
Abstract
Background
Apolipoprotein E epsilon 4 (APOE-ε4) carrier status is an established risk factor for Alzheimer’s disease (AD) dementia. It has also been linked with sleep disturbance in healthy older adults and increased insomnia risk. This association may be driven by the effect of APOE-ε4 on AD pathological change, itself associated with sleep abnormalities. To assess this relationship, we have evaluated post-mortem neuropathological findings in patients with and without cognitive impairment and AD pathology, who had extensive clinical assessment within 12 months of death.
Methods
This retrospective cohort study used UK Brain Banks Network data. Eligible subjects were aged over 50, with pre-mortem neuropsychiatry inventory scores of sleep disturbance (NPI-K), neurocognitive testing and functional cognitive status assessment (Clinical Dementia Rating scale). Neuropathological data included Thal phase, Braak stage and CERAD scores (measures of Aβ plaque distribution, tangle distribution and neuritic plaque density, respectively) combined to form the National Institute on Aging Alzheimer’s Association (NIA-AA) ABC score reflecting AD neuropathology. Participants with other significant intracerebral pathology or pathological features of non-AD dementia were excluded.
Multivariate linear regression was performed with NPIK Global Score (NPIK frequency score multiplied by severity score) as the dependent variable and APOE-ε4 heterozygosity or homozygosity as independent variables. Covariates included age, gender, APOE-ε2 status and ABC NPI measures reflecting depression and anxiety. Further models stratified by ABC score and functional cognitive status were also produced.
Results
Seven hundred twenty-eight records were identified. Two hundred two participants were included in the final analysis: mean (SD) age 84.0 (9.2) and MMSE 14.0 (11.8). Mean sleep disturbance scores were highest in ε4 homozygosity (n=11), 4.55 (5.4); intermediate in ε4 heterozygosity (n=95), 2.03 (4.0); and lowest in non-ε4 carriers (n=96), 1.36 (3.3). Within the full sample, controlling for pathological status, age, gender, depression, anxiety and CDR-SOB status, APOE-ε4 homozygosity was associated with sleep disturbance (β 2.53, p=0.034). APOE-ε4 heterozygosity was similarly associated in individuals without dementia (β 1.21, p=0.048).
Conclusion
These findings lend weight to the hypothesis that APOE-ε4 affects sleep by mechanisms independent of AD pathological change. Evaluation of those mechanisms would enhance understanding of sleep disturbance pathways and potentially provide treatment targets.
Similar content being viewed by others


Background
Relative to the common ε3 allele of apolipoprotein E (APOE ε3), the ε4 allele is an established risk factor for the development of sporadic and late-onset familial Alzheimer’s disease (AD) [1,2,3]. Within predominantly Caucasian populations, increasing allele dose is positively associated with AD risk, with ε4 heterozygosity conferring an odds ratio (OR) of approximately 3 and ε4 homozygosity an OR of approximately 14 [4]. APOE ε4 also lowers the age of onset in a similarly allele-number-dependent manner, with one allele advancing onset by 2–5 years and two by 5–10 years [5] although such relationships appear weaker in African-American populations [6,7,8].
Multiple mechanisms have been proposed through which APOE ε4 may exert these effects [9]. Apolipoprotein E4 (ApoE4) affects amyloid beta (Aβ) metabolism, predisposing to its extracellular deposition as amyloid plaques [10,11,12] and to more severe cerebral amyloid angiopathy [13,14,15,16]. Whilst effects on Aβ are hypothesised to represent the dominant pathway, proteolytic ApoE4 cleavage resulting from stress or injury also predisposes to tau hyperphosphorylation and neurofibrillary tangle (NFT) formation [17]. ApoE4 has further been associated with disruption to glucose metabolism [18,19,20], blood-brain barrier integrity [21], cerebrovascular function [22], lipid transport [23], synaptic function [24] and inflammatory responses [25] as well as neuronal toxicity and α-synuclein/TDP-43 pathologies [26, 27].
Multiple studies have also linked APOE ε4 to sleep disturbance: specifically, objective sleep disturbance in healthy older adults [28], an increased risk of insomnia [29] and obstructive sleep apnoea/sleep-disordered breathing in both adults [30, 31] and children [32]. Improved sleep was reported to attenuate the negative effect of ε4 on incident AD [33]. Additionally, ε4 has been proposed as a mediator of the relationship between sleep and cognitive decline, both obstructive sleep apnoea (OSA) and APOE ε4 impairing cognitive performance [34,35,36]. However, whilst possession of ε2 reduces the odds of developing AD [37, 38], this allele has been linked with increased likelihood of OSA [39].
Sleep disturbance, whilst traditionally associated with established AD disease [40,41,42], is detectable prior to the emergence of symptoms [43,44,45] and plays a potentially causative role in AD pathogenesis [46,47,48]. Therefore, APOE ε4 could influence AD incidence and progression through the effects of this allele on sleep. Establishing this categorically is complicated by the influence of ApoE4 on the pathological hallmarks of AD themselves detectable decades prior to symptomatic presentation with cognitive impairment [49] and also associated with sleep disturbance. Hippocampal and Entorhinal Cortex deposition of NFTs found in early Braak stages [50] have been associated with an increased likelihood of sleep disturbance [51]. AD pathology within the suprachiasmatic nuclei and the ventrolateral preoptic area has also been implicated in sleep disturbance [52, 53] with hippocampal Aβ burden in otherwise healthy adults correlating significantly with impairments in non-random eye movement (NREM) slow wave activity generation and showed a trend towards deterioration in macro-architectural sleep parameters [54].
Here, we have tested the hypothesis that APOE ε4 allele count increases sleep disturbance in people with and without cognitive impairment, independently of its influence on the two major hallmark AD pathologies (Aβ plaques and tau neurofibrillary tangles). We have controlled for the extent of AD pathological change by the gold standard of post-mortem neuropathological assessment [55] according the 2012 National Institute on Aging-Alzheimer’s Association Guidelines [56] and have excluded individuals with other significant intracerebral pathology.
Methods
Participants
This retrospective cohort study used data obtained for participants in the Brains for Dementia Research (BDR) Programme (https://www.brainsfordementiaresearch.org.uk/) and held on the UK Brain Banks Network (UKBBN) database. The database holds demographic and neuropathological details of donated brains, processed and assessed according to detailed and comprehensive post-mortem protocols, as well as clinical assessments undertaken prior to post-mortem as part of the BDR project established in 2007. This links 5 brain banks across the UK (London, Oxford, Newcastle, Bristol and Manchester) with common protocols for consent, tissue handling and quality indicators. Volunteers were recruited via posters, radio adverts, presentations to groups/clubs and signposted via Alzheimer’s Research UK and Alzheimer’s Society charities. The population comprises many healthy participants with a positive family history of dementia and also participants with a diagnosis of dementia [57]. See Table 1 for inclusion and exclusion criteria.
A spectrum of histopathological findings is represented, ranging from healthy tissue to marked AD neuropathological changes, in individuals both with and without clinical AD dementia. Included participants had full APOE genotyping.
Outcome measure
Sleep disturbance was measured by component K of the neuropsychiatric inventory (NPI-K) [58]. These score responses provided by an informant, caregiver or study partner including increased latency, increased wake time after sleep onset, wandering, early morning wakening, excessive daytime sleep and sleep-wake cycle disturbance. A global score was obtained by multiplying frequency and severity domains (see Table 2).
Neuropathologic data
Each participant had undergone post-mortem analysis of CERAD neuritic plaque stage, Thal Aβ plaque stage and Braak NFT stage allowing for calculation of the National Institute on Aging-Alzheimer’s Association ABC Score [56]. The combination of A, B and C score determine the extent of AD neuropathological change, designated “Not”, “Low”, “Intermediate” and “High”.
Statistical analysis
For data cleaning and analysis, we used R Studio v3.6.3 statistical software. Raw scores were used throughout. Demographic and clinical variables were tested across groups for normality and compared made using Kruskal-Wallis and Pearson chi-squared tests. All tests of significance were two-tailed with α = 0.05.
Descriptive statistic was produced with unadjusted means of sleep disturbance by APOE ε4 status. These were calculated for the whole population before being stratified by NIAA-AA ABC score and CDR status. A fourth unadjusted comparison stratified the whole population into four phenotypically separate groups categorised by low CDR (0/0.5) or high CDR (1/2/3) and low ABC score (none/low) or high ABC score (intermediate/high). These groups are termed ‘Healthy’ (low CDR and low ABC), ‘Other Cognitive Impairment’ (high CDR and low ABC), ‘Alzheimer’s Disease’ (low CDR and high ABC) and ‘Alzheimer’s dementia’ (high CDR and high ABC).
The primary outcome measure was determined by multivariate linear regression with NPI-K Sleep Disturbance Global Score as the dependent variable. Crude and adjusted analyses were performed including dummy variables reflecting APOE ε4 allele copy number, with 0 as reference. Covariates were introduced to an adjusted model to control for APOE ε2 allele number, age, gender, CDR sum of boxes (CDR-SOB) and neuropsychiatry inventory measures of depression and anxiety. Dummy variables were created to reflect NIAA-AA ABC neuropathological stages. All regression models were checked for multicollinearity with variance inflation factors < 10. The study is powered at 80% to detect an effect size of f2 = 0.088 (n = 202, α = 0.05).
As a post hoc sensitivity analysis, these regressions were repeated in groups stratified by NIAA-AA ABC score and CDR status.
Results
Participant selection
Initial database search yielded n = 728 BDR cases, of which n = 202 fulfilled our criteria for analysis (See Fig. 1).

Participant flow diagram
Baseline demographics
Selected participants had a mean age of 84.0 years (SD = 9.2), 51.0% were male, mean CDR 1.8 (SD = 1.3) and mean Mini-Mental State Examination (MMSE) score 14.0 (SD = 11.8). Baseline demographics of the study population stratified by APOE-ε4 allele count (non-ε4 carriers n = 96, ε4 heterozygotes n = 95, ε4 homozygotes n = 11) are shown in Table 3. There were statistically significant differences in AD ABC stage, mean MMSE and mean CDR scores.
Sleep disturbance by APOE ε4 status
Crude sleep disturbance scores in the cohort stratified by ε4 are shown in Table 4. There were statistically significant increases in all neuropsychiatry inventory measures of sleep disturbance between those with 2 vs 0 alleles. Severity, frequency and caregiver distress domains were also significantly higher in those with 2 vs 1 allele. There were increased caregiver distress scores only in those with 1 allele compared with 0.
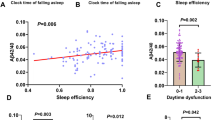
Positive trends between ε4 allele number and increasing mean NPIK sleep disturbance score were across the full-cohort irrespective of stratification by CDR status, ABC score of neuropathological change and clinical classification (Fig. 2).

Unadjusted NPIK sleep disturbance scores by APOE-E4 status. a Shows unadjusted global sleep disturbance scores and 95% confidence intervals by APOE ε4 status across the full population. Unadjusted global sleep disturbance scores are presented for the population stratified by NIAA-AA ABC Score (b) and CDR score (c). For unadjusted sleep scores by group status (healthy, other cognitive impairment, Alzheimer’s disease and Alzheimer’s dementia), see (d)
Primary analysis
Full multivariate linear regression revealed a statistically significant effect of APOE ε4 homozygosity on global scores of sleep disturbance (β 2.53, p=0.034) controlling for AD pathological status, ε2 carrier status, age, gender, depression, anxiety and CDR-SOB status. A positive trend was found for ε4 heterozygosity in both crude (β 0.67, p=0.221) and adjusted (β 0.41, p=0.471) analyses, although neither reached statistical significance (see Table 5).
Further, multivariate regression testing additional models were performed post hoc (Supplementary Material Table 1). Whilst the effect size estimates for APOE ε4 status within these models differed, overall trends and statistical significance remained unaltered. To further assess the independent effects of APOE ε4 status, further multivariate regressions were performed after stratification of the cohort by neuropathological change and CDR status (Tables 6 and 7). Positive trends between APOE ε4 status and sleep disturbance were seen in all stratified groups. Sleep disturbance was significantly associated with ε4 heterozygosity in the group without clinical dementia (CDR 0/0.5) (β 1.28, p=0.024) and with ε4 homozygosity in the relatively cognitively impaired group (CDR 1/2/3) (β 2.95, p=0.045).
Discussion
In this large, community-based cohort, APOE ε4 homozygosity was independently associated with sleep disturbance after controlling for the extent of AD neuropathological change, age, gender and affective symptoms, in individuals both with and without dementia. Homozygosity conferred a 2.53 (±1.18) mean point increase in NPI-K global sleep disturbance score. A non-significant trend towards an increased score was also noted with APOE ε4 heterozygosity, which conferred a 0.41-mean point increase in sleep disturbance score. Within the group without dementia (CDR 0/0.5), ε4 heterozygosity conferred a statistically significant increase in sleep disturbance score of 1.21 points.
Understanding of the relationship between ApoE status and sleep disturbance continues to evolve. Shortening of rapid-eye-movement (REM) sleep in individuals with MCI was significantly more apparent in carriers than non-carriers of the ε4 allele [59]. However, such differences extend beyond populations affected by cognitive impairment. Objective sleep disturbance in healthy adults as measured by polysomnography and actigraphy was found to be independently associated with the presence of the ε4 allele [28]. Furthermore, in a study that controlled for demographic variables, the ε4 allele was associated with insomnia in those both with and without psychiatric disorders [29]. Improved sleep attenuated the increased risk of AD development conferred by possession of ε4, in particular modifying its effect on neurofibrillary tangle formation [33]. Conversely, ε4 carriers with dementia were found to have slower rates of sleep disturbance progression than non-carriers [60] albeit in a small cohort. Our findings support previous findings indicating that people with one or more APOE ε4 alleles are likely to have more impaired sleep but add to these findings by controlling for the severity of Alzheimer’s disease pathology as determined neuropathologically. There are a range of potential explanations for these findings:
Apo E4-mediated AD pathological change
APOE ε4 is thought to influence AD pathology [9] through enhanced Aβ deposition [61], tau phosphorylation and neurotoxicity [62, 63], all of which may lead to sleep abnormalities [64]. For example, in participants with AD, APOE ε4 allele status influences CSF measures of tauopathy, itself associated with night-time behaviour disturbance [65]. We have found that ε4 influences sleep independently of Aβ and tau stage. Hence, as well as its impact on these hallmark features of AD, APOE ε4 may also influence AD risk through other neurotoxic pathways and/or loss of neuroprotective functions [9] that have the potential to influence sleep quality (Fig. 3).

Hypothetical mechanisms of APOE ε4-mediated sleep disturbance
Apo E4-mediated effects on melatonin
In a study of 85 patients with AD, APOE ε4 homozygosity compared with heterozygosity was associated with significantly reduced post-mortem CSF melatonin (32pg/ml ± 8 vs 71 ± 7, p=0.02) [66]. Reduced melatonin has been linked to sundown syndrome in dementia [67, 68], and replacement improves symptoms according to systematic review [69]. However, melatonin is up to five times higher in healthy adults compared with those with AD [66, 67], and it is plausible that APOE ε4 may be associated with reduced melatonin via secondary mechanisms linked to AD severity as opposed to direct effects.
Apo E4-mediated sleep disordered breathing
APOE ε4 was reported to be directly associated with OSA and symptomatic sleep disordered breathing [31], possession of this allele being associated with an approximate doubling of risk for apnoea-hypoxic index > 15 [OR 2.0 (1.2–3.5)] in adults and separately in children [32]. Two main mechanisms for a causal relationship were proposed. Firstly, an ε4-associated increase in respiratory/sleep centre tau or amyloid burden may drive centrally mediated sleep disordered breathing [31]. Alternatively, (or additionally) ApoE4 has a central role in lipid metabolism [70] mediating lipoprotein to cell surface receptor binding, increasing plasma low-density lipoprotein (LDL) levels and accelerating atherogenesis [71]. Centrally mediated sleep disordered breathing is recognised in a wide range of cerebral pathologies [72], including cerebrovascular pathology that might be exacerbated by possession of ε4. A further plausible mechanism, given that APOE ε4 predisposes to metabolic syndrome [73] and increased insulin resistance [74] would be through secondary increased obesity; however, ε4 carriers on average have a lower body mass index than do ε3 or ε2 carriers [75, 76]. A systematic review found no support for a causal association between APOE ε4 allele and OSA [OR 1.13 (0.86–1.47)] [77], but the authors commented that the studies were heterogeneous, may not have accommodated important gene-gene interactions and may have been underpowered.
Apo E4-mediated cerebral atrophy
Previous work linked possession of ε4 with accelerated age-related cortical thickness loss [78, 79]. This itself was associated with self-reported sleep disturbance in healthy community dwelling adults [80] and reduced objectively measured total sleep time, random eye movement, N2 and N3 stages of sleep in alcohol-use disorder [81].
Apo E4 effects on functional cerebral activity
Baseline activity within the Default Mode Network (DMN)—the distributed network of brain regions more active during rβ and characterised by high functional connectivity—is greater in APOE ε4 carriers than in non-carriers [82,83,84]. This overactivity was hypothesised to inhibit brain structures stimulating sleep initiation as described within the ‘failure to inhibit wakefulness’ hypothesis of sleep onset [85, 86]. Whilst the mechanism for this is uncertain, the findings extend to young adults aged 20–35, underlining a potential active role for ε4 outside of established AD pathology [83].
Study strengths and limitations
This study is subject to several limitations. Firstly, the sample, whilst deeply characterised, was limited by small numbers in the APOE-e4 homozygous group and relatively small numbers within each neuropathological group within the heterozygous group limiting power to detect full effects. Linked to this, p values of ‘statistically significant’ findings were close to 0.05. Reassuringly, however, the sleep disturbance signal was positively correlated with allele number. Participants in the BDR cohort are mostly from less socially and economically deprived parts of the UK [57] and are not therefore fully representative of the general population in the UK. Medication history was not comprehensive enough for inclusion in our analysis and may represent a significant confounder with prescribed sleep medications ameliorating or masking symptoms. Systemic illness that could have impacted on sleep disturbance may not have been detected post-mortem.
The use of the neuropsychiatry inventory sleep disturbance score as principal outcome measure is a further potential weakness. Whilst broad and encompassing a heterogeneous range of disorders, it relies on caregiver report and is therefore potentially subject to bias, e.g., false negative reports of subtle changes. However, it is well-validated, widely used and its reliance on a semi-objective caregiver as opposed to subjective personal reports also has advantages. Outcome scores for this study were obtained within 12 months of death. At the more distant end of this scale, pathological changes could have evolved between clinical data collection and autopsy; however, recall bias from retrospective data collection is eliminated.
Strengths include the categorisation of participants and quantification of AD changes based on the gold standard of post-mortem neuropathology with application of strict exclusion criteria, allowing for the effects of AD pathology to be largely determined in isolation. Data collected as part of the BDR project is standardised and collected as part of a detailed and comprehensive protocol. The population also reflected a range of AD pathology, with 38.2% of the study population recording ABC Scores of ‘Not’ or ‘Low’.
Conclusion
APOE ε4 homozygosity was associated with sleep disturbance, independent of AD pathological change and clinical functional status. Neuropathologically validated clinical studies often provide the first impetus in developing improved understanding of underlying mechanisms of neurological disease. There are a range of plausible mechanisms by which this effect of APOE ε4 may be exerted; further systematic testing of which would enhance understanding of sleep disturbance pathways and may subsequently provide treatment targets for this distressing symptom, also linked to AD progression.
Availability of data and materials
The dataset used and analysed in this current study are available from the first author on reasonable request.
Abbreviations
- AD:
-
Alzheimer’s disease
- APOE-ε4:
-
Apolipoprotein E epsilon 4
- BDR:
-
Brains for Dementia Research
- CDR:
-
Clinical Dementia Rating
- CDR-SOB:
-
Clinical Dementia Rating–sum of boxes
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DMN:
-
Default Mode Network
- LB:
-
Lewy body
- MMSE:
-
Mini-Mental State Examination
- NIA-AA:
-
National Institute on Aging and Alzheimer’s Association
- NPI-K:
-
Neuropsychiatry Inventory Measure of Sleep Disturbance
- NREM:
-
Non-random eye movement
- OR:
-
Odds ratio
- OSA:
-
Obstructive sleep apnoea
- SD:
-
Standard deviation
- TDP-43:
-
Transactive response DNA binding protein–43
- UK:
-
United Kingdom
References
Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43(8):1467–72.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–3.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349–56.
Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. 2011;16(9):903–7.
Sando SB, Melquist S, Cannon A, Hutton ML, Sletvold O, Saltvedt I, et al. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol. 2008;8:9.
Sahota A, Yang M, Gao S, Hui SL, Baiyewu O, Gureje O, et al. Apolipoprotein E-associated risk for Alzheimer’s disease in the African-American population is genotype dependent. Ann Neurol. 1997;42(4):659–61.
Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4, and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013;309(14):1483–92.
Tang MX, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, et al. The APOE-epsilon4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. JAMA. 1998;279(10):751–5.
Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–18.
Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(20):9649–53.
Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Verkkoniemi A, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333(19):1242–7.
Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol. 2009;65(6):650–7.
Rannikmäe K, Kalaria RN, Greenberg SM, Chui HC, Schmitt FA, Samarasekera N, et al. APOE associations with severe CAA-associated vasculopathic changes: collaborative meta-analysis. J Neurol Neurosurg Psychiatry. 2014;85(3):300–5.
Shinohara M, Murray ME, Frank RD, DeTure M, Yamazaki Y, Tachibana M, et al. Impact of sex and APOE4 on cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol. 2016;132(2):225–34.
Chalmers K, Wilcock GK, Love S. APOE epsilon 4 influences the pathological phenotype of Alzheimer’s disease by favouring cerebrovascular over parenchymal accumulation of A beta protein. Neuropathol Appl Neurobiol. 2003;29(3):231–8.
Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K, et al. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am J Neurodegener Dis. 2014;3(1):19–32.
Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q, et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24(10):2527–34.
Small GW, Mazziotta JC, Collins MT, Baxter LR, Phelps ME, Mandelkern MA, et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA. 1995;273(12):942–7.
Jagust WJ, Landau SM, AsDN I. Apolipoprotein E, not fibrillar β-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci. 2012;32(50):18227–33.
Zhao N, Liu CC, Van Ingelgom AJ, Martens YA, Linares C, Knight JA, et al. Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron. 2017;96(1):115–29.e5.
Halliday MR, Pomara N, Sagare AP, Mack WJ, Frangione B, Zlokovic BV. Relationship between cyclophilin a levels and matrix metalloproteinase 9 activity in cerebrospinal fluid of cognitively normal apolipoprotein e4 carriers and blood-brain barrier breakdown. JAMA Neurol. 2013;70(9):1198–200.
Schilling S, DeStefano AL, Sachdev PS, Choi SH, Mather KA, DeCarli CD, et al. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta-analysis. Neurology. 2013;81(3):292–300.
Hanson AJ, Banks WA, Hernandez Saucedo H, Craft S. Apolipoprotein E genotype and sex influence glucose tolerance in older adults: a cross-sectional study. Dement Geriatr Cogn Dis Extra. 2016;6(1):78–89.
Arendt T, Schindler C, Brückner MK, Eschrich K, Bigl V, Zedlick D, et al. Plastic neuronal remodeling is impaired in patients with Alzheimer’s disease carrying apolipoprotein epsilon 4 allele. J Neurosci. 1997;17(2):516–29.
Keene CD, Cudaback E, Li X, Montine KS, Montine TJ. Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr Opin Neurobiol. 2011;21(6):920–8.
Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014;127(6):811–24.
Wennberg AM, Tosakulwong N, Lesnick TG, Murray ME, Whitwell JL, Liesinger AM, et al. Association of apolipoprotein E ε4 with transactive response DNA-binding protein 43. JAMA Neurol. 2018;75(11):1347–54.
Drogos LL, Gill SJ, Tyndall AV, Raneri JK, Parboosingh JS, Naef A, et al. Evidence of association between sleep quality and APOE ε4 in healthy older adults: a pilot study. Neurology. 2016;87(17):1836–42.
Wang CC, Lung FW. The role of PGC-1 and Apoε4 in insomnia. Psychiatr Genet. 2012;22(2):82–7.
Gottlieb DJ, DeStefano AL, Foley DJ, Mignot E, Redline S, Givelber RJ, et al. APOE epsilon4 is associated with obstructive sleep apnea/hypopnea: the Sleep Heart Health Study. Neurology. 2004;63(4):664–8.
Kadotani H, Kadotani T, Young T, Peppard PE, Finn L, Colrain IM, et al. Association between apolipoprotein E epsilon4 and sleep-disordered breathing in adults. JAMA. 2001;285(22):2888–90.
Gozal D, Capdevila OS, Kheirandish-Gozal L, Crabtree VM. APOE epsilon 4 allele, cognitive dysfunction, and obstructive sleep apnea in children. Neurology. 2007;69(3):243–9.
Lim AS, Yu L, Kowgier M, Schneider JA, Buchman AS, Bennett DA. Modification of the relationship of the apolipoprotein E ε4 allele to the risk of Alzheimer disease and neurofibrillary tangle density by sleep. JAMA Neurol. 2013;70(12):1544–51.
Spira AP, Blackwell T, Stone KL, Redline S, Cauley JA, Ancoli-Israel S, et al. Sleep-disordered breathing and cognition in older women. J Am Geriatr Soc. 2008;56(1):45–50.
O'Hara R, Schröder CM, Kraemer HC, Kryla N, Cao C, Miller E, et al. Nocturnal sleep apnea/hypopnea is associated with lower memory performance in APOE epsilon4 carriers. Neurology. 2005;65(4):642–4.
Cosentino FI, Bosco P, Drago V, Prestianni G, Lanuzza B, Iero I, et al. The APOE epsilon4 allele increases the risk of impaired spatial working memory in obstructive sleep apnea. Sleep Med. 2008;9(8):831–9.
Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC Jr, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7(2):180–4.
Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39(1):17–23.
Uyrum E, Balbay O, Annakkaya AN, Gulec Balbay E, Silan F, Arbak P. The relationship between obstructive sleep apnea syndrome and apolipoprotein E genetic variants. Respiration. 2015;89(3):195–200.
Benca RM, Obermeyer WH, Thisted RA, Gillin JC. Sleep and psychiatric disorders. A meta-analysis. Arch Gen Psychiatry. 1992;49(8):651–68 discussion 69-70.
Weldemichael DA, Grossberg GT. Circadian rhythm disturbances in patients with Alzheimer’s disease: a review. Int. J Alzheimers Dis. 2010;2010:1.
Prinz PN, Peskind ER, Vitaliano PP, Raskind MA, Eisdorfer C, Zemcuznikov N, et al. Changes in the sleep and waking EEGs of nondemented and demented elderly subjects. J Am Geriatr Soc. 1982;30(2):86–93.
Ju YE, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70(5):587–93.
Lim AS, Kowgier M, Yu L, Buchman AS, Bennett DA. Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep. 2013;36(7):1027–32.
Sprecher KE, Koscik RL, Carlsson CM, Zetterberg H, Blennow K, Okonkwo OC, et al. Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology. 2017;89(5):445–53.
Mander BA, Winer JR, Jagust WJ, Walker MP. Sleep: a novel mechanistic pathway, biomarker, and treatment target in the pathology of Alzheimer’s disease? Trends Neurosci. 2016;39(8):552–66.
Fultz NE, Bonmassar G, Setsompop K, Stickgold RA, Rosen BR, Polimeni JR, et al. Coupled electrophysiological, hemodynamic, and cerebrospinal fluid oscillations in human sleep. Science. 2019;366(6465):628–31.
Ju YE, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology--a bidirectional relationship. Nat Rev Neurol. 2014;10(2):115–9.
Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119–28.
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389–404.
Ehrenberg AJ, Suemoto CK, França Resende EP, Petersen C, Leite REP, Rodriguez RD, et al. Neuropathologic correlates of psychiatric symptoms in Alzheimer’s disease. J Alzheimers Dis. 2018;66(1):115–26.
Wennberg AMV, Wu MN, Rosenberg PB, Spira AP. Sleep disturbance, cognitive decline, and dementia: a review. Semin Neurol. 2017;37(4):395–406.
Lu J, Greco MA, Shiromani P, Saper CB. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci. 2000;20(10):3830–42.
Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, et al. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. 2015;18(7):1051–7.
DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):32.
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1–13.
Francis PT, Costello H, Hayes GM. Brains for dementia research: evolution in a longitudinal brain donation cohort to maximize current and future value. J Alzheimers Dis. 2018;66(4):1635–44.
Cummings JL. The neuropsychiatric inventory: assessing psychopathology in dementia patients. Neurology. 1997;48(5 Suppl 6):S10–6.
Hita-Yanez E, Atienza M, Gil-Neciga E, Cantero JL. Disturbed sleep patterns in elders with mild cognitive impairment: the role of memory decline and ApoE epsilon4 genotype. Curr Alzheimer Res. 2012;9(3):290–7.
Yesavage JA, Friedman L, Kraemer H, Tinklenberg JR, Salehi A, Noda A, et al. Sleep/wake disruption in Alzheimer’s disease: APOE status and longitudinal course. J Geriatr Psychiatry Neurol. 2004;17(1):20–4.
Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology. 1996;46(6):1592–6.
Huang Y. Abeta-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2010;16(6):287–94.
Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103(15):5644–51.
Lloret M-A, Cervera-Ferri A, Nepomuceno M, Monllor P, Esteve D, Lloret A. Is Sleep Disruption a Cause or Consequence of Alzheimer's Disease? Reviewing Its Possible Role as a Biomarker. Int J Mol Sci. 2020;21(3):1168.
de Oliveira FF, Miraldo MC, de Castro-Neto EF, de Almeida SS, Matas SLA, Bertolucci PHF, et al. Associations of neuropsychiatric features with cerebrospinal fluid biomarkers of amyloidogenesis and neurodegeneration in dementia with Lewy bodies compared with Alzheimer’s disease and cognitively healthy people. J Alzheimers Dis. 2021;81(3):1295–309.
Liu RY, Zhou JN, van Heerikhuize J, Hofman MA, Swaab DF. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-epsilon4/4 genotype. J Clin Endocrinol Metab. 1999;84(1):323–7.
Mishima K, Tozawa T, Satoh K, Matsumoto Y, Hishikawa Y, Okawa M. Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep-waking. Biol Psychiatry. 1999;45(4):417–21.
Volicer L, Harper DG, Manning BC, Goldstein R, Satlin A. Sundowning and circadian rhythms in Alzheimer’s disease. Am J Psychiatry. 2001;158(5):704–11.
de Jonghe A, Korevaar JC, van Munster BC, de Rooij SE. Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. Are there implications for delirium? A systematic review. Int J Geriatr Psychiatry. 2010;25(12):1201–8.
Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis. 2014;72(Pt A):3–12.
Huang Y. Mechanisms linking apolipoprotein E isoforms with cardiovascular and neurological diseases. Curr Opin Lipidol. 2010;21(4):337–45.
Bassetti C, Aldrich MS. Sleep apnea in acute cerebrovascular diseases: final report on 128 patients. Sleep. 1999;22(2):217–23.
El-Lebedy D, Raslan HM, Mohammed AM. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc Diabetol. 2016;15:12.
Elosua R, Demissie S, Cupples LA, Meigs JB, Wilson PW, Schaefer EJ, et al. Obesity modulates the association among APOE genotype, insulin, and glucose in men. Obes Res. 2003;11(12):1502–8.
Tejedor MT, Garcia-Sobreviela MP, Ledesma M, Arbones-Mainar JM. The apolipoprotein E polymorphism rs7412 associates with body fatness independently of plasma lipids in middle aged men. PLoS One. 2014;9(9):e108605.
Volcik KA, Barkley RA, Hutchinson RG, Mosley TH, Heiss G, Sharrett AR, et al. Apolipoprotein E polymorphisms predict low density lipoprotein cholesterol levels and carotid artery wall thickness but not incident coronary heart disease in 12,491 ARIC study participants. Am J Epidemiol. 2006;164(4):342–8.
Thakre TP, Mamtani MR, Kulkarni H. Lack of association of the APOE epsilon 4 allele with the risk of obstructive sleep apnea: meta-analysis and meta-regression. Sleep. 2009;32(11):1507–11.
Espeseth T, Westlye LT, Fjell AM, Walhovd KB, Rootwelt H, Reinvang I. Accelerated age-related cortical thinning in healthy carriers of apolipoprotein E epsilon 4. Neurobiol Aging. 2008;29(3):329–40.
Fennema-Notestine C, Panizzon MS, Thompson WR, Chen CH, Eyler LT, Fischl B, et al. Presence of ApoE epsilon4 allele associated with thinner frontal cortex in middle age. J Alzheimers Dis. 2011;26(Suppl 3):49–60.
Sexton CE, Storsve AB, Walhovd KB, Johansen-Berg H, Fjell AM. Poor sleep quality is associated with increased cortical atrophy in community-dwelling adults. Neurology. 2014;83(11):967–73.
Zhang R, Tomasi D, Manza P, Shokri-Kojori E, Demiral SB, Feldman DE, et al. Sleep disturbances are associated with cortical and subcortical atrophy in alcohol use disorder. Transl Psychiatry. 2021;11(1):428.
Bondi MW, Houston WS, Eyler LT, Brown GG. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology. 2005;64(3):501–8.
Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, et al. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A. 2009;106(17):7209–14.
Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, et al. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343(7):450–6.
Espie CA, Broomfield NM, MacMahon KM, Macphee LM, Taylor LM. The attention-intention-effort pathway in the development of psychophysiologic insomnia: a theoretical review. Sleep Med Rev. 2006;10(4):215–45.
Marques DR, Gomes AA, Clemente V, Moutinho dos Santos J, Castelo-Branco M. Default-mode network activity and its role in comprehension and management of psychophysiological insomnia: a new perspective. New Ideas Psychol. 2015;36:30–7.
Acknowledgements
The Brains for Dementia Research (BDR) Programme (https://www.brainsfordementiaresearch.org.uk/) and UK Brain Banks Network (UKBBN) database for collecting and holding data used in this study.
Funding
JB receives funding from Alzheimer’s Research UK (supported by the Margaret Jost Fellowship and the Don Thoburn Memorial Scholarship), and the David Telling Charitable Trust and EC has received funding from BRACE and ARUK (Bristol & Bath Network).
Author information
Authors and Affiliations
Contributions
Project conception, data analysis and write-up: JB. Project oversight and review: SL, EC and LS. Data preparation and management: RC. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Brains for Dementia Research project has been approved by Newcastle & North Tyneside 1 Research Ethics Committee to function as a research tissue bank. Access to the participant data was provided by the UK Brain Banks Network data access Committee and the Brains for Dementia Research data access committee. The participants provided their written informed consent to participate in the Brains for Dementia Research programme.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Full Population Linear Regression – Additional Models.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Blackman, J., Love, S., Sinclair, L. et al. APOE ε4, Alzheimer’s disease neuropathology and sleep disturbance, in individuals with and without dementia. Alz Res Therapy 14, 47 (2022). https://doi.org/10.1186/s13195-022-00992-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-022-00992-y