
Abstract
Background
Monocyte Chemoattractant Protein-1 (MCP-1), a glial-derived chemokine, mediates neuroinflammation and may regulate memory outcomes among older adults. We aimed to explore the associations of plasma MCP-1 levels (alone and in combination with β-amyloid deposition—Aβ42/40) with overall and domain-specific cognitive evolution among older adults.
Methods
Secondary analyses including 1097 subjects (mean age = 75.3 years ± 4.4; 63.8% women) from the Multidomain Alzheimer Preventive Trial (MAPT). MCP-1 (higher is worse) and Aβ42/40 (lower is worse) were measured in plasma collected at year 1. MCP-1 in continuous and as a dichotomy (values in the highest quartile (MCP-1+)) were used, as well as a dichotomy of Aβ42/40. Outcomes were measured annually over 4 years and included the following: cognitive composite z-score (CCS), the Mini-Mental State Examination (MMSE), and Clinical Dementia Rating (CDR) sum of boxes (overall cognitive function); composite executive function z-score, composite attention z-score, Free and Cued Selective Reminding Test (FCSRT - memory).
Results
Plasma MCP-1 as a continuous variable was associated with the worsening of episodic memory over 4 years of follow-up, specifically in measures of free and cued delayed recall. MCP-1+ was associated with worse evolution in the CCS (4-year between-group difference: β = −0.14, 95%CI = −0.26, −0.02) and the CDR sum of boxes (2-year: β = 0.19, 95%CI = 0.06, 0.32). In domain-specific analyses, MCP-1+ was associated with declines in the FCSRT delayed recall sub-domains. In the presence of low Aβ42/40, MCP-1+ was not associated with greater declines in cognitive functions. The interaction with continuous biomarker values Aβ42/40× MCP-1 × time was significant in models with CDR sum of boxes and FCSRT DTR as dependent variables.
Conclusions
Baseline plasma MCP-1 levels were associated with longitudinal declines in overall cognitive and episodic memory performance in older adults over a 4-year follow-up. How plasma MCP-1 interacts with Aβ42/40 to determine cognitive decline at different stages of cognitive decline/dementia should be clarified by further research. The MCP-1 association on cognitive decline was strongest in those with amyloid plaques, as measured by blood plasma Aβ42/40.
Similar content being viewed by others

Introduction
Declines in cognitive function during aging is one of the most important public health challenges of the coming decades [1]. Early identification of older adults at risk of cognitive decline through the use of accessible and reliable biomarkers may inform timely intervention [2]. In this context, blood-born analytes have gained attention because of their feasibility, potential widespread use [2,3,4,5], and their association with cognitive outcomes and dementia onset in samples of older adults [6,7,8], suggesting that they may be predictors of cognitive function decline [9].
Immune dysregulation, characterized by chronic and exacerbated glial polarization, contributes to cognitive decline by promoting neurodegeneration (synaptic loss and neuronal death) [10]. In such dysregulated state, microglial cells release several inflammatory molecules [3] that contribute to microglial and astrocytic polarization, initiating a self-perpetuating cycle [11, 12]. Among them, the chemokine monocyte chemo-attractant protein-1—MCP-1, also known as C-C motif ligand 2 [13, 14], stands out given its tight relation to neuroimmune dysfunction [15]. This chemokine is an important regulator of monocyte/lymphocyte migration and infiltration into CNS through its interaction with CC-chemokine receptor 2 [16].
Although animal- [17, 18] and human-based [19,20,21,22,23,24] studies have linked increased CSF and plasma levels of MCP-1 with functional and brain structural changes associated with cognitive decline in older adults, most previous studies were cross-sectional and included either healthy subjects or people with a dementia diagnosis. No study investigated early stages of cognitive decline, such as people with spontaneous memory complaints and those with mild cognitive impairment [25].
Therefore, the main objective of the present study is to investigate the associations between plasma MCP-1 alone and overall and domain-specific cognitive evolution among community-dwelling older adults. Secondarily, we aimed to explore its interaction with plasma β-amyloid.
Methods
Study design and population
This observational longitudinal analysis uses data from the Multidomain Alzheimer Preventive Trial (MAPT, ClinicalTrials.gov [NCT00672685]), a randomized, multicenter, placebo-controlled trial conducted with community-dwelling older adults in France and Monaco. Participants were allocated into 4 groups, either receiving ω-3 polyunsaturated fatty acid (PUFA) supplementation, a multidomain intervention (based on cognitive training, nutritional counseling, and physical activity advice), both, or placebo. The intervention lasted for 3 years and was followed by an additional 2-year observational phase. Recruitment of participants started in May 2008 and ended in February 2011. Follow-up ended in April 2016.
Detailed description of the MAPT study can be found elsewhere [26, 27]. In summary, eligibility criteria comprised the following: age 70 years or older; not presenting major neurocognitive disorders, Mini-Mental State Examination [MMSE] score, ≥ 24; presenting at least 1 of the following: spontaneous memory concern, inability to perform 1 instrumental activity of daily living (ADL), or slow usual-pace walking speed (< 0.8 m/s). Participants were not included if they declared the use of ω-3 PUFA supplements during the 6 months before inclusion.
The population of the present study was composed of 1097 subjects with data on plasma MCP-1; among them, 429 individuals also had information on plasma β-amyloid. The present study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guideline [28].
MCP-1 and Aβ42/40 status
Plasma MCP-1 levels were measured at the MAPT 1-year visit using the fully automated immunoassay platform, ELLA (ProteinSimple/Bio-Techne, San Jose, CA, USA). MCP-1 levels were displayed as pg/mL. For analytical purposes, in the absence of cut-points to define plasma MCP-1 abnormal values, we used the quartiles in MCP-1 levels to define high values of the MCP-1 (MCP-1+; Q4 > 251 pg/mL) and MCP-1− (≤ Q4) groups.
We used the plasma Aβ42/40 ratio as a marker of β-amyloid deposition at 1-year visit [29]. Plasma samples were spiked with a known quantity of 15N-Aβ42 and 15N-Aβ40 for use as analytical internal standards. A full description of the immunoprecipitation methods applied has been previously described [30]. Briefly, Aβ42 and Aβ40 isoforms were simultaneously immunoprecipitated from 0.45 mL of plasma via a monoclonal anti-Aβ middomain antibody (HJ5.1, anti-Aβ13-28) conjugated to M-270 Epoxy Dynabeads (Invitrogen). Protein digestion into peptides was done using LysN endoprotease (Pierce). Liquid chromatography–mass spectrometry was performed as detailed elsewhere [30]. Plasma analyses were performed as targeted parallel reaction monitoring on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher) interfaced with an M-class nanoAcquity chromatography system (Waters). The precursor and product ion pairs used for analysis of Aβ isoforms were chosen as previously detailed [31, 32]. Derived integrated peak areas were analyzed using the Skyline software package [33]. Aβ42 and Aβ40 quantities (in pg/mL) were calculated by integrated peak area ratios to known concentrations of the internal standards. The plasma Aβ42/40 ratio was then calculated by dividing Aβ42 by Aβ40 and its normalized values were used to classify Aβ status (determined by Youden index as low (Aβ42/40+ ≤ 0.107) and normal (Aβ42/40− > 0.107), using β-amyloid load assessed by Positron Emission Tomography as the reference standard).
Specification of the assays used (limits of detection, coefficient of variation and linearity) are displayed in Additional file 1.
Outcome measures
Outcomes were assessed in the same visit in which MCP-1 and Aβ42/40 were measured (data collected before MCP-1 measurement were not used); outcomes were prospectively evaluated annually for 4 years. Overall cognitive performance was assessed using the following: a composite cognitive score (CCS) [27] based on four tests (the 10 orientation items of the MMSE, the Digit Symbol Substitution Test (DSST), free and total recall of the Free and Cued Selective Reminding Test (FCSRT), and the Category Naming Test), the MMSE score [34], and the Clinical Dementia Rating (CDR) sum of boxes [35].
Specific cognitive domains evaluated were as follows: (a) episodic memory (from the FCSRT) [36], (b) executive function (based on the composite of the z-scores of the Controlled Oral Word Association Test [37], the CNT [38], and the Trail Making Test-Part B [39]), and (c) attention (based on the composite of the z-scores of the DSST [40] and the Trail Making Test-Part A [39]). All these instruments were administered following standard procedures.
Potential confounders
Potential confounders consisted of age, sex, body mass index (BMI; kg/m2), MAPT group allocation, CDR status at baseline (CDR score 0; 0.5, or ≥ 1), 15-item Geriatric Depression Scale (GDS) [41], and apolipoprotein E (ApoE) ε4 genotype (carrier of at least one allele vs non-carrier). All confounders were measured at the 1-year MAPT visit.
Statistical analysis
Descriptive statistics (mean ± standard deviation or frequencies and percentages, as appropriate) were used for the characterization of the study population. Quantitative variables at baseline (the 1-year visit where plasma MCP-1 and Aβ were measured) were compared according to MCP-1 status by Student’s t tests, and categorical variables were compared using the χ2 tests.
Linear mixed-effects (LME) regression analyses (with random intercept and random slope for each participant) were performed to determine associations between baseline plasma MCP-1 (either continuous or dichotomous) and the annual rate of changes in the outcome measures. The LME models included the fixed effects of baseline plasma MCP-1, time, their interaction, and potential confounders (model 1: all the covariates described; model 2: all confounders except ApoE ε4 genotype) [42]. The rationale for removing ApoE ε4 genotype in a second model was the substantial number of subjects with missing information (110 out of 1097, 10% of the sample). Using similar adjusted models, we further investigated the joint associations of baseline MCP-1 (as a continuous variable) and Aβ42/40 status with the rate of change of cognitive outcomes by including a three-way interaction (Aβ42/40× MCP-1 × time) in the fixed effects, in addition to the main effects and the two-way interactions between these variables. For the categorical approach, we defined four groups: Aβ42/40−/MCP-1− (reference category), Aβ42/40−/MCP-1+, Aβ42/40+/MCP-1−, Aβ42/40+/MCP-1+). In the analyses of the CDR sum of boxes, CDR status at baseline was not included as a covariate.
In post-hoc analyses, we investigated wether MCP-1 associated with hippocampal volume changes (in cm3, assessed by magnetic resonance imaging—MRI) and β-amyloid deposition (cortical-to-cerebellar regional mean standardized uptake value ratio [SUVr] assessed by positron emission tomography—PET). A composite value computed as the mean of six predefined anatomically relevant cortical regions of interest (frontal, temporal, parietal, precuneus, anterior cingulate, and posterior cingulate [composite SUVr-cSUVr]) and hippocampus in isolation were used as outcome measures in age- and sex-adjusted models (using LME with random intercept at the center-nested participant’s level and random slope, and linear regression, respectively) given their putative associations with episodic memory [43]. Description of the MRI and PET methods can be found in and elsewhere and in Additional file 2 [26].
Results
Characterization of the sample
Table 1 shows the characteristics of the 1097 participants included in the present analyses (65.3% of the MAPT whole sample). Differences at baseline (1-year visit where plasma MCP-1 and Aβ were measured) between MAPT participants included in the present study and those not included are shown in Additional file 3. Mean MCP-1 was 244.96 ± 93.01 (Coefficient of variation: 2.18% ± 1.13%). In total, 274 subjects were classified as MCP-1+. These subjects were significantly older, presented higher BMI, worse scores in the CCS, the CDR sum of boxes, and FCSRT immediate free recall (IFR), compared to the MCP-1− group. Median (IQR) follow-up was 3.4 ± 1.0 years.
Evolution in cognitive outcomes according to MCP-1 plasma levels
As a continuous variable, baseline plasma MCP-1 levels were not prospectively associated to the rate of change of any of the overall cognitive performance outcomes. Regarding domain-specific cognitive functions, MCP-1 was associated with the worsening of episodic memory over 4 years of follow-up, specifically in the FCSRT: delayed free recall (DFR) (β = −0.003, 95%CI = −0.005, −0.001; p = 0.041) and delayed total recall (DTR) (β = −0.001, 95%CI = −0.003, −0.00005 p = 0.041). No further associations were found.
In the categorical approach, the MCP-1+ group showed a greater cognitive decline according to the CCS at the 4-year follow-up and the CDR sum of boxes score at the 2-year time-point, but this difference did not persist for 3-year and 4-year evolution (Table 2).
As displayed in Table 3, for domain-specific cognitive functions the MCP-1+ group showed a greater decline in the episodic memory domain (IFR, DFR, and DTR scores) compared to MCP-1− subjects. Whereas immediate total recall (ITR) declined significantly more in the MCP-1+ group over the first 2 years of follow-up (β = −0.68, 95%CI = −1.33, −0.02; p = 0.042), this association was not found onwards.
Removal of ApoE ε4 genotype status from the models slightly modified the results. CCS declines observed in both MCP-1 groups were no longer observed. Concerning memory outcomes in the FCSRT, associations remained significant, but the differences between groups for the IFR, DFR, and DTR scores were observed at 4 years of follow-up (Additional files 4 and 5).
Evolution in cognitive outcomes according to combined MCP-1 / Aβ42/40 plasma levels
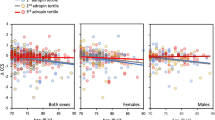
The Aβ42/40× MCP-1 × time interaction was significant in models with CDR sum of boxes and FCSRT DTR as dependent variables, indicating that greater levels in the MCP-1 might exacerbate the existing association between lower levels of Aβ42/40 and the worsening evolution in these two outcomes (CDR sum of boxes: β = −0.0005; 95% CI = −0.0009, −0.0002; p = 0.041; FCSRT DTR: β = 0.0005; 95% CI = 0.0002, 0.0009; p = 0.005). Figure 1 graphically displays the evolution in CDR sum of boxes and FCSRT DTR according to Aβ42/40 status at different levels of MCP-1.

Interactive longitudinal associations of Aβ42/40 and MCP-1 status on CDR Sum of boxes score
When combining Aβ42/40 and MCP-1 categories, both the Aβ42/40+/MCP-1− and the Aβ42/40+/MCP-1+ groups experienced significant worsening on the CCS score compared to the Aβ42/40−/MCP-1− group (between-group difference: β = −0.40, 95%CI = −0.59, −0.21; p < 0.001 and β = −0.41, 95%CI = −0.65, −0.17; p < 0.001, respectively), whereas only the Aβ42/40+/MCP-1− was associated with worsening on the MMSE score (between-group difference: β = −0.69, 95%CI = −1.23, −0.15; p = 0.012) (Table 4). Both the Aβ42/40+/MCP-1− and the Aβ42/40+/MCP-1+ groups showed significant progression in CDR sum of boxes score (β = 0.37, 95%CI = 0.06, 0.68; p = 0.019 and β = 0.54, 95%CI = 0.15, 0.93; p = 0.007 respectively) (Table 4).
Significant greater associations between Aβ42/40/MCP-1 status and executive function composite score were found only in the Aβ42/40+/MCP-1− group (β = −0.41, 95%CI = −0.62, −0.20; p < 0.001), whereas for the attention composite score, both Aβ42/40+/MCP-1− and Aβ42/40+/MCP-1+ groups showed a greater decline compared to the reference group (Table 4). With regard to the memory outcomes, the Aβ42/40+/MCP-1− group showed significant greater worsening in the performance across the four outcomes (between-group difference IFR: β = −4.22, 95%CI = −7.71, −2.17; p < 0.001, ITR: β = −3.14, 95%CI = −4.48, −1.81; p < 0.001, DFR: β = −1.90, 95%CI = −2.74, −1.06; p < 0.001 and DTR: β = −1.20, 95%CI = −1.69, −0.72; p < 0.001) compared to the Aβ42/40−/MCP-1− group over the 4 years of follow-up. Similarly, the Aβ42/40+/MCP-1+ experienced greater declines in the four FCSRT outcomes compared to the reference group (Table 5).
We did not find significant differences for any FCSRT outcome between Aβ42/40+/MCP-1− and Aβ42/40+/MCP-1+ (Table 5). Additional files 5 and 6 show the within-group evolution in the different outcomes. Sensitivity analyses removing ApoE ε4 genotype status from the models yielded similar results (Additional files 7 and 8).
Post hoc analyses
Based on the findings suggesting a domain-specific association between MCP-1 and episodic memory, we undertook exploratory analyses to test whether baseline plasma MCP-1 levels were associated with hippocampus volume changes and cSUVr and hippocampal β-amyloid load. MCP-1 was significantly associated with hippocampal atrophy (n = 299) when used as continuous (β = −0.0000005, 95%CI = −0.0000009, −0.00000007; p = 0.022), whereas in the categorical approach, the MCP-1+ group did not show a greater hippocampal atrophy over the follow-up (mean = 966.1 ± 111 days), compared to MCP-1− (between-group difference: β = −0.04, 95%CI = −0.10, 0.01; p = 0.107).
With regard to cross-sectional PET-scan β-amyloid load (n = 193), continuous plasma MCP-1 was neither cross-sectionally associated with hippocampal (β = −0.00008, 95%CI = −0.0002, 0.00008; p = 0.299) or cSUVr (β = −0.0001, 95%CI = −0.0004, 0.0001; p = 0.337), whereas in the categorical approach, MCP-1+ was associated with lower hippocampal SUVr (β = −0.04, 95%CI = −0.07, −0.002; p = 0.036), but not with cSUVr (β = −0.00002, 95%CI = −0.0001, 0.0001; p = 0.797).
Discussion
Our results showed that higher levels of MCP-1 were associated with greater overtime declines on both overall and domain-specific cognitive functions. Domain-specific analysis revealed that higher plasma MCP-1 levels were consistently associated with more pronounced decreases in the episodic memory performance; exploratory analysis found MCP-1 was notably associated with overtime hippocampal atrophy, suggesting MCP-1 association with cognition might be mediated by changes in the hippocampus structure. The association between MCP-1 and cognitive decline was strongest in those with amyloid plaques, as measured by blood plasma Aβ42/40.
Our findings linking higher plasma MCP-1 and cognitive decline are compatible with available research describing its pathophysiological involvement in neurodegeneration as well as with results of recent animal- and human-based studies addressing its association with cognitive decline [44]. It is possible that peripheral immunologic alterations mirror CNS neuroinflammatory processes [45, 46], what would allow to monitor cognitive decline-related pathophysiological changes occurring before overt clinical manifestations [47]. In fact, it has been proposed that peripheral-mediated processes might not only constitute a reflection of CNS inflammation, but also exacerbate CNS glial cell activation. This occurs in the presence of an age- and inflammation-related increase in blood-brain barrier permeability, allowing the leakage of circulating inflammatory factors and immune cells into CSF and brain parenchyma [48]. In this sense, MCP-1, given its potential role as a regulator of the migration and infiltration into the CNS, might have a prominent utility as a neuroinflammatory marker [16], even when measured in plasma.
Previous murine-based studies have suggested that aging is characterized by increased circulating MCP-1 and that elevated MCP-1 is associated with age-related decline in neurogenesis and subsequent worse performance in memory domains [17, 18]. Contrary to our findings, the first human study exploring longitudinal associations of MCP-1 with Alzheimer’s disease (AD) progression showed that CSF but not plasma MCP-1 was elevated in prodromal AD compared to non-prodromal AD individuals with mild-cognitive impairment (MCI), and that CSF MCP-1 was associated to greater cognitive decline in subjects developing AD [34]. Notwithstanding, Lee et al. showed increasing levels of plasma MCP-1 along the AD continuum (from healthy controls to severe AD dementia—CDR = 3) in an outpatient sample of older adults. They further showed that, among MCI and AD participants, MCP-1 levels were associated with 2-year declines in cognitive function evaluated through the MMSE [24]. In line with our findings, Bettcher et al. found associations of plasma MCP-1 levels with episodic memory cross-sectionally in cognitively impaired subjects [20] and longitudinally [23] in asymptomatic older adults; no associations were observed with other cognitive functions. Our findings, alongside Bettcher et al.’s results, suggest the MCP-1-cognitive function associations may be specific to episodic memory function in older adults. The results of our exploratory, post hoc neuroimaging analyses are compatible with previous research linking neuroinflammatory processes and hippocampal volumes in different samples of older adults [49]; since changes in hippocampal volume are linked to episodic memory performance [43], it is possible that the MCP-1-episodic memory associations might be mediated by hippocampal atrophy.
Surprisingly, the magnitude of the associations with cognitive decline was higher in the Aβ42/40+/MCP-1− compared to the Aβ42/40+/MCP-1+. Although statistically non-significant, this unexpected association deserves further investigations since it raises questions about the role of MCP-1 on cognitive function in the context of β-amyloid deposition, especially on episodic memory. To the best of our knowledge, the sole study investigating the combined interaction of MCP-1 and β-amyloid [19] showed that, in the presence of abnormal CSF tau protein and Aβ42/P-tau ratio, increased levels of CSF MCP-1 exacerbated cognitive decline among MCI older adults. Indeed, while we found a modest worsening of both the CDR sum of boxes score and a measure of episodic memory when MCP-1 and Aβ42/40 levels were used as continuous, our study failed in finding the added value of MCP-1 when combined with Aβ42/40 in the categorical approach. Recent research has revealed that, distinctly to in pre-clinical stages, in MCI and AD, microglial function is impaired [50, 51], exacerbating Aβ deposition [52, 53], tau pathology [54, 55], and synaptic/neuronal loss [53, 56].
In any case, the conflicting findings about the potential interactions between MCP-1 and β-amyloid deposition to determine cognitive declines ask for further research in this topic. Specifically, the exploration of the role of MCP-1 in determining cognitive declines at different stages of cognitive decline/dementia progression should be subject of further research [57]. Importantly, our study expands findings of available studies to a sample of older adults in early stages of cognitive decline for the first time.
Other mechanisms besides β-amyloid deposition are involved in neuroinflammation-mediated cognitive impairment. Indeed, mechanistic research has shown the potential of MCP-1 to promote tau-phosphorylation and neurofibrillary tangle formation [54, 58], a core feature of AD that has been lately shown to be to be more closely related to cognitive evolution than β-amyloid dyshomeostasis in the brain [59]. Potentially, tau-phosphorylation and the formation of intracellular neurofibrillary tangles might partially explain associations between MCP-1 and cognitive outcomes in older adults. Unfortunately, we did not have available data on tau-related markers. This topic needs further investigations.
Strengths and limitations
Our study presents strengths: its longitudinal nature with a relatively long follow-up and several time-points of data collection, which allow us to know the trajectories of the different cognitive outcome measures; its large sample size compared to previous investigations; and the assessment of both overall and domain-specific cognitive functions.
Nevertheless, our findings should be interpreted considering several limitations. We included a group of relatively healthy and highly educated older adults who participated in a randomized clinical trial; therefore, results may differ in observational studies with demographically diverse populations and generalization of results should be cautious. However, the interventions of the MAPT study did not have significant effects on cognitive function [27]; moreover, all our analyses were adjusted to MAPT group allocation, minimizing potential bias. Although the sample size was substantially reduced in the analyses exploring the combination of MCP-1 and Aβ42/40, which might have led to a reduction in statistical power, we still had 429 subjects with available data in both markers, a larger sample than the population of previous investigation [19]. Although the ability of peripheral MCP-1 to reflect its CNS levels is not fully established since this chemokine may be expressed by different tissues, there is some evidence showing MCP-1 levels in plasma and CSF are strongly correlated [46].
Conclusion
The current study showed that higher plasma MCP-1 levels were associated with declines in both overall and episodic memory cognitive performances in older adults over a 4-year follow-up. Whether MCP-1 levels in the context of low-plasma Aβ42/40 ratio may confer an increased risk for cognitive decline remains an open question. Further research is needed to examine the potential associations between longitudinal changes in plasma MCP-1 and cognitive evolution in population-based studies to clarify the validity of this chemokine as a marker of cognitive decline at different disease stages. In addition, the clarification of the directionality and the mechanisms underlying the association between neuroinflammation and cognitive decline at different stages of the disease should be the subject of future research.
Availability of data and materials
The data that support the findings of this study are available from MAPT Study Group but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of Nicola Coley (nicola.coley@inserm.fr).
References
Scheltens P, Strooper BD, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397(10284):1577–90.
Morgan AR, Touchard S, Leckey C, O’Hagan C, Nevado-Holgado AJ, NIMA Consortium, et al. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimers Dement. 2019;15(6):776–87.
Park J-C, Han S-H, Mook-Jung I. Peripheral inflammatory biomarkers in Alzheimer’s disease: a brief review. BMB Rep. 2020;53(1):10–9.
Lai KSP, Liu CS, Rau A, Lanctôt KL, Köhler CA, Pakosh M, et al. Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J Neurol Neurosurg Psychiatry. 2017;88(10):876–82.
Alawode DOT, Heslegrave AJ, Ashton NJ, Karikari TK, Simrén J, Montoliu-Gaya L, et al. Transitioning from cerebrospinal fluid to blood tests to facilitate diagnosis and disease monitoring in Alzheimer’s disease. J Intern Med. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/joim.13332. Cited 2021 May 24.
Brickman AM, Manly JJ, Honig LS, Sanchez D, Reyes-Dumeyer D, Lantigua RA, et al. Plasma p-tau181, p-tau217, and other blood-based Alzheimer’s disease biomarkers in a multi-ethnic, community study. Alzheimers Dement. Available from: https://alz-journals.onlinelibrary.wiley.com/doi/abs/10.1002/alz.12301. Cited 2021 Mar 17.
Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2020;324(8):772–81.
Verberk IMW, Hendriksen HMA, van Harten AC, Wesselman LMP, Verfaillie SCJ, van den Bosch KA, et al. Plasma amyloid is associated with the rate of cognitive decline in cognitively normal elderly: the SCIENCe project. Neurobiol Aging. 2020;89:99–107.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–9.
Albornoz EA, Woodruff TM, Gordon R. Inflammasomes in CNS diseases. Exp Suppl. 2018;108:41–60.
Chen W, Gao Q, Han S, Pan F, Fan W. The CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal transition by cooperatively activating STAT3-Twist signaling. Tumour Biol. 2015;36(2):973–81.
Elsaafien K, Korim WS, Setiadi A, May CN, Yao ST. Chemoattraction and recruitment of activated immune cells, central autonomic control, and blood pressure regulation. Front Physiol. 2019;10:984.
Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry Clin Neurosci. 1997;51(3):135–8.
Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, et al. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152(1):307–17.
Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23(21):7922–30.
Sozzani S, Zhou D, Locati M, Rieppi M, Proost P, Magazin M, et al. Receptors and transduction pathways for monocyte chemotactic protein-2 and monocyte chemotactic protein-3. Similarities and differences with MCP-1. J Immunol. 1994;152(7):3615–22.
Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, et al. The aging systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477(7362):90–4.
Villeda SA, Wyss-Coray T. The circulatory systemic environment as a modulator of neurogenesis and brain aging. Autoimmun Rev. 2013;12(6):674–7.
Westin K, Buchhave P, Nielsen H, Minthon L, Janciauskiene S, Hansson O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS One. 2012;7(1):e30525.
Bettcher BM, Fitch R, Wynn MJ, Lalli MA, Elofson J, Jastrzab L, et al. MCP-1 and eotaxin-1 selectively and negatively associate with memory in MCI and Alzheimer’s disease dementia phenotypes. Alzheimers Dement (Amst). 2016;3:91–7.
Kimura A, Yoshikura N, Hayashi Y, Inuzuka T. Cerebrospinal fluid C-C motif chemokine ligand 2 correlates with brain atrophy and cognitive impairment in Alzheimer’s disease. J Alzheimers Dis. 2018;61(2):581–8.
Fuh J-L, Lee W-J, Liao Y-C, Wang S-J. P1-108: Plasma MCP-1 is associated with a faster decline of cognitive function in MCI and dementia due to Alzheimer’s disease. Alzheimers Dement. 2015;11(7S_Part_8):P380.
Bettcher BM, Neuhaus J, Wynn MJ, Elahi FM, Casaletto KB, Saloner R, et al. Increases in a pro-inflammatory chemokine, MCP-1, are related to decreases in memory over time. Front Aging Neurosci. 2019;11:25.
Lee W-J, Liao Y-C, Wang Y-F, Lin I-F, Wang S-J, Fuh J-L. Plasma MCP-1 and cognitive decline in patients with Alzheimer’s disease and mild cognitive impairment: a two-year follow-up study. Sci Rep. 2018;8(1):1280.
Galimberti D, Fenoglio C, Lovati C, Venturelli E, Guidi I, Corrà B, et al. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol Aging. 2006;27(12):1763–8.
Vellas B, Carrie I, Gillette-Guyonnet S, Touchon J, Dantoine T, Dartigues JF, et al. MAPT study: a multidomain approach for preventing Alzheimer’s disease: design and baseline data. J Prev Alzheimers Dis. 2014;1(1):13–22.
Andrieu S, Guyonnet S, Coley N, Cantet C, Bonnefoy M, Bordes S, et al. Effect of long-term omega 3 polyunsaturated fatty acid supplementation with or without multidomain intervention on cognitive function in elderly adults with memory complaints (MAPT): a randomised, placebo-controlled trial. Lancet Neurol. 2017;16(5):377–89.
Vandenbroucke JP, von Elm E, Altman DG, Gøtzsche PC, Mulrow CD, Pocock SJ, et al. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE): explanation and elaboration. Int J Surg. 2014;12(12):1500–24.
Giudici KV, de Souto Barreto P, Guyonnet S, Li Y, Bateman RJ, Vellas B, et al. Assessment of plasma amyloid-β42/40 and cognitive decline among community-dwelling older adults. JAMA Netw Open. 2020;3(12):e2028634.
Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA, et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93(17):e1647–59.
Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, Schneider T, et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13(8):841–9.
Mawuenyega KG, Kasten T, Sigurdson W, Bateman RJ. Amyloid-beta isoform metabolism quantitation by stable isotope-labeled kinetics. Anal Biochem. 2013;440(1):56–62.
Pino LK, Searle BC, Bollinger JG, Nunn B, MacLean B, MacCoss MJ. The Skyline ecosystem: informatics for quantitative mass spectrometry proteomics. Mass Spectrom Rev. 2020;39(3):229–44.
Folstein MF, Folstein SE, McHugh PR. ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–98.
Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–72.
Grober E, Buschke H, Crystal H, Bang S, Dresner R. Screening for dementia by memory testing. Neurology. 1988;38(6):900–3.
Mapstone M. Neuropsychological assessment, 4th edition. Neurology. 2005;64(6):1103.
Cardebat D, Doyon B, Puel M, Goulet P, Joanette Y. Formal and semantic lexical evocation in normal subjects. Performance and dynamics of production as a function of sex, age and educational level. Acta Neurol Belg. 1990;90(4):207–17.
Llinàs-Reglà J, Vilalta-Franch J, López-Pousa S, Calvó-Perxas L, Torrents Rodas D, Garre-Olmo J. The trail making test. Assessment. 2017;24(2):183–96.
Jaeger J. Digit symbol substitution test: the case for sensitivity over specificity in neuropsychological testing. J Clin Psychopharmacol. 2018;38(5):513–9.
D’Ath P, Katona P, Mullan E, Evans S, Katona C. Screening, detection and management of depression in elderly primary care attenders. I: The acceptability and performance of the 15 item Geriatric Depression Scale (GDS15) and the development of short versions. Fam Pract. 1994;11(3):260–6.
Maezawa I, Maeda N, Montine TJ, Montine KS. Apolipoprotein E-specific innate immune response in astrocytes from targeted replacement mice. J Neuroinflammation. 2006;3(1):10.
Rizzolo L, Narbutas J, Van Egroo M, Chylinski D, Besson G, Baillet M, et al. Relationship between brain AD biomarkers and episodic memory performance in healthy aging. Brain Cogn. 2021;148:105680.
Bettcher BM, Johnson SC, Fitch R, Casaletto KB, Heffernan KS, Asthana S, et al. Cerebrospinal fluid and plasma levels of inflammation differentially relate to cns markers of Alzheimer’s disease pathology and neuronal damage. J Alzheimers Dis. 2018;62(1):385–97.
Britschgi M, Wyss-Coray T. Immune cells may fend off Alzheimer disease. Nat Med. 2007;13(4):408–9.
Sun Y-X, Minthon L, Wallmark A, Warkentin S, Blennow K, Janciauskiene S. Inflammatory markers in matched plasma and cerebrospinal fluid from patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2003;16(3):136–44.
Zhang R, Miller RG, Madison C, Jin X, Honrada R, Harris W, et al. Systemic immune system alterations in early stages of Alzheimer’s disease. J Neuroimmunol. 2013;256(1–2):38–42.
Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–6.
Cabinio M, Saresella M, Piancone F, LaRosa F, Marventano I, Guerini FR, et al. Association between hippocampal shape, neuroinflammation, and cognitive decline in Alzheimer’s disease. J Alzheimers Dis. 2018;66(3):1131–44.
Serpente M, Bonsi R, Scarpini E, Galimberti D. Innate immune system and inflammation in Alzheimer’s disease: from pathogenesis to treatment. Neuroimmunomodulation. 2014;21(2–3):79–87.
Guillot-Sestier M-V, Town T. Let’s make microglia great again in neurodegenerative disorders. J Neural Transm (Vienna). 2018;125(5):751–70.
Yamamoto M, Horiba M, Buescher JL, Huang D, Gendelman HE, Ransohoff RM, et al. Overexpression of monocyte chemotactic protein-1/CCL2 in β-amyloid precursor protein transgenic mice show accelerated diffuse β-amyloid deposition. Am J Pathol. 2005;166(5):1475–85.
Kiyota T, Yamamoto M, Xiong H, Lambert MP, Klein WL, Gendelman HE, et al. CCL2 accelerates microglia-mediated Aβ oligomer formation and progression of neurocognitive dysfunction. PLoS One. 2009;4(7) Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2703798/. Cited 2021 May 25.
Joly-Amado A, Hunter J, Quadri Z, Zamudio F, Rocha-Rangel PV, Chan D, et al. CCL2 overexpression in the brain promotes glial activation and accelerates tau pathology in a mouse model of tauopathy. Front Immunol. 2020;11:997.
Vaz M, Domingues C, Trindade D, Barra C, Oliveira JM, Rosa IM, et al. IL-8 and MCP-1 impact on tau phosphorylation and phosphatase activity. Curr Alzheimer Res. 2020;17(11):985–1000.
Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–51.
Shen X-N, Niu L-D, Wang Y-J, Cao X-P, Liu Q, Tan L, et al. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: a meta-analysis and systematic review of 170 studies. J Neurol Neurosurg Psychiatry. 2019;90(5):590–8.
Nordengen K, Kirsebom B-E, Henjum K, Selnes P, Gísladóttir B, Wettergreen M, et al. Glial activation and inflammation along the Alzheimer’s disease continuum. J Neuroinflammation. 2019;16(1):46.
Desikan RS, McEvoy LK, Thompson WK, Holland D, Brewer JB, Aisen PS, et al. Amyloid-β--associated clinical decline occurs only in the presence of elevated P-tau. Arch Neurol. 2012;69(6):709–13.
Acknowledgements
MAPT Study Group
Principal investigator: Bruno Vellas (Toulouse); Coordination: Sophie Guyonnet ; Project leader: Isabelle Carrié ; CRA: Lauréane Brigitte ; Investigators: Catherine Faisant, Françoise Lala, Julien Delrieu, Hélène Villars ; Psychologists: Emeline Combrouze, Carole Badufle, Audrey Zueras ; Methodology, statistical analysis and data management: Sandrine Andrieu, Christelle Cantet, Christophe Morin; Multidomain group: Gabor Abellan Van Kan, Charlotte Dupuy, Yves Rolland (physical and nutritional components), Céline Caillaud, Pierre-Jean Ousset (cognitive component), Françoise Lala (preventive consultation). The cognitive component was designed in collaboration with Sherry Willis from the University of Seattle, and Sylvie Belleville, Brigitte Gilbert and Francine Fontaine from the University of Montreal.
Co-Investigators in associated centers: Jean-François Dartigues, Isabelle Marcet, Fleur Delva, Alexandra Foubert, Sandrine Cerda (Bordeaux); Marie-Noëlle-Cuffi, Corinne Costes (Castres); Olivier Rouaud, Patrick Manckoundia, Valérie Quipourt, Sophie Marilier, Evelyne Franon (Dijon); Lawrence Bories, Marie-Laure Pader, Marie-France Basset, Bruno Lapoujade, Valérie Faure, Michael Li Yung Tong, Christine Malick-Loiseau, Evelyne Cazaban-Campistron (Foix); Françoise Desclaux, Colette Blatge (Lavaur); Thierry Dantoine, Cécile Laubarie-Mouret, Isabelle Saulnier, Jean-Pierre Clément, Marie-Agnès Picat, Laurence Bernard-Bourzeix, Stéphanie Willebois, Iléana Désormais, Noëlle Cardinaud (Limoges); Marc Bonnefoy, Pierre Livet, Pascale Rebaudet, Claire Gédéon, Catherine Burdet, Flavien Terracol (Lyon), Alain Pesce, Stéphanie Roth, Sylvie Chaillou, Sandrine Louchart (Monaco); Kristel Sudres, Nicolas Lebrun, Nadège Barro-Belaygues (Montauban); Jacques Touchon, Karim Bennys, Audrey Gabelle, Aurélia Romano, Lynda Touati, Cécilia Marelli, Cécile Pays (Montpellier); Philippe Robert, Franck Le Duff, Claire Gervais, Sébastien Gonfrier (Nice); Yannick Gasnier and Serge Bordes, Danièle Begorre, Christian Carpuat, Khaled Khales, Jean-François Lefebvre, Samira Misbah El Idrissi, Pierre Skolil, Jean-Pierre Salles (Tarbes).
MRI group: Carole Dufouil (Bordeaux), Stéphane Lehéricy, Marie Chupin, Jean-François Mangin, Ali Bouhayia (Paris); Michèle Allard (Bordeaux); Frédéric Ricolfi (Dijon); Dominique Dubois (Foix); Marie Paule Bonceour Martel (Limoges); François Cotton (Lyon); Alain Bonafé (Montpellier); Stéphane Chanalet (Nice); Françoise Hugon (Tarbes); Fabrice Bonneville, Christophe Cognard, François Chollet (Toulouse).
PET scans group: Pierre Payoux, Thierry Voisin, Julien Delrieu, Sophie Peiffer, Anne Hitzel, (Toulouse); Michèle Allard (Bordeaux); Michel Zanca (Montpellier); Jacques Monteil (Limoges); Jacques Darcourt (Nice).
Medico-economics group: Laurent Molinier, Hélène Derumeaux, Nadège Costa (Toulouse).
Biological sample collection: Bertrand Perret, Claire Vinel, Sylvie Caspar-Bauguil (Toulouse).
Safety management: Pascale Olivier-Abbal
DSA Group:
Sandrine Andrieu, Christelle Cantet, Nicola Coley
Funding
The MAPT study was supported by grants from the Gérontopôle of Toulouse, the French Ministry of Health (PHRC 2008, 2009), Pierre Fabre Research Institute (manufacturer of the omega-3 supplement), ExonHit Therapeutics SA, and Avid Radiopharmaceuticals Inc. The data sharing activity was supported by the Association Monegasque pour la Recherche sur la maladie d’Alzheimer (AMPA) and the INSERM-University of Toulouse III UMR 1295 Unit. The present work was performed in the context of the Inspire Program, a research platform supported by grants from the Region Occitanie/Pyrénées-Méditerranée (Reference number: 1901175) and the European Regional Development Fund (ERDF) (Project number: MP0022856). This study received funds from Alzheimer Prevention in Occitania and Catalonia (APOC Chair of Excellence - Inspire Program).
Author information
Authors and Affiliations
Consortia
Contributions
JLSS directed and contributed to all aspects of this manuscript and wrote the initial draft. KGV, PSB, JD and BV contributed to the conception, study design, theoretical background, and data interpretation. RJB and YL processed biological samples. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Eligible subjects provided written informed consent prior to inclusion. The MAPT Study Group was authorized by the French Health Authority and approved by the Advisory Committee for the Protection of Persons participating in Biomedical Research of Toulouse.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Description of features of ELLA System for plasma MCP-1 determination.
Additional file 2.
Neuroimaging procedures. Description of neuroimaging techniques used in the present study.
Additional file 3.
Differences between included and excluded participants. Differences in baseline characteristics between MAPT study participants included and excluded in the present study.
Additional file 4.
Evolution in overall cognitive outcomes, executive function and attention according to plasma MCP-1 status (excluding ApoE ε4 genotype). Mixed-effect linear regression analysis for variation in overall cognitive outcomes, executive function and attention over time according to plasma MCP-1 status among community-dwelling older adults (excluding ApoE ε4 genotype).
Additional file 5.
Evolution in overall memory outcomes according to plasma MCP-1 status (excluding ApoE ε4 genotype). Mixed-effect linear regression analysis for variation in memory outcomes over time according to plasma MCP-1 status among community-dwelling older adults (excluding ApoE ε4 genotype).
Additional file 6.
Within group evolution in overall cognitive outcomes, executive function and attention according to plasma MCP-1 status (excluding ApoE ε4 genotype). Within group evolution in memory outcomes over time according to plasma MCP-1 status among community-dwelling older adults (excluding ApoE ε4 genotype).
Additional file 7.
Within group evolution in memory outcomes, executive function and attention according to plasma MCP-1 status (excluding ApoE ε4 genotype). Mixed-effect linear regression analysis for variation in memory outcomes over time according to plasma MCP-1 status among community-dwelling older adults (excluding ApoE ε4 genotype).
Additional file 8.
Within group evolution in memory outcomes, executive function and attention according to plasma MCP-1 status (excluding ApoE ε4 genotype). Mixed-effect linear regression analysis for variation in overall cognitive outcomes, executive function and attention over time according to combined plasma Aβ42/40+ and MCP-1 status among community-dwelling older adults (excluding ApoE ε4 genotype).
Additional file 9.
Within group evolution in memory outcomes, executive function and attention according to plasma MCP-1 status (excluding ApoE ε4 genotype). Mixed-effect linear regression analysis for variation in memory outcomes over time according to combined plasma Aβ42/40+ and MCP-1 status among community-dwelling older adults (excluding ApoE ε4 genotype).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sanchez-Sanchez, J.L., Giudici, K.V., Guyonnet, S. et al. Plasma MCP-1 and changes on cognitive function in community-dwelling older adults. Alz Res Therapy 14, 5 (2022). https://doi.org/10.1186/s13195-021-00940-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-021-00940-2