rate of early A. limnaeus embryos may suggest other reasons for maternal packaging of sod1 that are more consistent with survival of environmental stress, perhaps playing a role in their unique tolerance of oxygen deprivation.
All organisms possess cellular mechanisms that help maintain homeostasis in the face of environmental stress; many of these mechanisms are activated in the cellular stress response (CSR). A minimal stress proteome (MSP) was described by Kultz [71] that included 44 proteins and protein families that participate in the CSR. Analysis of the 1–2 cell stage transcriptomes from A. limnaeus and D. rerio suggests a similar number of genes represented in the MSP that are maternally packaged (Table 6). These transcripts represent 2% or less of the transcriptome and less than 1% of the most abundant 25% of transcripts. Based on this admittedly limited evaluation, neither species appears to be particularly enriched for potentially stress-responsive gene expression. However, the nature of the most abundant transcripts from this list is different in the two species. While mitochondrial genes critical for metabolism are highly abundant in both species, they dominate the most abundant transcripts in D. rerio, while in A. limnaeus the two most highly abundant MSP genes are a molecular chaperone and a thioredoxin (Additional file 9). These data suggest some potentially important functional differences in the stress-responsive transcripts that are maternally packaged in these two species that warrant future attention.
Slow developmental rate
A relatively slow developmental rate is a general character of all Atherinid fishes including the cyprinodonts [72]. Even within the Aplocheiloid fishes (annual killifish and their relatives), embryos of annual killifish tend to develop much more slowly compared to closely related non-annual relatives. Recently, rates of cleavage were found to be significantly slower in annual killifish compared to non-annual killifish in three lineages that are thought to have independently evolved the annual life history [73]. Thus, when compared to rapidly developing species such as zebrafish, the transcriptome of 1–2 cell stage embryos of A. limnaeus may provide clues to the molecular underpinnings of slow development in Atherinids and specifically for annual killifishes.
A simple comparison of the D. rerio and A. limnaeus transcriptomes suggests a higher abundance of cell-cycle-associated transcripts in zebrafish. For example, of the top 20 unique and highly abundant transcripts in zebrafish, there are 4 cyclins, 2 histones and a number of other transcripts that are expected to be high in proliferating cells. In contrast, these transcripts are not found among the top 20 unique and highly abundant transcripts in A. limnaeus. In addition, 9 of the 20 most differentially expressed transcripts between D. rerio and A. limnaeus are mitochondrially encoded and are statistically more abundant in D. rerio. While these transcripts may be maternally provisioned, it has also been demonstrated that active transcription of the mitochondrial genome occurs very early in development and well before activation of the nuclear genome [74]. Thus, the observed differences in mitochondrially derived transcripts could arise either through maternal provisioning, differences in the rates of mitochondrial transcription or perhaps both. While the abundance rank of these transcripts is high in both D. rerio and A. limnaeus, they are generally 2–4 times more abundant in the D. rerio transcriptome. This higher representation of mitochondrially encoded transcripts is consistent with a more active transcription of the mitochondrial genome in D. rerio, compared to A. limnaeus, and this may perhaps lead to higher rates of mitochondrial activity.
One remarkable difference between embryos of either species is in the profile of transcripts encoding for zinc finger proteins. A total of 462 zinc finger protein transcripts were present in A. limnaeus compared to 165 in D. rerio. Many of the genes in both datasets corresponded to counts less than 100 FPKM. However, the RNA-binding protein, zinc finger protein 36, C3H1 type-like 2 (LOC106511212 or Zfp36l2 in other species), was particularly high in A. limnaeus with a gene count of approximately 3500 FPKM, while in D. rerio, the paralogs of this gene (zfp36l1 and zfp36l2) have counts of 8 and 36 FPKM, respectively. The gene zfp36l2 is shared among all vertebrates and is known to promote mRNA decay through interactions with the 3’-untranslated region (UTR) [75]. This protein has been described as part of a common mechanism that can induce cellular quiescence through post-transcriptional regulation of mRNA stability. Recently, it has been shown that the mRNAs degraded through this mechanism code for proteins that promote cell-cycle progression into the S-phase [76]. The high level of expression for this particular transcript in A. limnaeus could be a mechanism for slowing progression through the cell cycle during development. It is also reasonable to hypothesize that this transcript could play a critical role in the regulation of developmental arrest associated with diapause in this species.
The homeobox transcription factor, nanog, is known to play a role in the maintenance of pluripotency in a number of animal models [77]. However, in the Medaka, nanog acts instead as a critical regulator of cell proliferation during early development that supports cell-cycle progression into the S-phase [77]. Transcripts for nanog are abundant (1011 FPKM) in fertilized embryos of zebrafish [17]. When the protein sequence for D. rerio nanog (AEZ64150.1) was compared to protein sequences of A. limnaeus (NCBI BLASTp) the best resulting match was LOC106530841, which annotated as homeobox protein DLX-1-like (36% identity, e value = 3e−31). Interestingly, expression of this transcript in A. limnaeus is much lower (317 FPKM) than that of nanog in zebrafish. The apparent altered role for nanog in fish development and the lower abundance in embryos of A. limnaeus point to another potential mechanism that could slow cell-cycle progression and potentially contribute to the ability of these embryos to arrest development in diapause.
Evolutionary biologists have been discussing the potential importance of developmental rate and conducting laboratory selection experiments to alter this rate for decades [78–82]. However, mechanistic studies that can explain differences in developmental rate are lacking with only a few studies focusing on heterozygosity in a variety of metabolic enzyme isoforms [83–85]. Thus, the differences pointed out here between A. limnaeus and D. rerio may be the first attempt to evaluate, on a global level, the potential cellular mechanisms for altering developmental rate. We do not have enough data at this point to infer causality, but this study certainly points to a number of likely candidates for future evaluation.
Regulation of developmental trajectory through alternative mRNA splice variants
There are 57 exons that are differentially expressed in the two alternative developmental trajectories in A. limnaeus. These exons are found in genes with a myriad of functions including DNA/RNA binding, protein degradation, intracellular signaling, post-transcriptional and post-translational modifications, the cellular stress response, cytoskeletal and transport properties, as well as cellular adhesion. Most of the exons (86%) are more abundant in diapause-destined embryos and absent or only rarely expressed in escape-bound embryos. It is important to note that the presence or absence of an exon changes not the gene dosage, but rather likely alters the regulation or activity of the gene product. Given the wide range of cellular pathways in which these gene products function, changes in their regulation could have profound effects on cellular and organismal physiology. GO analysis indicates an enrichment in genes with roles in a number of signaling pathways that are known to regulate rates of cell proliferation. It is especially interesting that the insulin/IGF pathway is enriched, given its known role in regulating diapause in C. elegans and arthropods [86–88]. This may indicate a role for IGF signaling in the regulation of diapause in A. limnaeus which would suggest a perhaps universal role for IGF signaling during animal diapause. It is also interesting that genes important for glycolysis are represented because diapausing embryos of A. limnaeus are known to be poised for anaerobic metabolism compared to escape embryos [89]. A role for alternative splicing in the regulation of fish physiology is supported by previous work on estrogen receptors in killifish exposed to estrogenic compounds [19].
While it is beyond the scope of this paper to explore every gene that is represented in this list, one of the mRNA splice variants highly packaged in diapause-bound embryos (over 60-fold higher than in escape-bound embryos) is found within the gene phkb (phosphorylase kinase, beta subunit). A recent study of this protein suggests it is a modulator of hsp 27 [90], one of the most abundant transcripts in the A. limnaeus 1–2 cell stage transcriptome. In the milkfish, expression of this gene was reduced following exposure to thermal and salinity stress [91]. In the Medaka, phkb is a key regulator of glycogen synthesis and contributes to calcium and insulin signaling pathways [92]. Given the noted importance of insulin signaling to the regulation of diapause across a variety of animal species, this transcript holds exceptional promise as a target for future functional studies and suggests a potential avenue for alteration of developmental trajectory by incubation temperature.
The discovery of these differentially packaged mRNA variants suggests that alternative splicing rather than differential gene expression may be critical for determining maternal influence on developmental trajectory in A. limnaeus. This is a novel result and suggests a subtle role for changes in gene regulation in the control of diapause in this species. Future studies will be required to test for the influence and function of these genes in response to temperature and other factors that are known to affect developmental trajectory.
The small non-coding RNA transcriptome
A high diversity of sncRNAs, such as demonstrated here, is common in early developmental stages of animals and is likely important for the proper regulation of gametogenesis and fertilization [93, 94]. To our knowledge this is the first report of the sncRNA transcriptome of a 1–2 cell stage fish embryo. The profiles of abundance, diversity and sequence length distribution presented here for A. limnaeus are similar to those reported for the zebrafish 256-cell stage embryo [41]. However, it is worthy of note that few mature miRNA sequences were identified here in A. limnaeus, while a great variety of miRNAs are present in zebrafish at the 256-cell stage and in later developmental stages of A. limnaeus (Romney and Podrabsky, unpublished observations). This may indicate that mature miRNAs are not a common or major component of the maternally packaged transcriptome in fish embryos.
In C. elegans, miRNAs are known to be key regulators of the developmental switch associated with entrance into the diapause-like dauer state [95]. Recent studies suggest diapause-specific miRNAs in flesh flies as well [96]. In addition, miRNAs are critical for reactivation of delayed implanting mouse embryos [97, 98]. The lack of miRNA diversity in 1–2 cell stage embryos of A. limnaeus, and the fact that no miRNA transcripts appear to be differentially expressed in association with either developmental trajectory suggest that maternal control over entrance into diapause is not likely mediated through any currently described miRNA. It is possible that some of the novel sequences discussed below may act as miRNAs, but more work is needed to explore this possibility.
The high diversity of sncRNAs identified in embryos of A. limnaeus falls into two basic categories: those having many sequence variants with low abundance and those with few sequence variants that are highly abundant. While a functional role for the low abundance sequences cannot be ruled out, it is also possible that these are the products of RNA degradation [99]. However, it is important to note that degradation is a perfectly acceptable means to generate biologically active molecules and thus these sequences should not be summarily dismissed. However, given the large number of sequences involved, we have chosen to focus on the highly abundant transcripts that have few variants. We propose a role for these RNAs similar to sncRNA such as endogenous small interfering RNAs (endo-siRNAs) or regulatory genes derived from longer RNAs such as tRNAs, rRNAs and small nucleolar RNAs (snoRNAs). Understanding the role of these potentially novel sncRNAs could elucidate gene regulatory pathways that support alternative developmental trajectories in this species.
The most abundant sncRNA sequences annotated as antisense RNAs, specifically the ST7 antisense RNA 1 conserved region 1 (RF02179). In humans, this long non-coding RNA is involved in the cellular response to DNA damage [100] and is described as a tumor suppressor gene [101, 102], but there is a paucity of information available. When aligned in an antisense orientation in the A. limnaeus genome these sequences map to RNA genes, zinc finger proteins, as well as a gene annotated as synaptophysin like (involved in synaptic vesicular transport). When mapped in a sense direction, the sequences align to regions in rRNA genes suggesting they could be generated from excision of internal spacer RNAs from rRNA transcripts. Small RNAs derived from rRNAs and tRNAs have recently been described as important regulators of cell differentiation [93, 99]. It is particularly interesting that these sncRNA sequences may target zinc finger proteins, which appear to be enriched in the transcriptome of A. limnaeus. Obviously, the complete picture of the significance of these sncRNAs cannot be determined by expression profile alone, and functional studies are needed to draw additional conclusions.
Only 4 sncRNAs were packaged into embryos at statistically different levels in embryos developing on the two developmental trajectories. All of these sequences are novel, and thus, we currently do not know enough about their biology to make valid predictions for their mode of action or potential targets. However, if we make the assumption they are acting as antisense RNAs to block translation or induce degradation of mRNA targets, a cohesive story emerges that is consistent with a role for these sncRNAs in the regulation of cell proliferation in escape embryos and the potential for chromatin remodeling control of gene expression in diapause-bound embryos. For instance, three of the potential target sequences for sncRNAs that are more abundant in the escape-bound embryos are mRNA transcripts that encode for proteins known to regulate cell-cycle progression. One sequence is antisense to an intron in a zinc finger protein 646-like sequence that contains a SFP1 domain. SFP1 in yeast is a known transcriptional repressor that regulates ribosomal protein expression and blocks the G2/M transition of the cell cycle [103, 104]. Another sequence could specifically target two A. limnaeus genes. One target, RNA-binding motif, single-stranded-interacting protein 3 (RBMS3), is known to bind the c-Myc promotor and reduce cell proliferation through alteration of β-catenin expression in at least two types of human cancer [105, 106]. Another target for this sncRNA could be the transcript for endosialin-1, also known as TEM-1 in humans. In humans and mice, high TEM-1 expression is associated with pericyte proliferation and angiogenesis and acts through platelet-derived growth factor receptor pathways that lead to activation of immediate early genes like c-fos [107, 108]. The function of endosialin-1 is currently not understood during early development and has not been characterized in A. limnaeus or any other fish. Reduction of this protein could reduce proliferation if it acts as it does in mammals. However, what we know about this protein is restricted to its role in pericyte cells of adult mammals, and it is possible that a context-dependent role for endosialin-1 during early development could be critical for other essential functions such as cell migration or differentiation. The important aspect of this protein is that it contains the functional domains necessary to interact with cell proliferation signaling pathways and thus has the potential for regulation of cell proliferation or perhaps migration. Another potential targeted mRNA transcript in escape-bound embryos codes for a neurexin-2-like protein. The transcript for this protein is maternally packaged in Xenopus and is highly expressed during early development in zebrafish, although its function is unknown [109, 110], and thus the role that this protein may play in determination of developmental trajectory in A. limnaeus remains unclear. The one sncRNA sequence that is upregulated in diapause-bound embryos targets an uncharacterized protein with SANT and reverse transcriptase domains. Proteins with SANT domains are known to be highly expressed in proliferating cells especially during early development and have a role in the regulation of chromatin organization through histone acetylation [111, 112]. Thus, reduction of this protein could have large-scale effects on gene expression. Unfortunately, we do not know when or where in the developing embryo that these sncRNAs may be active, and future studies to inhibit or modify their expression are needed to evaluate their potential role in altering developmental trajectory in A. limnaeus.
Conclusion
This study reports for the first time the maternally packaged transcriptome of an annual killifish and represents one of only a handful of similar studies on early vertebrate embryos. The data support a transcriptomic poise that could support the unique characters of development in A. limnaeus compared to other fishes. Surprisingly, the transcriptomes of A. limnaeus and D. rerio are more unique than similar, even at the 1–2 cell stage. Unlike the transcriptomes of other teleost species, A. limnaeus embryos contain an abundance of gene transcripts that support the slow developmental rates and stress tolerance specific to their life history. For the first time, splice variants of mRNAs are identified that are differentially packaged into oocytes in a vertebrate with alternative developmental trajectories. In addition, the sncRNAs that are differentially packaged suggest an important mechanism for post-transcriptional control that may contribute to the regulation of developmental trajectory, and support the unique embryonic characters in this lineage. Studies such as this are essential to broaden our understanding of the evolution of developmental and phenotypic plasticity.
Methods
Animals
Adult A. limnaeus were housed in the PSU aquatic vertebrate facility and cared for according to standard laboratory methods established for this species [113] that were approved by the PSU Institutional Animal Care and Use Committee (PSU Protocol #33). This laboratory strain of A. limnaeus was originally collected near the town of Quisiro in Venezuela [32] and has been cultured continuously in the laboratory since 1995. Embryos were collected twice a week from 42 pairs of fish during controlled spawning events. Clutches of embryos (20–200 embryos/clutch) were kept separate by spawning date and mating pair. Approximately 2 h after each spawning event (1–2 cell stage), all but 10–15 embryos from each clutch were flash-frozen in liquid nitrogen. The remaining embryos were used to determine the proportion of diapause and escape embryos produced by that spawning pair on that date. These embryos were transferred into embryo medium and maintained at 25 °C in darkness [113] for 17–21 days post-fertilization (dpf) when they were scored as escape- or diapause-bound embryos using previously established criteria [5]. Total RNA samples were prepared from pools of embryos (30–50 embryos per sample) representing 6 females producing either 100% diapausing or escape embryos (n = 6 pooled samples for each trajectory, see Additional file 1 for details).
RNA extraction
Total RNA was extracted from whole embryos using TRIzol reagent (Life Technologies; 50 µl/embryo) according to the manufacturer’s instructions for tissues containing polysaccharides. RNA concentration and purity were assessed by UV absorbance at 260 nm and calculating A260/A280 ratios (Tecan Infinite M200 Pro with NanoQuant plate, Switzerland). RNA integrity was assessed by agarose gel electrophoresis. RNA samples were maintained at −80 °C until use.
Poly-A RNA sequencing
cDNA libraries were prepared using the Illumina TruSeq RNA Sample Preparation Kit (v2, Illumina, San Diego, CA, USA) following the manufacturer’s instructions with 1 µg of total RNA as starting material. The purified cDNA libraries were quantified by qPCR and their quality confirmed on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) using a DNA 1000 chip. The libraries were sequenced (100 nt paired-end reads, 4 samples multiplexed per lane on the flow cell) on an Illumina HiSeq 2000 at the Oregon Health & Science University (OHSU).
sncRNA sequencing
sncRNA libraries were prepared using the Illumina TruSeq small RNA sample preparation kit following the manufacturer’s instructions with 1 µg of total RNA as starting material. The RNA samples used to prepare these libraries are the same samples used to prepare the poly-A RNA-sequencing libraries. Eleven cycles of PCR amplification were used for library production. Libraries were quantified by qPCR and quality checked on an Agilent Bioanalyzer 2100 using a DNA 1000 chip. The libraries were sequenced (100 cycles, paired-end, 12 samples multiplexed per flow cell lane) at OHSU using an Illumina HiSeq 2000.
Bioinformatics pipeline
A variety of software packages were used to process and analyze the transcriptomic data. CLC genomics workbench (version 6.5) for a Mac Pro (2 × 3.06 GHz 6-Core Intel Xeon and 64 GB RAM) was used for genomic mapping and generation of count data for the sncRNA libraries. The rest of the analyses were performed in a UNIX environment on the Portland State University computing cluster (Dell PowerEdge R730; 2× Intel(R) Xeon(R) CPU E5-2695 v3 with 28 cores @ 2.30 GHz and 256 GB RAM). Differential gene expression and exon usage analyses were performed using the R Bioconductor packages DESeq2 and DEXSeq [114, 115]. Gene ontologies were assigned using software tools on the PANTHER Web site [116].
Analysis of poly-A RNA sequence data
Sequence quality was initially assessed using FastQC, version 0.10.1, [117] to ensure high-quality data. Sequence reads were filtered on quality scores and trimmed for the presence of adapter sequences using Trimmomatic [118] with the settings “ILLUMINACLIP:2:30:7:1:true,” “SLIDINGWINDOW:5:15,” “LEADING:20,” “TRAILING:20” and “MINLEN: 25.” Quality reads were mapped to the A. limnaeus genome 1.0 using the very fast local preset in Bowtie2 [119]. Preserved paired reads after trimming were aligned in paired-end mode, and any orphaned mates after trimming were aligned in single-end mode. Reads that aligned to the A. limnaeus genome with 0 mismatches were used for expression analyses. Gene counts (union mode) were generated for all samples using the summarizeOverlaps function of the GenomicAlignments package from Bioconductor [120] and the NCBI A. limnaeus genome annotation Release 100 [33]. Count matrices were filtered for genes with 1 or more normalized counts summed across all replicates. Ontologies of overrepresented genes were determined using PANTHER software with Bonferroni-corrected P value <0.01 [116].
Gene abundance (FPKM) and differential expression analysis were performed using DESeq2 in the R Bioconductor package. Differential gene expression between diapause- and escape-destined embryos was determined on gene count data using the negative binomial distribution and estimations of mean–variance dependence [114]. Differential exon usage was tested using DEXSeq [115]. In both cases, differential expression was evaluated using a Benjamini–Hochberg multiple comparisons adjusted FDR of 10%.
Analysis of sncRNA sequence data
Sequence files were preliminarily examined using FastQC, version 0.10.1 [117], to explore sequencing quality. Small RNA reads were trimmed for quality and adapters with Trimmomatic using the settings “ILLUMINACLIP:2:30:7:1:true,” “SLIDINGWINDOW:5:15,” “LEADING:20,” “TRAILING:20” and “MINLEN: 15.” Trimmed reads that aligned to the A. limnaeus genome with 0 mismatches were retained and counted using the “annotate and merge” function in CLC workbench (version 6.5; CLCbio, Arhus, Denmark). Read counts were normalized across all libraries by DESeq2 to generate a catalog of small RNAs per treatment (average of 2 counts per million or greater) as well as differential expression based on a log2 fold change of 1 or greater using the Benjamini–Hochberg multiple comparisons adjusted FDR of 10%.
Sequences were annotated by alignment to small RNA databases allowing up to two mismatches to sequences for Danio rerio, Fugu rubripes, Tetraodon nigroviridis and Oryzias latipes using miRBase (v.21), Rfam (v. 12.1) and to all D. rerio piRNAs downloaded from Ensembl (Zebrafish V10). Small RNA sequences were mapped to the A. limnaeus genome using Bowtie and evaluated for proximity to annotated genes. For those sncRNA sequences annotated as miRNAs, consensus genomic sequences were determined by alignment to the A. limnaeus genome with perfect matches. Precursor stem-loop secondary structures were predicted from expanded regions of 80–100 nucleotides upstream in the 5′ direction using the Vienna package RNAfold prediction tool in Geneious software (R 8.1.6).
Comparative transcriptomics
Orthologous transcript pairs between A. limnaeus and D. rerio were identified by using NCBI BLASTn to align all RNA sequences of one species to the other species and vice versa. Transcript pairs were determined as reciprocal best hits (RBH) when the sequence alignment between two transcripts resulted in the best matching score for both comparisons. The resultant list was cross-referenced to the maternally packaged transcripts dataset of Harvey et al. for D. rerio [17], and thus gene names for both species are based on the zebrafish annotation for this analysis.
Abbreviations
- cDNA:
-
complementary DNA
- FPKM:
-
fragments per kilobase of transcript per million mapped reads
- GO:
-
gene ontology
- lncRNA:
-
long non-coding RNA
- miRNA:
-
micro-RNA
- mRNA:
-
messenger RNA
- PCR:
-
polymerase chain reaction
- piRNA:
-
piwiRNA
- qPCR:
-
quantitative polymerase chain reaction
- rRNA:
-
ribosomal RNA
- sncRNA:
-
small non-coding RNA
- snRNA:
-
small nuclear RNA
- snoRNAs:
-
small nucleolar RNA
- tRNA:
-
transfer RNA
References
Wourms JP. The developmental biology of annual fishes I. Stages in the normal development of Austrofundulus myersi Dahl. J Exp Zool. 1972;182:143–68.
Wourms JP. The developmental biology of annual fishes III. Pre-embryonic and embryonic diapause of variable duration in the eggs of annual fishes. J Exp Zool. 1972;182:389–414.
Wourms JP. The developmental biology of annual fish II. Naturally occurring dispersion and reaggregation of blastomeres during the development of annual fish eggs. J Exp Zool. 1972;182:169–200.
Wagner JT, Podrabsky JE. Gene expression patterns that support novel developmental stress buffering in embryos of the annual killifish Austrofundulus limnaeus. EvoDevo. 2015;6:2.
Podrabsky JE, Garrett IDF, Kohl ZF. Alternative developmental pathways associated with diapause regulated by temperature and maternal influences in embryos of the annual killifish Austrofundulus limnaeus. J Exp Biol. 2010;213:3280–8.
Podrabsky J, Riggs C, Wagner J. Tolerance of Environmental Stress. In: Berois N, García G, De Sá R, editors. Annual fishes: life history strategy, diversity, and evolution. Boca Raton: CRC Press; 2016. p. 159–84.
Baroux C, Autran D, Gillmor C, Grimanelli D, Grossniklaus U. The maternal to zygotic transition in animals and plants. Cold Spring Harb Symp Quant Biol. 2008;sqb-2008.
Tadros W, Lipshitz HD. The maternal-to-zygotic transition: a play in two acts. Development. 2009;136:3033–42.
Shen-Orr SS, Pilpel Y, Hunter CP. Composition and regulation of maternal and zygotic transcriptomes reflects species-specific reproductive mode. Genome Biol. 2010;11:1.
Bowerman B. Maternal control of pattern formation in early Caenorhabditis elegans embryos. Curr Top Dev Biol. 1998;39:73.
Pelegri F. Maternal factors in zebrafish development. Dev Dyn. 2003;228:535–54.
White JA, Heasman J. Maternal control of pattern formation in Xenopus laevis. J Exp Zool B. 2008;310:73–84.
Aanes H, Winata CL, Lin CH, Chen JP, Srinivasan KG, Lee SG, Lim AY, Hajan HS, Collas P, Bourque G. Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res. 2011;21:1328–38.
Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van Dongen S, Inoue K, Enright AJ, Schier AF. Zebrafish MiR-430 Promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312:75–9.
Kane DA, Kimmel CB. The zebrafish midblastula transition. Development. 1993;119:447–56.
Mishima Y, Giraldez AJ, Takeda Y, Fujiwara T, Sakamoto H, Schier AF, Inoue K. Differential regulation of germline mRNAs in soma and germ cells by zebrafish miR-430. Curr Biol. 2006;16:2135–42.
Harvey SA, Sealy I, Kettleborough R, Fenyes F, White R, Stemple D, Smith JC. Identification of the zebrafish maternal and paternal transcriptomes. Development. 2013;140:2703–10.
Paranjpe SS, Jacobi UG, van Heeringen SJ, Veenstra GJ. A genome-wide survey of maternal and embryonic transcripts during Xenopus tropicalis development. BMC Genom. 2013;14:762.
Cotter KA, Nacci D, Champlin D, Yeo AT, Gilmore TD, Callard GV. Adaptive significance of ERα splice variants in killifish (Fundulus heteroclitus) resident in an estrogenic environment. Endocrinology. 2016;157:2294–308.
Guilgur LG, Prudêncio P, Sobral D, Liszekova D, Rosa A, Martinho RG. Requirement for highly efficient pre-mRNA splicing during Drosophila early embryonic development. Elife. 2014;3:e02181.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Hannon GJ. RNA interference. Nature. 2002;418:244–51.
Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–4.
Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RHA. MicroRNA expression in zebrafish embryonic development. Science. 2005;309:310–1.
Zhao Y, Srivastava D. A developmental view of microRNA function. Trends Biochem Sci. 2007;32:189–97.
Barrey E, Saint-Auret G, Bonnamy B, Damas D, Boyer O, Gidrol X. Pre-microRNA and mature microRNA in human mitochondria. PLoS ONE. 2011;6:e20220.
Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15:423–37.
Murphy WJ, Collier GE. A molecular phylogeny for Aplocheiloid fishes (Atherinomorpha, Cyprinodontiformes): the role of vicariance and the origins of annualism. Mol Biol Evol. 1997;14:790–9.
Taphorn DC, Thomerson JE. A revision of the South American cyprinodont fishes of the genera Rachovia and Austrofundulus, with the description of a new genus. Acta Biol Venez. 1978;9:377–452.
Weitzman SH, Wourms JP. South American cyprinodont fishes allied to Cynolebias with the description of a new species of Austrofundulus from Venezuela. Copeia. 1967;1967:89–100.
Hrbek T, Taphorn DC, Thomerson JE. Molecular phylogeny of Austrofundulus Myers (Cyprinodontiformes: Rivulidae), with revision of the genus and the description of four new species. Zootaxa. 2005;825:1–39.
Podrabsky JE, Hrbek T, Hand SC. Physical and chemical characteristics of ephemeral pond habitats in the Maracaibo basin and Llanos region of Venezuela. Hydrobiologia. 1998;362:67–78.
Wagner J, Warren W, Minx P, Podrabsky J. Austrofundulus limnaeus 1.0 draft genome assembly with annotation. National Center for Biotechnology Information. 2015. http://www.ncbi.nlm.nih.gov/genome/?term=txid52670[orgn].
Podrabsky JE, Tingaud-Sequeira A, Cerdà J. Metabolic dormancy and responses to environmental desiccation in fish embryos. In: Lubzens E, Cerda J, Clark M, editors. Dormancy and resistance in harsh environments. Berlin: Springer; 2010. p. 203–26.
Furness AI, Lee K, Reznick DN. Adaptation in a variable environment: phenotypic plasticity and bet-hedging during egg diapause and hatching in an annual killifish. Evolution. 2015;69:1461–75.
Podrabsky JE, Riggs CL, Duerr JM. Anoxia tolerance during vertebrate development—insights from studies on the annual killifish Austrofundulus limnaeus. In: Padilla P (ed) Anoxia. Rijeka: InTech; 2012. p. 3–24.
Podrabsky J, Romney A, Culpepper K. Alternative developmental pathways. In: Berois N, García G, De Sá R, editors. Annual fishes. Life history strategy, diversity, and evolution. Boca Raton: CRC Press; 2016. p. 63–73.
Carroll SB, Grenier JK, Weatherbee SD. From DNA to diversity: molecular genetics and the evolution of animal design. New York: Wiley; 2013.
Gould SJ. Ontogeny and phylogeny–revisited and reunited. BioEssays. 1992;14:275–9.
Irie N, Kuratani S. Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nat Commun. 2011;2:248.
Wei C, Salichos L, Wittgrove CM, Rokas A, Patton JG. Transcriptome-wide analysis of small RNA expression in early zebrafish development. RNA. 2012;18:915–29.
Tingaud-Sequeira A, Lozano J-J, Zapater C, Otero D, Kube M, Reinhardt R, Cerdà J. A rapid transcriptome response is associated with desiccation resistance in aerially-exposed killifish embryos. PLoS ONE. 2013;8:e64410.
Lanes CF, Bizuayehu TT, de Oliveira Fernandes JM, Kiron V, Babiak I. Transcriptome of Atlantic cod (Gadus morhua L.) early embryos from farmed and wild broodstocks. Mar Biotechnol (NY). 2013;15:677–94.
Bai J, Solberg C, Fernandes JM, Johnston IA. Profiling of maternal and developmental-stage specific mRNA transcripts in Atlantic halibut Hippoglossus hippoglossus. Gene. 2007;386:202–10.
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–4.
Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, Eddy SR, Floden EW, Gardner PP, Jones TA, Tate J, Finn RD. RFAM 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43:D130–37.
Pang KC, Stephen S, Engström PG, Tajul-Arifin K, Chen W, Wahlestedt C, Lenhard B, Hayashizaki Y, Mattick JS. RNAdb—a comprehensive mammalian noncoding RNA database. Nucleic Acids Res. 2005;33:D125–30.
Vesterlund L, Jiao H, Unneberg P, Hovatta O, Kere J. The zebrafish transcriptome during early development. BMC Dev Biol. 2011;11:1.
Pauli A, Valen E, Lin MF, Garber M, Vastenhouw NL, Levin JZ, Fan L, Sandelin A, Rinn JL, Regev A. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012;22:577–91.
Machado BE, Podrabsky JE. Salinity tolerance in diapausing embryos of the annual killifish Austrofundulus limnaeus is supported by exceptionally low water and ion permeability. J Comp Physiol B. 2007;177:809–20.
Podrabsky JE, Lopez JP, Fan TWM, Higashi R, Somero GN. Extreme anoxia tolerance in embryos of the annual killifish Austrofundulus limnaeus: insights from a metabolomics analysis. J Exp Biol. 2007;210:2253–66.
Krone PH, Lele Z, Sass JB. Heat shock genes and the heat shock response in zebrafish embryos. Biochem Cell Biol. 1997;75:487–97.
Krebs RA, Feder ME. Hsp70 and larval thermotolerance in Drosophila melanogaster: how much is enough and when is more too much? J Insect Physiol. 1998;44:1091–101.
Feder ME, Cartaño NV, Milos L, Krebs RA, Lindquist SL. Effect of engineering Hsp70 copy number on Hsp70 expression and tolerance of ecologically relevant heat shock in larvae and pupae of Drosophila melanogaster. J Exp Biol. 1996;199:1837–44.
King AM, MacRae TH. The small heat shock protein p26 aids development of encysting Artemia embryos, prevents spontaneous diapause termination and protects against stress. PLoS ONE. 2012;7:e43723.
King AM, Toxopeus J, MacRae TH. Artemin, a diapause-specific chaperone, contributes to the stress tolerance of Artemia franciscana cysts and influences their release from females. J Exp Biol. 2014;217:1719–24.
Rutherford S, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–42.
Queitsch C, Sangster TA, Lindquist S. Hsp90 as a capacitor of phenotypic variation. Nature. 2002;417:618–24.
Yeyati PL, Bancewicz RM, Maule J, van Heyningen V. Hsp90 selectively modulates phenotype in vertebrate development. PLoS Genet. 2007;3:0431–47.
Mao L, Shelden E. Developmentally regulated gene expression of the small heat shock protein Hsp27 in zebrafish embryos. Gene Expr Patterns. 2006;6:127–33.
Rupik W, Jasik K, Bembenek J, Widłak W. The expression patterns of heat shock genes and proteins and their role during vertebrate’s development. Comp Biochem Physiol A. 2011;159:349–66.
Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–6.
Hightower LE, Norris CE, Di Iorio PJ, Fielding E. Heat shock responses of closely related species of tropical and desert fish. Am Zool. 1999;39:877–88.
Norris CE, Brown MA, Hickey E, Weber LA, Hightower LE. Low-molecular-weight heat shock proteins in a desert fish (Poeciliopsis lucida): Homologs of human Hsp27 and Xenopus Hsp30. Mol Biol Evol. 1997;14:1050–61.
Podrabsky JE, Somero GN. Changes in gene expression associated with acclimation to constant temperatures and fluctuating daily temperatures in an annual killifish Austrofundulus limnaeus. J Exp Biol. 2003;207:2237–54.
Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol. 2003;284:H549–58.
Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998;273:18092–8.
Anderson SN, Podrabsky JE. The effects of hypoxia and temperature on metabolic aspects of embryonic development in the annual killifish Austrofundulus limnaeus. J Comp Physiol B. 2014;184:355–70.
Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 2003;66:1499–503.
Lin C-T, Tseng W-C, Hsiao N-W, Chang H-H, Ken C-F. Characterization, molecular modelling and developmental expression of zebrafish manganese superoxide dismutase. Fish Shellfish Immunol. 2009;27:318–24.
Kultz D. Molecular and evolutionary basis of the cellular stress response. Annu Rev Physiol. 2004;67:225–57.
Parenti LR. A phylogenetic and biogeographic analysis of cyprinodontiform fishes (teleostei, Atherinomorpha). Bull Am Mus Nat Hist. 1981;168:335–557.
Dolfi L, Ripa R, Cellerino A. Transition to annual life history coincides with reduction in cell cycle speed during early cleavage in three independent clades of annual killifish. EvoDevo. 2014;5:32.
Heyn P, Kircher M, Dahl A, Kelso J, Tomancak P, Kalinka AT, Neugebauer KM. The earliest transcribed zygotic genes are short, newly evolved, and different across species. Cell Rep. 2014;6:285–92.
Zhang L, Prak L, Rayon-Estrada V, Thiru P, Flygare J, Lim B, Lodish HF. ZFP36L2 is required for self-renewal of early burst-forming unit erythroid progenitors. Nature. 2013;499:92–6.
Galloway A, Saveliev A, Łukasiak S, Hodson DJ, Bolland D, Balmanno K, Ahlfors H, Monzón-Casanova E, Mannurita SC, Bell LS. RNA-binding proteins ZFP36L1 and ZFP36L2 promote cell quiescence. Science. 2016;352:453–9.
Camp E, Sánchez-Sánchez AV, García-España A, DeSalle R, Odqvist L, Enrique O’Connor J, Mullor JL. Nanog regulates proliferation during early fish development. Stem Cells. 2009;27:2081–91.
Marien D. Selection for developmental rate in Drosophila pseudoobscura. Genetics. 1958;43:3.
Strathmann RR, Staver JM, Hoffman JR. Risk and the evolution of cell-cycle durations of embryos. Evolution. 2002;56:708–20.
Prasad N, Shakarad M, Gohil V, Sheeba V, Rajamani M, Joshi A. Evolution of reduced pre-adult viability and larval growth rate in laboratory populations of Drosophila melanogaster selected for shorter development time. Genet Res. 2000;76:249–59.
Chippindale AK, Alipaz JA, Chen H-W, Rose MR. Experimental evolution of accelerated development in Drosophila. 1. Developmental speed and larval survival. Evolution. 1997;51:1536–51.
Hubbs CL. The structural consequences of modifications of the developmental rate in fishes, considered in reference to certain problems of evolution. Am Nat. 1926;60:57–81.
DiMichele L, Westerman ME. Geographic variation in developmental rate between populations of the teleost Fundulus heteroclitus. Mar Biol. 1997;128:1–7.
DiMichele L, Powers DA. Allozyme variation, developmental rate, and differential mortality in the teleost Fundulus heteroclitus. Physiol Zool. 1991;64:1426–43.
Danzmann RG, Ferguson MM, Allendorf FW, Knudsen KL. Heterozygosity and developmental rate in a strain of rainbow trout (Salmo gairdneri). Evolution. 1986;40:86–93.
Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–6.
Sim C, Kang DS, Kim S, Bai X, Denlinger DL. Identification of FOXO targets that generate diverse features of the diapause phenotype in the mosquito Culex pipiens. Proc Natl Acad Sci USA. 2015;112:3811–6.
Sim C, Denlinger D. Insulin signaling and the regulation of insect diapause. Front Phsyiol. 2013;4:1–10.
Chennault T, Podrabsky JE. Aerobic and anaerobic capacities differ in embryos of the annual killifish Austrofundulus limnaeus that develop on alternate developmental trajectories. J Exp Zool A. 2010;313A:587–96.
Chebotareva NA, Makeeva VF, Bazhina SG, Eronina TB, Gusev NB, Kurganov BI. Interaction of Hsp27 with native phosphorylase kinase under crowding conditions. Macromol Biosci. 2010;10:783–9.
Hu Y-C, Kang C-K, Tang C-H, Lee T-H. Transcriptomic analysis of metabolic pathways in milkfish that respond to salinity and temperature changes. PLoS ONE. 2015;10:e0134959.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62.
García-López J, Alonso L, Cárdenas DB, Artaza-Alvarez H, de Dios Hourcade J, Martínez S, Brieño-Enríquez MA, del Mazo J. Diversity and functional convergence of small noncoding RNAs in male germ cell differentiation and fertilization. RNA. 2015;21:946–62.
Bourc’his D, Voinnet O. A small-RNA perspective on gametogenesis, fertilization, and early zygotic development. Science. 2010;330:617–22.
Rougvie AE, Moss EG. Developmental transitions in C. elegans larval stages. Curr Top Dev Biol. 2013;105:153–80.
Reynolds JA, Peyton JT, Denlinger DL. Changes in microRNA abundance may regulate diapause in the flesh fly, Sarcophaga bullata. Insect Biochem Mol Biol. 2017;84:1–14.
Cheong AW, Pang RT, Liu W-M, Kottawatta KSA, Lee K-F, Yeung WS. MicroRNA Let-7a and dicer are important in the activation and implantation of delayed implanting mouse embryos. Hum Reprod. 2014;29:750–62.
Liu W-M, Pang RT, Cheong AW, Ng EH, Lao K, Lee K-F, Yeung WS. Involvement of microRNA lethal-7a in the regulation of embryo implantation in mice. PLoS ONE. 2012;7:e37039.
Harding JL, Horswell S, Heliot C, Armisen J, Luscombe NM, Zimmerman LB, Miska EA, Hill CS. Small RNA profiling of Xenopus embryos reveals novel miRNAs and a new class of small RNAs derived from intronic transposable elements. Genome Res. 2014;24:96–106.
Liu Q, Sun S, Yu W, Jiang J, Zhuo F, Qiu G, Xu S, Jiang X. Altered expression of long non-coding RNAs during genotoxic stress-induced cell death in human glioma cells. J Neurooncol. 2015;122:283–92.
Vincent JB, Petek E, Thevarkunnel S, Kolozsvari D, Cheung J, Patel M, Scherer SW. The RAY1/ST7 tumor-suppressor locus on chromosome 7q31 represents a complex multi-transcript system. Genomics. 2002;80:283–94.
Haverty PM, Lin E, Tan J, Yu Y, Lam B, Lianoglou S, Neve RM, Martin S, Settleman J, Yauch RL. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature. 2016;533:333–7.
Xu Z, Norris D. The SFP1 gene product of Saccharomyces cerevisiae regulates G2/M transitions during the mitotic cell cycle and DNA-damage response. Genetics. 1998;150:1419–28.
Marion RM, Regev A, Segal E, Barash Y, Koller D, Friedman N, O’Shea EK. Sfp1 is a stress-and nutrient-sensitive regulator of ribosomal protein gene expression. Proc Natl Acad Sci USA. 2004;101:14315–22.
Chen J, Kwong DLW, Zhu C-L, Chen L-L, Dong S-S, Zhang L-Y, Tian J, Qi C-B, Cao T-T, Wong AMG. RBMS3 at 3p24 inhibits nasopharyngeal carcinoma development via inhibiting cell proliferation, angiogenesis, and inducing apoptosis. PLoS ONE. 2012;7:e44636.
Liang Y-N, Liu Y, Meng Q, Li X, Wang F, Yao G, Wang L, Fu S, Tong D. RBMS3 is a tumor suppressor gene that acts as a favorable prognostic marker in lung squamous cell carcinoma. Med Oncol. 2015;32:1–9.
Tomkowicz B, Rybinski K, Sebeck D, Sass P, Nicolaides NC, Grasso L, Zhou Y. Endosialin/TEM-1/CD248 regulates pericyte proliferation through PDGF receptor signaling. Cancer Biol Ther. 2010;9:908–15.
Maia M, DeVriese A, Janssens T, Moons M, Lories RJ, Tavernier J, Conway EM. CD248 facilitates tumor growth via its cytoplasmic domain. BMC Cancer. 2011;11:162.
Zeng Z, Sharpe CR, Simons JP, Górecki DC. The expression and alternative splicing of alpha-neurexins during Xenopus development. Int J Dev Biol. 2003;50:39–46.
Rissone A, Monopoli M, Beltrame M, Bussolino F, Cotelli F, Arese M. Comparative genome analysis of the neurexin gene family in Danio rerio: insights into their functions and evolution. Mol Biol Evol. 2007;24:236–52.
Mo X, Kowenz-Leutz E, Laumonnier Y, Xu H, Leutz A. Histone H3 tail positioning and acetylation by the c-Myb but not the v-Myb DNA-binding SANT domain. Genes Dev. 2005;19:2447–57.
Boyer LA, Langer MR, Crowley KA, Tan S, Denu JM, Peterson CL. Essential role for the SANT domain in the functioning of multiple chromatin remodeling enzymes. Mol Cell. 2002;10:935–42.
Podrabsky JE. Husbandry of the annual killifish Austrofundulus limnaeus with special emphasis on the collection and rearing of embryos. Environ Biol Fish. 1999;54:421–31.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–17.
Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016;44:D336–42.
Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010.
Bolger AA, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics. 2014;30:2114–20.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80.
Authors’ contributions
ALR designed and performed the experiment and analyzed the data. JEP aided in the design and analysis of the experiment. ALR and JEP both contributed to the writing of the manuscript. Both authors read and approved the final manuscript.
Acknowledgements
The authors thank Will Patterson for his help in supporting the bioinformatics analyses.
Competing interests
The authors declare that they have no competing interests.
Data availability
The original datasets are publically available via NCBI’s Sequence Read Archive (SRA): https://www.ncbi.nlm.nih.gov/bioproject/PRJNA272154. The accession identity of each NCBI biosample used in this study can be found in Additional file 1.
Ethics approval and consent to participate
Adult A. limnaeus were housed according to standard laboratory methods established for this species [113] under approval of the PSU IACUC Protocol #33.
Funding
This work was supported by National Science Foundation Grant IOS 1354549 to JEP.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
13227_2017_69_MOESM1_ESM.xlsx
Additional file 1. Details of RNA samples and the resulting cDNA sequencing libraries prepared from diapause- and escape-bound embryos.
13227_2017_69_MOESM5_ESM.xlsx
Additional file 5. GO analysis of top 100 most abundant transcripts in the 1–2 cell stage transcriptomes of A. limnaeus and D. rerio.
13227_2017_69_MOESM6_ESM.xlsx
Additional file 6. Alignment information of sncRNAs for A. limnaeus, regarding the nearest gene and its position within the gene or genomic region.
13227_2017_69_MOESM7_ESM.xlsx
Additional file 7. Most abundant sncRNAs in A. limnaeus 1–2 cell stage transcriptome and the annotations of their genomic alignments.
13227_2017_69_MOESM8_ESM.xlsx
Additional file 8. Summary of genome alignments for most abundantly expressed sncRNA in the A. limnaeus 1–2 cell stage transcriptome.
13227_2017_69_MOESM9_ESM.xlsx
Additional file 9. Number of genes designated in the minimal stress proteome identified in the A. limnaeus 1–2 cell stage transcriptome.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Romney, A.L., Podrabsky, J.E. Transcriptomic analysis of maternally provisioned cues for phenotypic plasticity in the annual killifish, Austrofundulus limnaeus . EvoDevo 8, 6 (2017). https://doi.org/10.1186/s13227-017-0069-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13227-017-0069-7