
Abstract
Background
Genome-wide association studies (GWAS) have identified a number of genetic variants for Alzheimer’s disease (AD). However, most GWAS were conducted in individuals of European ancestry, and non-European populations are still underrepresented in genetic discovery efforts. Here, we performed GWAS to identify single nucleotide polymorphisms (SNPs) associated with amyloid β (Aβ) positivity using a large sample of Korean population.
Methods
One thousand four hundred seventy-four participants of Korean ancestry were recruited from multicenters in South Korea. Discovery dataset consisted of 1190 participants (383 with cognitively unimpaired [CU], 330 with amnestic mild cognitive impairment [aMCI], and 477 with AD dementia [ADD]) and replication dataset consisted of 284 participants (46 with CU, 167 with aMCI, and 71 with ADD). GWAS was conducted to identify SNPs associated with Aβ positivity (measured by amyloid positron emission tomography). Aβ prediction models were developed using the identified SNPs. Furthermore, bioinformatics analysis was conducted for the identified SNPs.
Results
In addition to APOE, we identified nine SNPs on chromosome 7, which were associated with a decreased risk of Aβ positivity at a genome-wide suggestive level. Of these nine SNPs, four novel SNPs (rs73375428, rs2903923, rs3828947, and rs11983537) were associated with a decreased risk of Aβ positivity (p < 0.05) in the replication dataset. In a meta-analysis, two SNPs (rs7337542 and rs2903923) reached a genome-wide significant level (p < 5.0 × 10−8). Prediction performance for Aβ positivity increased when rs73375428 were incorporated (area under curve = 0.75; 95% CI = 0.74–0.76) in addition to clinical factors and APOE genotype. Cis-eQTL analysis demonstrated that the rs73375428 was associated with decreased expression levels of FGL2 in the brain.
Conclusion
The novel genetic variants associated with FGL2 decreased risk of Aβ positivity in the Korean population. This finding may provide a candidate therapeutic target for AD, highlighting the importance of genetic studies in diverse populations.
Similar content being viewed by others

Background
Genetic factors play an important role in the pathogenesis of Alzheimer’s disease (AD) because heritability is estimated to be 58%–79% [1]. In addition to APOE ɛ4, recent genome-wide association studies (GWAS) have discovered a number of genetic risk variants for AD [2, 3]. However, a large proportion of AD heritability is still unexplained.
Accumulation of amyloid-beta (Aβ) in the brain is the earliest pathogenic process in AD, followed by tau deposition, neurodegeneration, and cognitive impairment [4]. Therefore, detecting individuals with Aβ deposition is of utmost importance for the prevention and early treatment of AD [5]. Previous studies have evaluated the genetic basis of Aβ deposition using positron emission tomography (PET) imaging [6,7,8,9,10] and identified several novel Aβ associated genetic variants outside the APOE region from European ancestry [11]. However, as each ancestry has a distinct genetic background, replication of the novel genetic findings in different populations is challenging. A number of previous studies failed to replicate European GWAS findings in other ethnic populations [12,13,14,15]. Furthermore, it should be noted that most previous GWAS were conducted in individuals of European ancestry, and non-European populations are underrepresented in genetic discovery efforts [16,17,18].
In this study, using a large sample of the Korean population, we conducted a GWAS to identify single nucleotide polymorphisms (SNPs) associated with Aβ deposition in the brain. We identified novel SNPs for Aβ deposition and demonstrated their associations in an independent cohort of the Korean population. Then, we assessed the topography of Aβ deposition related to the novel SNP. Furthermore, we developed an Aβ prediction model incorporating the novel SNP.
Materials and methods
Participants
For the discovery dataset, total 1214 participants of Korean ancestry were recruited from 14 referral hospitals in South Korea from January 2013 to July 2019. Among them, 923 participants were recruited from the Samsung Medical Center, 201 participants were recruited from a multicenter study of the Korean Brain Aging Study for the Early Diagnosis and Prediction of AD (KBASE-V) [19], and 90 participants were recruited from a multicenter study of Clinical Research Platform based on Dementia Cohort.
For the replication dataset, we used data from 306 participants of Korean ancestry from the biobank of the Chronic Cerebrovascular Disease consortium, recruited from 2016 to 2018. This was part of the ongoing Biobank Innovation for chronic Cerebrovascular disease With ALZheimer’s disease Study (BICWALZS) and the Center for Convergence Research of Neurological Disorders.
For the discovery and replication dataset, we included participants (i) who were diagnosed with amnestic mild cognitive impairment (aMCI), AD dementia (ADD), or were cognitively unimpaired (CU) based on detailed neuropsychological tests [20,21,22], and (ii) who underwent amyloid PET imaging. Participants with aMCI met the following criteria, modified from Peterson’s criteria [23]: (i) normal activities of daily living; (ii) objective memory impairment on verbal or visual memory test, below the 16th percentile of age- and education-matched norms; and (iii) did not have dementia. Those with ADD satisfied the core clinical criteria for probable ADD according to the National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer’s Disease and Related Disorders Association [21]. We excluded participants if they had (i) a causative genetic mutation for AD, such as PSEN1, PSEN2, and APP; (ii) structural abnormalities detected on brain MRI, such as severe cerebral ischemia, territorial infarction, or brain tumors; and (iii) other medical or psychiatric diseases that may cause cognitive impairment. All participants provided written informed consent, and the study was approved by the Institutional Review Board of each center.
Genotyping and imputation
Participants were genotyped using the Illumina Asian Screening Array BeadChip (Illumina, CA, USA) for discovery data and Affymetrix customized Korean chips (Affymetrix, CA, USA) for replication data. Only SNP markers were analyzed. We conducted QC using PLINK software (version 1.9) [24]. Participants were excluded based on the following criteria: (i) call rate < 95%, (ii) mismatch between reported and genetically inferred sex, (iii) deviation from each population parameter, (iv) excess heterozygosity rate (5 standard deviation from the mean), and (v) in cases of related pairs (identified with identity by descent ≥ 0.125) within and between the discovery and replication datasets.
SNPs were excluded based on the following criteria: (i) call rate < 98%, (ii) minor allele frequency (MAF) < 1%, and (iii) a p value < 1.0 × 10−6 for the Hardy-Weinberg equilibrium test. After QC, genome-wide imputation was performed using the Minimac4 software with all available reference haplotypes from HRC-r1.1 on the University of Michigan Imputation Server [25, 26]. For post-imputation QC, we excluded SNPs based on the following criteria: (i) poor imputation quality (r2 ≤ 0.8) and (ii) MAF ≤ 1%. Finally, a total of 4,906,407 SNPs was analyzed.
Amyloid PET acquisition and image analysis
Amyloid PET images were obtained using a Discovery STE PET/CT scanner (GE Medical Systems, Milwaukee, WI, USA). PET images were acquired for 20 min, starting at 90 min after intravenous injection of either 18F-florbetaben or 18F-flutemetamol. Aβ positivity or negativity was determined by well-trained nuclear physicians using visual assessments for florbetaben and flutemetamol [27, 28] PET. Briefly, positivity for tracer uptake was assessed in four cortical regions (lateral temporal, frontal, parietal, and posterior cingulate cortices) for florbetaben PET and five cortical regions (lateral temporal, frontal, parietal, posterior cingulate cortices, and striatum) for flutemetamol PET. Amyloid PET positivity was defined as having at least one cortical region with evidence of positive uptake.
A subset of participants in the discovery cohort (n = 824) and the replication cohort (n = 260) had amyloid PET data available for PET image analysis. For PET image analysis, we performed the following preprocessing using Statistical Parametric Mapping software 12 (SPM, http://www.fil.ion/uc.ac.uk/spm) running on MATLAB (MathWorks 2014b): (1) co-registration of PET to T1-weighted structural MRI, (2) structural MRI segmentation and calculation of transformation matrix, (3) normalization of PET to a Montreal Neurological Institute (MNI) space, and (4) spatial smoothing with a Gaussian kernel of 8-mm full width at half maximum. To calculate the standardized uptake value ratio (SUVR) for each PET image, we used two reference regions (the cerebellar cortex for florbetaben and pons for flutemetamol). The masks of reference regions were obtained from the GAAIN website (http://www.GAAIN.org).
Statistical analysis
GWAS analysis
Logistic regression analysis was performed to determine the association between SNPs and Aβ positivity controlling for age, sex, and the first three principal components (PC) of the genetic ancestry, expressed as Aβ positivity = β0 + β1 age + β2 sex + β3 PC1 + β4 PC2 + β5 PC3 + β6 SNP (additive model, coded as 0, 1, and 2 according to the number of minor alleles). Reported p values were two-tailed, and we defined a p value less than 5.0 × 10−8 as being statistically significant and less than 1.0 × 10−5 or 1.0 × 10−6 as being statistically suggestive based on previous studies [29,30,31]. We assessed genomic inflation according to a previous study [32]. For the replication analysis, reported p values were two-tailed, and a p value less than 0.05, was considered statistically significant. Furthermore, considering the small size of the replication dataset, we performed a permutation test to infer the statistical significance of SNPs from the null distribution. We recalculated the t values of SNPs from logistic regression analysis of randomly shuffled Aβ positivity (10,000 permutations). We calculated the fraction of permutations that showed a more significant association than the observed t values of SNPs derived from the original dataset.
To check if SNPs were associated with Aβ positivity independent of APOE genotype, we performed a conditional analysis by further adjusting for APOE genotype. We also performed a p value based meta-analysis and calculated the summary effect size by averaging the study specific effect sizes, with weights reflecting the standard errors from the study specific effect sizes.
Effects of the newly identified SNPs
After identifying associated SNPs, we calculated the risk of the identified SNPs on Aβ deposition in all participants and at each cognitive level (CU, aMCI, and ADD). We also examined whether Aβ associated SNPs are associated with ADD risk using CU and ADD participants using the following logistic model: ADD = β0 + β1 age + β2 sex + β3 education + β4 identified SNPs.
Next, using the previously reported cut-off values for Aβ positivity (SUVR 0.6 for flutemetamol [33], and SUVR 1.4 for florbetaben [34]), we also performed logistic regression to evaluate whether the identified SNPs were associated with Aβ deposition based on SUVR cut-off values.
Furthermore, we performed voxel-wise PET image analysis to determine which regional Aβ deposition is associated with SNPs after adjusting for the effects of age, sex, genetic PCs, APOE genotype, and PET tracer type. T static maps were thresholded by p < 0.001 with cluster size > 20 when uncorrected for multiple tests or p < 0.05 when corrected for multiple tests using family-wise rate.
To test the clinical utility of the newly identified SNPs, we developed multivariable logistic models to predict Aβ positivity in each individual. To evaluate the performance of the logistic model, we measured the area under curve (AUC) from the receiver operating characteristic curve analysis. For internal validation, we conducted a 10-fold cross-validation with 100 repeats using the discovery data. We reported the mean AUC with 95% confidence interval (CI) of the model. As an external validation, parameters estimated from the discovery data were used to test the Aβ prediction performance in the replication data. We used R software (http://www.r-project.org) and MATLAB for the statistical analyses and results visualization.
Finally, we characterized the function of the identified SNPs by leveraging bioinformatic tools and previously reported results. First, we checked whether MAF of SNPs in our data was similar to that in the East Asian population using the 1000 Genomes Project dataset [35]. To evaluate the genotype-specific expression of identified SNPs in human brain tissues, we performed cis-expression quantitative trait loci (cis-eQTL) analysis through the Genotype-Tissue Expression portal (https://gtexportal.org) [36]. We reported genes that showed significant expression changes in the brain tissues (p < 0.05).
Results
Participants
After QC of genotype data, a total of 1190 (383 CU, 330 aMCI, and 477 ADD) and 284 participants (46 CU, 167 aMCI, and 71 ADD) remained available for the discovery and replication data, respectively. Table 1 shows the baseline demographics for the two datasets (discovery and replication data).
GWAS analysis
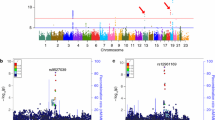
A quantile-quantile plot of p values revealed no genomic inflation (λ = 1.008) (Fig. 1a). In the discovery data, we identified 61 genome-wide significant SNPs on chromosome 19 (p < 5.0 × 10−8) (Fig. 1b). However, all significant SNPs fell within the 500 kb region surrounding APOE and lost genome-wide significance when we adjusted for the APOE ɛ4 allele (Table S1). Outside of the APOE region, 38 SNPs on chromosomes 1, 7, 8, 12, and 22 (p < 1.0 × 10−5), and nine SNPs on chromosome 7 (< 1.0 × 10−6) showed genome-wide suggestive significance (Table S2). Among the nine SNPs, four were associated with Aβ positivity (p < 0.05) in the replication dataset (Table 2). The permutation test of all four SNPs showed t-values lower than the lowest 5% of 10,000 permutations (Table 2, Figure S1).

a Q-Q plot. b Manhattan plot of GWAS analysis. c Regional association plot of rs73375428. The dotted line in the Manhattan plot indicate the genome-wide significance level (p = 5.0 × 10−8, gray dotted line) and the genome-wide suggestive level (p = 1.0 × 10−6, red dotted line; p = 1.0 × 10−5, blue dotted line), respectively. Regional association plot was modified from the SNiPA (single nucleotide polymorphism annotator) (https://snipa.helmholtz-muenchen.de/snipa3). GWAS, genome-wide association study; MAF, minor allele frequency; Q-Q plot, quantile-quantile plot; SNP, single nucleotide polymorphism; SPDYE18, speedy/RINGO cell cycle regulator family member E18; PMS2P9, PMS1 homolog2 mismatch repair system component pseudogene 9; FAM185BP, family with sequence similarity 185 member A pseudogene; SPDYE17, speedy/RINGO cell cycle regulator family member E17; 1-UPK3BP1-PMS2P11, uroplakin 3B pseudogene 1- PMS1 homolog2 mismatch repair system component pseudogene11; FGL2, fibrinogen-like protein 2; CCDC146, coiled-coil domain containing 146; GSAP, gamma secretase activating protein; GCNT1P5, glucosaminyl transferase 1 pseudogene 5
Of the four SNPs, rs11983537 was genotyped while the remaining were imputed. Imputation qualities of the identified SNPs were high (mean r2 0.97 ± 0.02). Of note, two of the four SNPs (rs73375428 and rs2903923) showed genome-wide significant associations (p < 5.0 × 10−8) in the meta-analysis of the discovery and replication datasets (Table 2). When we adjusted for the effect of the APOE ɛ4 allele, all four SNPs were associated with Aβ positivity in the replication datasets (p < 0.05) (Table 2). Since the identified four SNPs showed high linkage disequilibrium (mean r2 0.95 ± 0.05) with each other, we selected rs73375428 for subsequent analyses because it showed the most significant association in the primary analysis of the discovery dataset.
Effects of the newly identified SNPs
In the logistic model, the APOE ɛ4 allele was associated with a 5-fold higher risk of Aβ positivity (odds ratio [OR] = 5.330; 95% CI = 4.188–6.788; p < 0.001) and rs73375428 was associated with a 2-fold lower risk of Aβ positivity (OR = 0.519; 95% CI = 0.404–0.666; p < 0.001). When we adjusted the effect of diagnosis (CU, aMCI, and ADD), the effect of rs73375428 remained significant (OR = 0.556; 95% CI = 0.406–0.666; p < 0.001). In the subgroup analysis, the association of rs73375428 with Aβ positivity was significant in the CU and aMCI groups but not in the ADD group, while the association of APOE ɛ4 was significant across all cognitive states (Table 3). When we defined Aβ positivity based on SUVR, rs73375428 was also associated with a decreased risk of Aβ positivity in both discovery (OR = 0.608; 95% CI = 0.523–0.707; p < 0.001) and replication (OR = 0.551; 95% CI = 0.408–0.744; p = 0.047) datasets (Table S3).
In the voxel-wise PET image analysis, APOE ɛ4 was associated with increased Aβ deposition on the wide cortical areas of the frontal, parietal, and temporal lobes. The SNP rs73375428 was associated with decreased Aβ deposition in the precuneus, lateral parietal, and medial frontal areas, independent of age, sex, genetic PCs, APOE ɛ4, and PET tracer type (Fig. 2).

Results of voxel-wise PET image analysis. T static maps showing a decreased Aβ deposition in participants with the minor allele of the rs73375428 variant (first row: thresholded by uncorrected p < 0.001 with cluster size > 20; second row: thresholded by family-wise rate-corrected p < 0.05) and b increased Aβ deposition in participants with APOE ɛ4 allele (thresholded by family-wise rate corrected p < 0.05). X and Z are based on MNI coordinates. Aβ, amyloid β; MNI, Montreal Neurological Institute
We additionally analyzed the risk of APOE ɛ4 and rs73375428 on the clinical diagnosis of ADD. APOE ɛ4 significantly increased ADD risk (OR = 3.413; 95% CI = 2.63–4.42; p < 0.001) independent of age, sex, education, and rs73375428; and rs73375428 significantly decreased ADD risk (OR = 0.579; 95% CI = 0.421–0.795; p < 0.001) independent of age, sex, education, and APOE ɛ4.
We developed prediction models to test the clinical utility of the APOE ɛ4 allele and newly identified SNP (rs73375428) in predicting Aβ positivity. In the 10-fold cross-validation with 100 repetitions, the model (model 1) including only clinical factors (age, sex, and level of education) showed an AUC of 0.506 (95% CI = 0.500–0.512). After incorporating the APOE ɛ4 allele in the model (model 2), the prediction performance significantly increased (AUC = 0.723; 95% CI = 0.717–0.729). Moreover, when the model included rs73375428 (model 3), the prediction performance further increased (AUC = 0.749; 95% CI = 0.743–0.755) (Fig. 3). When each model, trained in the discovery data, was tested in the replication data, the highest AUC was also observed in the model including both APOE ɛ4 and rs73375428 (model 1 AUC = 0.509, model 2 AUC = 0.693, model 3 AUC = 0.714).

ROC curves for the prediction of Aβ positivity. Solid lines indicate the mean of AUC and dotted lines indicate 95% CIs of AUC. Each model is developed by the multivariate logistic regression. Aβ, amyloid β; AUC, area under curve; CF, clinical factors; ROC, receiver-operating characteristic
Cis-eQTL analysis
rs73375428 was located in the intron of the coiled-coil domain containing the 146 (CCDC146) gene (Fig. 1c). After identifying three additional SNPs with high LD (r2 > 0.7) (rs11983537, rs6978259, and rs3828947), we performed cis-eQTL analysis using the GTEx database. We found that two SNPs (rs73375428 and rs6978259) had significant cis-eQTL effects on the fibrinogen-like protein 2 (FGL2) gene in the brain cortex. Furthermore, a greater dosage of minor allele in SNPs was associated with decreased expression of FGL2 in the brain cortex (rs73375428, normalized effect size [NES] = -0.175, p = 0.02; rs6978259, NES = -0.176, p = 0.01).
Association of previously reported Aβ risk loci from European populations with Aβ positivity in the Korean population
Among the 16 Aβ-associated SNPs reported by Yan et al. [11], no SNP outside the APOE region showed significant association with Aβ positivity and only MAGEF1 (OR = 0.810, p = 0.058) locus showed marginal association in our cohort (Table S4). Based on the public dataset (1000 Genomes Project phase 3) [35], the frequency of the previously reported SNPs differed between Europeans and East Asians, while our cohort (Korean) showed similar allele frequencies to that of East Asians (Table S4).
Discussion
We performed GWAS to identify genetic factors associated with Aβ deposition in the brain using the largest amyloid PET imaging and GWAS data collected from multicenters in South Korea. We identified four novel SNPs (rs73375428, rs2903923, rs3828947, and rs11983537) on chromosome 7, which were associated with a decreased risk of Aβ positivity in the brain at the suggestive level (< 1.0 × 10−6). These associations were also observed in the independent cohort (p < 0.05). Having a minor allele in rs73375428 (G) was associated with a 2-fold decreased risk of Aβ positivity (OR = 0.519) and decreased Aβ deposition in the precuneus, lateral parietal, and medial frontal areas. Incorporating rs73375428, in addition to age, sex, education, and APOE e4, better predicted Aβ positivity. The minor allele of rs73375428 was associated with decreased expression levels of FGL2 in the brain.
We identified four novel SNPs (rs73375428, rs2903923, rs3828947, and rs11983537) associated with a decreased risk of Aβ positivity in the brain. In the discovery dataset, nine SNPs showed genome-wide suggestive significance (< 1.0 × 10−6), of which four SNPs were associated with a decreased risk of Aβ positivity (p < 0.05) in an independent cohort. Although the significance of four novel SNPs was at the suggestive level, meta-analysis of the discovery and replication datasets showed that two SNPs (rs73375428 and rs2903923) reached a genome-wide significance level (p < 5.0 × 10−8). Furthermore, the obtained OR of rs73375428 for Aβ positivity was 0.519, which was strong compared with the ORs of previously reported Aβ- or ADD-associated SNPs (Aβ-associated SNPs OR from 0.84 to 1.2 [13]). In our cohort, about 30% of CU participants carried one or more minor alleles in rs73375428 (MAF of 0.160). This is in accordance with the previously reported MAF of rs73375428 in the East Asian population (MAF of 0.131) [35], which indicates that the samples used in this study were not biased and may reflect the East Asian population. In the subgroup analysis, the identified SNP (rs73375428) decreased the risk of Aβ positivity in the CU and aMCI group but not in the ADD group. This finding may suggest that in the course of AD spectrum, the effect of rs73375428 diminishes in the dementia stage.
Further imaging analysis and prediction model for Aβ positivity showed consistent results. PET image analysis showed that the participants with minor allele in rs73375428 had less Aβ deposition in the precuneus, lateral parietal, and medial frontal areas. These areas are part of the default mode network, typical regions where Aβ deposits in AD [37]. Identifying patients with Aβ deposition is of the utmost importance in predicting the prognosis and selecting patients for clinical trials of anti-Aβ therapy [38]. Currently available diagnostic tools for measuring Aβ are either invasive (cerebrospinal fluid examination) or expensive (PET), hampering their widespread application in clinical practice [39]. We demonstrated that genetic data (APOE ɛ4 and rs73375428) obtained from blood samples with clinical information could predict Aβ positivity with an AUC of 0.749. Furthermore, we demonstrated that the prediction performance improved when rs73375428 was included in the model in addition to age, sex, and APOE ɛ4, suggesting the clinical utility of rs73375428.
The identified SNPs were associated with decreased expression of FGL2 in the brain cortex. Although further specific biological mechanistic studies are required, this result suggests that FGL2 may be a possible link between rs73375428 and decreased Aβ deposition in the brain. FGL2 is a membrane-bound or secreted protein expressed by immune cells that have either coagulation activity [40, 41] or immune-suppressive functions [42, 43]. A previous study demonstrated that FGL2 expression is associated with brain tumor progression through the immune system [44]. FGL2 was also associated with AD. One prior study demonstrated that when human microglia were exposed to Aβ peptide, FGL2 expression in microglia was reduced more than six-fold as an inflammatory response to Aβ peptide [45]. Furthermore, Taguchi et al. obtained brain samples from both patients with AD and controls of Japanese population and demonstrated that FGL2 was upregulated in the AD hippocampus as compared to controls [46]. Given these previous observations, we speculated that participants with minor alleles of rs73375428 could have reduced the risk of Aβ deposition in the brain through decreased expression of FGL2, which reflects the reactive inflammatory response (e.g., Aβ clearance) to Aβ peptide. More functional studies are necessary to elucidate the role of FGL2 in AD pathogenesis.
Our results showed some evidence for ethnic similarity and differences in genetic variants associated with Aβ. As expected, variants in the APOE locus exhibited a significant association with Aβ deposition in the brain, confirming that the APOE variants are important risk factors for AD across various ethnicities [47]. However, there were some ethnic differences. We observed a stronger effect of the variant in APOE (rs429358) on Aβ positivity in the Korean population than that in the European population (Korean, OR = 5.275; European, OR = 1.197 [11]). This is similar to the results in previous studies of the East Asian population, in which the effect of APOE ɛ4 on AD risk was stronger in Han Chinese [48] and Japanese [47] than in the European population. Furthermore, outside the APOE locus, previously reported Aβ associated SNPs in European ancestry data were not replicated [11] in our cohort. Ethnic differences in the effect size and significance might be attributed to the differences in allele frequency and LD pattern across different populations [12]. Indeed, we observed heterogeneity in the allele frequency between the European and Korean cohorts (Table S4). Furthermore, epigenomic patterns, lifestyle, education attainment, and other non-genetic factors may also account for differences across populations. However, it should be noted that the lack of replication might also be a result of insufficient sample size of our cohort. Nevertheless, these findings suggest that the discovery from GWAS in one population may not be applicable to other populations. Therefore, continuous efforts of population-specific and trans-ethnic studies are necessary to accurately discover risk genetic variants.
Limitations
This study has several limitations. First, the statistical significance of the novel SNP was at the genome-wide suggestive level, and the sample size of the replication dataset was small. Furthermore, although associations between four SNPs and Aβ (p < 0.05) were found in the independent dataset, the statistical significance disappeared after correction for multiple tests of nine SNPs. However, our study might present true findings for the following reasons: (i) nine suggestive SNPs at a more conservative p-value (< 1.0 × 10−6) showed high LD with each other, which might reduce the number of independent tests to one; (ii) the permutation test of the four SNPs showed that if the null hypothesis was true, the chance of observing our findings would be extremely small for a given sample size; (iii) two SNPs (rs73375428 and rs2903923) showed genome-wide significant associations in the meta-analysis; and (iv) the biological relevance of FGL2 association with the identified SNPs in the brain tissue suggests a potential AD-associated gene. Nevertheless, our findings should be interpreted with caution and replicated in larger independent datasets. Second, imputation was performed using a large reference panel of mixed populations rather than the Korean population. However, we conducted a strict post-imputation QC, excluding SNPs with poor imputation quality (r2 ≤ 0.8) or low frequency (MAF < 1%). As a result, the imputation qualities of the identified SNPs were high (mean r2 0.97 ± 0.02). Third, the cis-eQTL dataset was obtained from healthy populations and not from subjects with AD. Furthermore, the causality of the identified SNPs and FGL2 expression could not be evaluated in the current analysis. Functional studies using gene editing are necessary to determine the association between the identified SNPs and FGL2. Fourth, GWAS was conducted using Aβ positivity, determined by the visual assessment not by quantitative Aβ SUVR. Since this study was conducted using large data obtained from multiple cohorts, some data were not available for SUVR analysis. However, the visual assessment of Aβ positivity has high correlations with histopathological findings of Aβ deposition in the brain [49, 50], and it is more widely used in the clinical practice.
Conclusions
We identified novel SNPs that reduce the risk of Aβ deposition in the brain and suggested a possible role of FGL2 in AD pathogenesis. This finding may provide a candidate therapeutic target for AD, highlighting the importance of genetic studies in diverse populations.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Aβ:
-
Amyloid-beta
- AD:
-
Alzheimer’s disease
- ADD:
-
Alzheimer’s disease dementia
- aMCI:
-
Amnestic mild cognitive impairment
- AUC:
-
Area under curve
- BICWALZS:
-
Biobank Innovation for chronic Cerebrovascular disease With ALZheimer’s disease Study
- CCDC146:
-
Coiled-coil domain containing the 146
- CI:
-
Confidence interval
- cis-eQTL:
-
Cis-expression quantitative trait loci
- CU:
-
Cognitive unimpaired
- FGL2:
-
Fibrinogen-like protein 2
- GWAS:
-
Genome-wide association studies
- MAF:
-
Minor allele frequency
- MNI:
-
Montreal Neurological Institute
- NES:
-
Normalized effect size
- OR:
-
Odds ratio
- PC:
-
Principal component
- PET:
-
Positron emission tomography
- SNP:
-
Single nucleotide polymorphism
- KBASE-V:
-
Korean Brain Aging Study for the Early Diagnosis and Prediction of AD
References
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–74. https://doi.org/10.1001/archpsyc.63.2.168.
Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30. https://doi.org/10.1038/s41588-019-0358-2.
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–13. https://doi.org/10.1038/s41588-018-0311-9.
Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–16. https://doi.org/10.1016/S1474-4422(12)70291-0.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–92. https://doi.org/10.1016/j.jalz.2011.03.003.
Apostolova LG, Risacher SL, Duran T, Stage EC, Goukasian N, West JD, et al. Associations of the top 20 Alzheimer disease risk variants with brain amyloidosis. JAMA Neurol. 2018;75(3):328–41. https://doi.org/10.1001/jamaneurol.2017.4198.
Raghavan NS, Dumitrescu L, Mormino E, Mahoney ER, Lee AJ, Gao Y, et al. Association between common variants in RBFOX1, an RNA-binding protein, and brain amyloidosis in early and preclinical Alzheimer disease. JAMA Neurol. 2020;77(10):1288–98. https://doi.org/10.1001/jamaneurol.2020.1760.
Li J, Zhang Q, Chen F, et al. Genetic interactions explain variance in cingulate amyloid burden: an AV-45 PET genome-wide association and interaction study in the ADNI cohort. Biomed Res Int. 2015;2015:647389.
Ramanan VK, Risacher SL, Nho K, Kim S, Shen L, McDonald BC, et al. GWAS of longitudinal amyloid accumulation on 18F-florbetapir PET in Alzheimer’s disease implicates microglial activation gene IL1RAP. Brain. 2015;138(10):3076–88. https://doi.org/10.1093/brain/awv231.
Ramanan VK, Risacher SL, Nho K, et al. APOE and BCHE as modulators of cerebral amyloid deposition: a florbetapir PET genome-wide association study. Mol Psychiatry. 2014;19(3):351–7. https://doi.org/10.1038/mp.2013.19.
Yan Q, Nho K, Del-Aguila JL, et al. Genome-wide association study of brain amyloid deposition as measured by Pittsburgh Compound-B (PiB)-PET imaging. Mol Psychiatry. 2021;26(1):309–21.
Adeyemo A, Rotimi C. Genetic variants associated with complex human diseases show wide variation across multiple populations. Public Health Genomics. 2010;13(2):72–9. https://doi.org/10.1159/000218711.
Li YR, Keating BJ. Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. Genome Med. 2014;6(10):91. https://doi.org/10.1186/s13073-014-0091-5.
Carlson CS, Matise TC, North KE, Haiman CA, Fesinmeyer MD, Buyske S, et al. Generalization and dilution of association results from European GWAS in populations of non-European ancestry: the PAGE study. PLoS Biol. 2013;11(9):e1001661. https://doi.org/10.1371/journal.pbio.1001661.
Kunkle BW, Schmidt M, Klein H-U, et al. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta-analysis. JAMA Neurol. 2021;78(1):102–13.
Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. 2016;538(7624):161–4. https://doi.org/10.1038/538161a.
Petrovski S, Goldstein DB. Unequal representation of genetic variation across ancestry groups creates healthcare inequality in the application of precision medicine. Genome Biol. 2016;17:1–3.
Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51(4):584–91. https://doi.org/10.1038/s41588-019-0379-x.
Hwang J, Jeong JH, Yoon SJ, Park K, Kim EJ, Yoon B, et al. Clinical and biomarker characteristics according to clinical spectrum of Alzheimer’s disease (AD) in the validation cohort of Korean Brain Aging Study for the Early Diagnosis and Prediction of AD. J Clin Med. 2019;8(3):341. https://doi.org/10.3390/jcm8030341.
Lee DY, Lee KU, Lee JH, Kim KW, Jhoo JH, Kim SY, et al. A normative study of the CERAD neuropsychological assessment battery in the Korean elderly. J Int Neuropsychol Soc. 2004;10(1):72–81. https://doi.org/10.1017/S1355617704101094.
McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–9. https://doi.org/10.1016/j.jalz.2011.03.005.
Kang SH, Park YH, Lee D, Kim JP, Chin J, Ahn Y, et al. The cortical neuroanatomy related to specific neuropsychological deficits in Alzheimer’s continuum. Dement Neurocogn Disord. 2019;18(3):77–95. https://doi.org/10.12779/dnd.2019.18.3.77.
Petersen RC. Mild cognitive impairment. N Engl J Med. 2011;364(23):2227–34. https://doi.org/10.1056/NEJMcp0910237.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. https://doi.org/10.1086/519795.
Fuchsberger C, Abecasis GR, Hinds DA. minimac2: faster genotype imputation. Bioinformatics. 2015;31(5):782–4. https://doi.org/10.1093/bioinformatics/btu704.
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44(8):955–9. https://doi.org/10.1038/ng.2354.
Thurfjell L, Lilja J, Lundqvist R, Buckley C, Smith A, Vandenberghe R, et al. Automated quantification of 18F-flutemetamol PET activity for categorizing scans as negative or positive for brain amyloid: concordance with visual image reads. J Nucl Med. 2014;55(10):1623–8. https://doi.org/10.2967/jnumed.114.142109.
Curtis C, Gamez JE, Singh U, Sadowsky CH, Villena T, Sabbagh MN, et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 2015;72(3):287–94. https://doi.org/10.1001/jamaneurol.2014.4144.
Stranger BE, Stahl EA, Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011;187(2):367–83. https://doi.org/10.1534/genetics.110.120907.
Nishino J, Ochi H, Kochi Y, Tsunoda T, Matsui S. Sample size for successful genome-wide association study of major depressive disorder. Front Genet. 2018;9:227. https://doi.org/10.3389/fgene.2018.00227.
Stein MB, McCarthy MJ, Chen C-Y, et al. Genome-wide analysis of insomnia disorder. Mol Psychiatry. 2018;23(11):2238–50. https://doi.org/10.1038/s41380-018-0033-5.
Devlin B, Roeder K, Wasserman L. Genomic control, a new approach to genetic-based association studies. Theor Popul Biol. 2001;60(3):155–66. https://doi.org/10.1006/tpbi.2001.1542.
Vandenberghe R, Van Laere K, Ivanoiu A, et al. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann Neurol. 2010;68(3):319–29. https://doi.org/10.1002/ana.22068.
Bullich S, Seibyl J, Catafau AM, Jovalekic A, Koglin N, Barthel H, et al. Optimized classification of 18F-Florbetaben PET scans as positive and negative using an SUVR quantitative approach and comparison to visual assessment. Neuroimage Clin. 2017;15:325–32. https://doi.org/10.1016/j.nicl.2017.04.025.
Consortium GP. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. https://doi.org/10.1038/nature15393.
Carithers LJ, Moore HM. The genotype-tissue expression (GTEx) project. Biopreserv Biobank. 2015;13(5):307–8. https://doi.org/10.1089/bio.2015.29031.hmm.
Palmqvist S, Schöll M, Strandberg O, et al. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat Commun. 2017;8:1–13.
Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–62. https://doi.org/10.1016/j.jalz.2018.02.018.
Fargo KN, Carrillo MC, Weiner MW, Potter WZ, Khachaturian Z. The crisis in recruitment for clinical trials in Alzheimer’s and dementia: an action plan for solutions. Alzheimers Dement. 2016;12(11):1113–5. https://doi.org/10.1016/j.jalz.2016.10.001.
McGilvray ID, Lu Z, Wei AC, et al. Murine hepatitis virus strain 3 induces the macrophage prothrombinase fgl-2 through p38 mitogen-activated protein kinase activation. J Biol Chem. 1998;273(48):32222–9. https://doi.org/10.1074/jbc.273.48.32222.
Rabizadeh E, Cherny I, Lederfein D, Sherman S, Binkovsky N, Rosenblat Y, et al. The cell-membrane prothrombinase, fibrinogen-like protein 2, promotes angiogenesis and tumor development. Thromb Res. 2015;136(1):118–24. https://doi.org/10.1016/j.thromres.2014.11.023.
Chan CW, Kay LS, Khadaroo RG, et al. Soluble fibrinogen-like protein 2/fibroleukin exhibits immunosuppressive properties: suppressing T cell proliferation and inhibiting maturation of bone marrow-derived dendritic cells. J Immunol. 2003;170(8):4036–44. https://doi.org/10.4049/jimmunol.170.8.4036.
Yan J, Kong L-Y, Hu J, et al. FGL2 as a multimodality regulator of tumor-mediated immune suppression and therapeutic target in gliomas. J Natl Cancer Inst. 2015;107:djv137.
Yan J, Zhao Q, Gabrusiewicz K, et al. FGL2 promotes tumor progression in the CNS by suppressing CD103+ dendritic cell differentiation. Nat Commun. 2019;10:1–15.
Walker DG, Link J, Lue LF, Dalsing-Hernandez JE, Boyes BE. Gene expression changes by amyloid β peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol. 2006;79(3):596–610. https://doi.org/10.1189/jlb.0705377.
Taguchi K, Yamagata HD, Zhong W, Kamino K, Akatsu H, Hata R, et al. Identification of hippocampus-related candidate genes for Alzheimer’s disease. Ann Neurol. 2005;57(4):585–8. https://doi.org/10.1002/ana.20433.
Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA. 1997;278(16):1349–56. https://doi.org/10.1001/jama.1997.03550160069041.
Wu P, Li H-L, Liu Z-J, Tao QQ, Xu M, Guo QH, et al. Associations between apolipoprotein E gene polymorphisms and Alzheimer’s disease risk in a large Chinese Han population. Clin Interv Aging. 2015;10:371–8. https://doi.org/10.2147/CIA.S73396.
Sabri O, Sabbagh MN, Seibyl J, Barthel H, Akatsu H, Ouchi Y, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: phase 3 study. Alzheimers Dement. 2015;11(8):964–74. https://doi.org/10.1016/j.jalz.2015.02.004.
Salloway S, Gamez JE, Singh U, Sadowsky CH, Villena T, Sabbagh MN, et al. Performance of [18F] flutemetamol amyloid imaging against the neuritic plaque component of CERAD and the current (2012) NIA-AA recommendations for the neuropathologic diagnosis of Alzheimer’s disease. Alzheimers Dement. 2017;9(1):25–34. https://doi.org/10.1016/j.dadm.2017.06.001.
Acknowledgements
Not applicable.
Funding
This research was supported by a National Research Foundation of Korea grant, funded by the Korean government (MSIP) (2018R1A1A3A04079255 and 2014M3C7A1064752); the ICT Creative Consilience program (IITP-2020-0-01821) supervised by the Institute for Information & Communications Technology Planning & Evaluation (IITP); the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health and Welfare, Republic of Korea (HU21C0111 and HI19C1132); the Ministry of Science and ICT, Republic of Korea (2019RIA5A2026045); and the Korea Centers for Disease Control and Prevention (2018-ER6202-01 and 4845-303).
Author information
Authors and Affiliations
Contributions
H.R.K., H.H.W., and H.J.K. contributed to the study design, data collection, data analysis, and drafting the manuscript. S.H.J. contributed to the data collection, data analysis, and revising the manuscript. J.K., H.J., S.H.K., S.H., J.P.K., S.K., J.H.J., S.J.Y., K.W.P., E.J.K., B.Y., J.W.J., J.Y.H., S.H.C., Y.N., K.W.K., S.E.K., J.S.L., N.Y.J., Y.L., B.C.K., S.J.S., C.H.H., D.L.N., and S.W.S. contributed to the data collection, data interpretation, and revising the manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All participants provided written informed consent, and the study was approved by the Institutional Review Board at all participating institutions.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Significant (p value< 5.0×10-8) SNPs associated with Aβ positivity. Table S2. Suggestive SNPs associated with Aβ positivity. Table S3. Association of genome-wide suggestive SNPs (p<1.0×10-6 ) with Aβ positivity based on SUVR. Table S4. Association of previously reported Aβ risk loci from European populations with Aβ positivity in the Korean population.
Additional file 2: Figure S1.
Histogram of t-values obtained from the permutations. Red dotted lines indicate the lowest 5% of the 10,000 permutations. Red arrows indicate the observed t-value obtained from the original dataset.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kim, HR., Jung, SH., Kim, J. et al. Identifying novel genetic variants for brain amyloid deposition: a genome-wide association study in the Korean population. Alz Res Therapy 13, 117 (2021). https://doi.org/10.1186/s13195-021-00854-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-021-00854-z