
Abstract
Background
The neuroprotective role of interleukin (IL)-33 is supported by numerous preclinical studies, but it remains uninvestigated in clinical studies of Alzheimer’s disease (AD). We aimed to examine the association between human blood levels of IL-33 and cognitive preservation in amnestic mild cognitive impairment (aMCI) and AD.
Methods
A total of 100 participants (26 controls, 35 aMCI patients, and 39 AD patients) completed two Mini-Mental State Examinations (MMSEs) over a 1-year interval. In all 100 participants at the second MMSE, we examined the plasma levels of IL-33, IL-β, IL-1 receptor agonist (IL-1RA), beta amyloid (Aβ), and tau and apolipoprotein E (ApoE) genotyping; we also performed Hopkins Verbal Learning Test, Trail Making Test, forward and backward digit span, and Clinical Dementia Rating.
Results
IL-33 expression showed a positive trend among controls (1/26 = 3.8%), aMCI (9/35 = 25.7%), and AD (17/39 = 43.6%) (trend analysis: P < 0.001). Patients expressing IL-33 preserved their cognitive function compared with IL-33 non-expressing patients (1-year ΔMMSE, 0.16 ± 1.6 vs − 1.5 ± 2.6; P = 0.006). The cognitive preservation was not associated with the lower levels of Aβ, tau, and ApoE ε4, while higher levels of ApoE ε4 and phosphorylated tau were indeed associated with cognitive decline. The aMCI patients with AD conversion during study period had higher proportion of IL-33(−) than non-AD converters (90.9% vs 53.3%, P = 0.04).
Conclusions
IL-33 or its associated signaling pathways may represent a new treatment paradigm for aMCI and AD.
Similar content being viewed by others


Background
The main pathophysiology of Alzheimer’s disease (AD) involves the accumulation of insoluble forms of amyloid-beta (Aβ) peptide into plaques and the aggregation of the microtubule protein tau into neurofibrillary tangles [1,2,3,4,5]. In addition to the amyloid and tau hypotheses, substantial evidence suggests that innate immune system-mediated actions drive and exacerbate AD pathogenesis [6,7,8,9,10]. Importantly, in the preclinical stages of AD, neuroinflammation triggers a vicious cycle of microglial activation, release of pro-inflammatory factors, and neuronal damage. Exploration of innate immune-mediated mechanisms and the use of immunomodulation as a disease-modification strategy have been promising in the preclinical research of AD [11]. Animal studies have shown that targeting the innate immune molecules or their respective signaling pathways may substantially ameliorate AD-related pathology [11,12,13,14].
Interleukin (IL)-33 is a member of the IL-1 family and broadly expressed in stromal and barrier tissue, including oligodendrocytes and astrocytes in the central nervous system (CNS) [15, 16]. Although IL-33 is originally thought to be a cellular alarmin released from nuclear stores after tissue damage, new in vivo data found that astrocyte-derived IL-33 is the key molecule promoting synapse refinement by microglia during CNS development [17]. Increasing evidence also indicates the critical role of IL-33 in shaping type 1, type 2, and regulatory immune responses [15]. Supporting IL-33 as a therapeutic target in AD comes from animal and human cellular and genetic studies [18,19,20]. An animal study showed that peripheral IL-33 administration reduced soluble Aβ levels and amyloid plaque deposition and reversed synaptic plasticity impairment and cognitive decline in AD mouse models [18]. Another animal study reported that IL-33 deficiency caused tau abnormality, neurodegeneration, and AD-like symptoms in aged mice [19]. Consistent with these findings, a human genetic study showed that IL-33 expression is reduced in the brains of individuals with AD [21]. Another human study reported that when compared with the mild cognitive impairment (MCI) patients with subsequent AD conversion, the MCI patients without AD conversion had higher levels of IL-33+ cells that were also positively correlated with hippocampus volumes [20]. These findings suggested the potential therapeutic role of IL-33 in AD.
To date, no human study has examined the association between IL-33 and cognitive preservation in MCI and AD. Here, we showed that < 50% of MCI and AD patients had peripheral IL-33 expression. Also, the IL-33-expressing patients preserved their cognitive function over 1-year period compared with the patients without IL-33 expression. The cognitive preservation was not associated with the levels of Aβ and tau protein, the risk factors of AD. In contrast, higher apolipoprotein E (ApoE) ε4 expression and higher levels of phosphorylated tau 181 (p-Tau) were associated with rapid cognitive decline. Moreover, IL-33 non-expression was associated with AD conversion in the MCI patients. Collectively, this is the first human study supporting the association between the peripheral IL-33 expression and cognitive preservation in MCI and AD.
Methods
Subjects and study design
The protocol was approved by the Institutional Review Board for the Protection of Human Subjects at the Tri-Service General Hospital (TSGHIRB 1-107-05-111). A total of 109 participants aged between 64 and 88 years were recruited between January 2015 and December 2018 at the memory clinic at the Tri-Service General Hospital of the National Defense Medical Center, Taiwan. Individuals were eligible if they had negative findings on physical and neurological examinations, laboratory tests (creatinine, fasting blood sugar, free-thyroxine 4, high-sensitivity thyroid stimulating hormone, vitamin B12, folic acid, serologic test for syphilis, and routine blood tests), and neuroimaging examinations (brain computed tomography or magnetic resonance imaging).
Participants underwent a baseline Mini-Mental Status Examination (MMSE) at recruitment. After 1-year follow-up, the following cognitive tests were performed, including MMSE, Clinical Dementia Rating (CDR), short-form Geriatric Depression Scale (GDS-S), Hopkins Verbal Learning Test (HVLT), forward and backward digit span, Trail Making Test Part A (TMTA), and Hachinski Ischemia Scale (HIS).
Individuals were excluded if they had the following: (a) a history of major or uncontrolled medical condition, such as heart failure, sepsis, liver cirrhosis, renal failure, chronic obstructive pulmonary disease, and poorly controlled diabetes (hemoglobin A1c > 8.5), myocardial infarction, or malignancy; (b) substance abuse; (c) a history of major neurological disorders, such as stroke or Parkinson’s disease; (d) GDS-S > 9 or modified Rankin Scale scores > 3; and (e) a history of major psychiatric condition that can impair cognition, such as major depressive disorder, bipolar disorder, or schizophrenia.
Participants were allocated to the control group, MCI due to AD (aMCI) group, or AD group based on the results of HVLT, MMSE, CDR, and HIS and the recommendations from the National Institute on Aging-Alzheimer’s Association (NIA-AA) workgroups on diagnostic guidelines for AD and aMCI [22, 23]. Normal controls were required to satisfy the following: (a) no active neurological or psychiatric disorders; (b) no psychotropic drugs; (c) MMSE > 26 (middle school), MMSE > 22 (primary school), and MMSE > 19 (illiteracy); and (d) CDR score = 0. In addition to NIA-AA criteria [22], aMCI was required to satisfy the following criteria: (a) CDR = 0.5; (b) MMSE > 26 (middle school), MMSE > 22 (primary school), and MMSE > 19 (illiteracy); (c) HIS ≤ 3; and (d) HVLT ≤ 22 [24]. In addition to NIA-AA criteria [23], AD was required to satisfy the following criteria: (a) CDR ≥ 0.5; (b) MMSE ≤ 26 (middle school), MMSE ≤ 22 (primary school), and MMSE ≤ 19 (illiteracy); (c) HIS ≤ 3; and (d) HVLT ≤ 19 [24].
Preparation of plasma samples
Fasting blood was drawn using 9-mL K3-EDTA tubes (455036, Greiner Bio-one GmbH, Kremsmünster, Austria), which were gently inverted three times immediately following blood collection. Blood samples were then centrifuged at a relative centrifugal force (2300g) for 10 min (4 °C) using a swing-out (bucket) rotor (5202R, Eppendorf, Hamburg, Germany). Each 0.4-mL plasma sample was transferred to a fresh 2.0-mL tube (CryzoTraq, Ziath, Cambridge, UK). All plasma samples were stored in 0.5 mL aliquots at − 80 °C within 8 h of blood collection. For the measurements of the cytokine levels, the plasma samples were thawed on ice, and 50-μL aliquots were prepared and stored at − 80 °C.
Plasma levels of Aβ and tau protein
Immunomagnetic reduction (IMR), an ultra-sensitive analytical assay method, can reliably assay ultra-low concentrations of human blood biomarkers, including Aβ1-40, Aβ1-42, total tau (t-Tau), and p-Tau181 [25]. For each plasma sample, the levels of Aβ1-40, Aβ1-42, t-Tau, and p-Tau181 were assayed using IMR kits (MF-AB0-0060, MF-AB2-0060, MF-TAU-0060, and MF-PT1-0060, MagQu Co., New Taipei City, Taiwan). For each assay, 40 μL (Aβ1-40, t-Tau, and p-Tau181) or 60 μL (Aβ1-42) of plasma was mixed with 80 or 60 μL of reagent, respectively. Each reported biomarker concentration represents the average of duplicated measurements. An IMR analyzer (XacPro-S, MagQu Co., New Taipei City, Taiwan) was used for all assays. The reliability of IMR measurements ranged from 0.17 to 1000 pg/mL for Aβ1-40, 0.77 to 30,000 pg/mL for Aβ1-42, 0.026 to 3000 pg/mL for t-Tau, and 0.0196 to 1000 pg/mL for p-Tau181. The intra-assay or inter-assay coefficient of assay variation using IMR was within the range of 7 to 10% for high-concentration quality control samples of Aβ1-40, Aβ1-42, t-Tau, or p-Tau181. For low-concentration quality control samples of Aβ1-40, Aβ1-42, t-Tau, or p-Tau181 using IMR, the intra-assay or inter-assay coefficient of assay variation was within the range of 10 to 15%. For each kind of biomarker, two batches of reagent were used. The quality of each batch of reagents was well controlled by monitoring particle size, particle concentration, and bioactivity. The variation in these reagent properties between batches is lower than 10%.
Plasma levels of cytokines
A multiplex bead array assay was used to examine plasma levels of cytokines. The detailed procedures for detection of soluble cytokines by multiplex bead array assays have been previously reported [26, 27]. Three cytokines (IL-1β, IL-1 receptor antagonist (RA), and IL-33) were determined by using a customized human cytokine magnetic bead panel (Bio-Rad; Yu-Shing Biotech., Ltd., Taipei, Taiwan) according to the manufacturer’s instructions (Bio-Rad; Genmall Biotechnology Co., Ltd., Taipei, Taiwan). The median fluorescence intensities were collected on a Bio-Plex 200 instrument (Bio-Rad) using Bio-Plex Manager software version 6.0 (Bio-Rad). Study samples were tested in duplicate, and the duplicate measurements were averaged for statistical analysis. Standard curves were created from duplicate values, and all samples were analyzed as single determinations. All analyses were performed in one batch using kits from the same production lot.
ApoE genotyping
To efficiently obtain genetic information from samples collected from Taiwanese patients of Han Chinese ethnicity, the Taiwan Biobank (TWB) designed the TWB genotype array, based on the Affymetrix Axiom genotyping platform. The TWB genotype array enabled good-quality genotyping. Two single-nucleotide polymorphisms (SNPs, rs429358 and rs7412) defining ApoE isoforms were genotyped using the TWB array.
Statistical analysis
Categorical variables were analyzed using Pearson’s chi-square test, and continuous variables were analyzed using Student’s t test or the Mann-Whitney test. Trend analysis was analyzed using the Cochran-Armitage test. Spearman’s rank-order correlation analysis was carried out to examine the association between MMSE score and IL-33 levels. All tests were two-sided, and P < 0.05 was considered significant. Error bars represent mean ± standard deviation. All statistical analyses were performed using SPSS software version 25.0 (IBM SPSS, IBM Corp., Armonk, NY, USA) and GraphPad Prism software version 8.0 (GraphPad Software, San Diego, CA, USA).
Results
The demographics and cognitive performance
A total of 100 individuals fulfilled the study criteria and completed the second MMSE, of which 26 were healthy controls (HC), 35 were aMCI, and 39 were AD. We first examined the group differences in demographics and cognitive performance (Table 1). The patient group (aMCI plus AD) was older (P < 0.001) and had higher education level (P = 0.02) than the control group. Their performance on all of the cognitive tests (all P < 0.001) was poorer than that of healthy controls.
We examined the demographic and cognitive differences between aMCI and AD patients. Compared with aMCI patients, AD patients showed poorer performance on the first (P < 0.001) and second MMSE (P < 0.001), HVLT (P = 0.005), backward digit span (P = 0.001), TMTA (P = 0.007), and CDR (P < 0.001). The groups did not differ in female proportion, age, body mass index, and education levels.
The peripheral levels of IL-33 in HC, aMCI, and AD
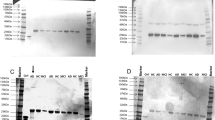
We next sought to examine the peripheral levels of IL-33 among the three groups. The lowest limit of detection for IL-33 was 0.2 pg/mL. Among the 100 samples, the IL-33 levels were detectable only in 28 samples. We analyzed IL-33 data as a binary variable. IL-33(+) indicates detectable IL-33 levels and IL-33(−) undetectable. The proportion of IL-33(+) and the plasma concentration of IL-33 in each group are shown in Fig. 1. The three groups had significantly different proportion of IL-33(+) (P = 0.002), and HC, aMCI, and AD showed a positive linear trend (3.8%, 25.7%, 43.6%; P < 0.001).

Patients with aMCI or AD had higher proportion of peripheral IL-33 expression with linear trend. Pearson’s Chi-Square test: P = 0.002; Cochran–Armitage test for linear trend analysis: P < 0.001. Abbreviations: aMCI amnestic mild cognitive impairment, AD Alzheimer’s disease, HC healthy controls, IL interleukin
The peripheral levels of Aβ1-42, Aβ1-40, t-Tau, p-Tau, IL-1β, and IL-1RA
To explore group differences in AD-related biomarkers and cytokine levels, we examined the peripheral levels of Aβ1-42, Aβ1-40, t-Tau, p-Tau, IL-1β, and IL-1RA in the three groups (Table 2). As expected, the patient group had higher levels of Aβ1-42 (17.3 ± 1.0 vs 16.9 ± 0.7, P = 0.038), t-Tau (26.6 ± 5.1 vs 23.8 ± 3.9, P = 0.014), and p-Tau (4.1 ± 0.9 vs 3.7 ± 0.7, P = 0.035) than the control group. The levels of these biomarkers were higher in AD than in aMCI patients, but not statistically significant. The other two IL-1 families (IL-1β and IL-1RA) were 100% detectable in the control and the patient groups. The patient group had higher levels of IL-1β (0.12 ± 0.11 vs 0.07 ± 0.04, P = 0.039) and IL-1RA (96 ± 79 vs 63 ± 31, P = 0.045) compared with the control group. The comparisons between aMCI and AD in IL-1β and IL-1RA levels were not statistically significant.
The association between IL-33 expression and cognitive preservation
To determine whether IL-33 expression might be a protective factor for aMCI and AD patients, we examined the association between IL-33 expression and 1-year change in MMSE. The aMCI and AD patients were divided into two groups: IL-33(+) and IL-33(−). The IL-33(+) patients did not differ from the IL-33(−) patients in female proportion (76.9% vs 66.7%, P = 0.357), age (77.0 ± 8.6 vs 76.6 ± 8.5, P = 0.848), education levels (8.4 ± 5.1 vs 9.0 ± 4.8, P = 0.632), and BMI (24.0 ± 3.5 vs 24.6 ± 3.4, P = 0.455).
Figure 2 shows that the IL-33(+) patients significantly preserved their general cognitive function compared with the IL-33(−) patients (1-year ΔMMSE of IL-33(+) vs IL-33(−), 0.16 ± 1.6 vs − 1.5 ± 2.6, P = 0.006). We then examined whether the IL-33(+) patients had lower levels of Aβ or tau that may contribute to their cognitive preservation. Figure 2 shows that the IL-33(+) patients did not have lower levels of Aβ1-42, Aβ1-40, t-Tau, or p-Tau. Instead, the IL-33(+) patients had higher levels of Aβ1-40 than the IL-33(−) patients (53.5 ± 4.3 vs 51.3 ± 4.1, P = 0.035). When analyzing Aβ1-42/Aβ1-40 ratio, group difference did not reach significance (IL-33(+) vs IL-33(−), 0.33 ± 0.04 vs 0.34 ± 0.03, P = 0.130). We also examined whether the IL-33(+) patients had lower expression of ApoE ε4. The proportion of ApoE ε4 expression did not differ significantly between groups [IL-33(+) vs IL-33(−), 15.4% vs 22.9%, P = 0.442].

Patients with IL-33 expression may preserve cognitive function although having high levels of Aβ1-40. Error bars indicate standard deviation. Abbreviations: Aβ amyloid β, MMSE Mini Mental Status Examination, p-Tau phosphorylated Tau 181, t-Tau total Tau
Figure 3 illustrates the Spearman correlation analysis for the association between the follow-up MMSE and the IL-33 levels in the patient group. A significant positive correlation was observed in the patient group (aMCI + AD) (rho = 0.429, P = 0.029). The patients with aMCI and the patients with AD also showed positive correlations, but they were not statistically significant.

The positive correlation between the levels of IL-33 and MMSE in the patient group with 95% confidence intervals. Abbreviations: aMCI amnestic mild cognitive impairment, AD Alzheimer’s disease, IL interleukin
We next assessed the levels of IL-1β and IL-1RA between IL-33(+) and IL-33(−) patients. We found that the IL-33(+) patients had higher levels of IL-1β than IL-33(−) patients (0.15 ± 0.14 vs 0.10 ± 0.10, P = 0.011). The levels of IL-1RA was also higher in IL-33(+) patients than in IL-33(−) patients but without statistical significance (96.07 ± 40.24 vs 95.64 ± 94.02, P = 0.189).
The association between cognitive preservation and Aβ, tau, and ApoE ε4
Aβ, tau, and ApoE ε4 are well-known risk factors of AD, and thereby, we sought to examine whether cognitive preservation was associated with ApoE ε4 non-expression or lower levels of Aβ and tau. The patient group was divided into two groups: ApoE ε4(+) and ApoE ε4(−). Additionally, the patient group was divided into two categories—high and low—according to the calculated mean value of f Aβ1-42, Aβ1-40, t-Tau, and p-Tau. Figure 4 shows that ApoE ε4 expression (P = 0.009) and higher levels of p-Tau (P = 0.038) were significantly associated with cognitive decline compared with ApoE ε4 non-expression and lower levels of p-Tau, respectively. ApoE ε4 non-expression and lower levels of Aβ1-42, Aβ1-40, t-Tau, and p-Tau were not associated with cognitive preservation.

Cognitive preservation was not associated with ApoE4, p-Tau, Aβ1-42, Aβ1-40, and t-Tau, while ApoE ε4 expression and high levels of p-Tau had significantly cognitive decline than their comparators. a MMSE change between ApoE4 expression vs ApoE4 non-expression. b MMSE change between high levels of p-Tau vs low levels. c MMSE change between high levels of Aβ1-42 vs low levels. d MMSE change between high levels of Aβ1-40 vs low levels. e MMSE change between high levels of t-Tau vs low levels. Error bars indicate standard deviation. Abbreviations: Aβ amyloid β, MMSE Mini Mental State Examination, p-Tau phosphorylated Tau 181, t-Tau total Tau
IL-33(−) between aMCI with AD conversion vs aMCI without AD conversion
Finally, we sought to test whether IL-33(−) patients may have higher risk of AD conversion. The aMCI patients without any decline between the first and second MMSE were defined as non-AD converter, while the AD patients with first MMSE > 27 (middle school), > 23 (primary school), and > 20 (illiteracy) were defined as AD converters. For example, an AD patient with 9-year education level had first MMSE score of 27 and second MMSE score of 24 which were allocated into AD converter. Thus, we identified 11 AD converters and 15 non-AD converters. We next examined the proportion of IL-33(−) between AD converters and non-AD converters. Our results showed that AD converters had higher proportion of IL-33(−) than non-AD converters (90.9% vs 53.3%, P = 0.04).
Discussion
The CNS has the highest levels of IL-33 expression in all human organs [15,16,17], and recent basic and preclinical studies have reported its extended physiological and pathophysiological role in CNS development [17, 19], recovery [16, 28, 29], and disease [15, 18, 21]. Here, we further our understanding of the IL-33 in human AD research. The main findings of this study were as follows: (1) most of healthy controls did not have detectable levels of peripheral IL-33; (2) the IL-33 expression showed positive linear trend between healthy controls, aMCI, and AD; (3) the IL-33-expressing patients preserved their cognitive function over 1-year period; (4) the cognitive preservation was not associated with the levels of Aβ and tau and the expression of ApoE ε4; and (5) the aMCI patients with subsequent AD conversion had higher proportion of IL-33 non-expression.
In our study, the aMCI and AD patients had higher peripheral levels of Aβ1-42, t-Tau, and p-Tau than controls, indirectly reflecting their central neurodegenerative conditions. The aMCI and AD patients lacking IL-33 expression revealed significantly cognitive decline, while the patients with IL-33 expression preserved their cognitive function over 1-year period. This finding was consistent with the bidirectional relationship between IL-33 deficiency and neurodegeneration in several studies, including the following: (1) mice lacking IL-33 had persistent inflammation and severe neurodegeneration in retinal detachment [30]; (2) IL-33 deficiency mice failed to repair deoxyribonucleic acid damage of aged neuron, resulting in neurodegeneration and tau abnormality [19]; (3) mice lacking IL-33 were found to have impaired recovery after CNS injury [16]; and (4) IL-33 treatment rescued contextual memory deficits in AD mouse models [18]. Collectively, our study provided the first human evidence that linking IL-33 to neurodegeneration in the aMCI and AD patients.
Comparisons between our study and the animal studies with manipulation of IL-33 revealed inconsistent findings. An animal study showed that peripheral administration of IL-33 could reduce soluble Aβ levels and reverse cognitive decline in AD mouse models [18]. Our study design was observational in nature, and we could not observe the effects of exogenous IL-33 administration in rescuing AD mice-related brain neuropathology [18]. Our aMCI and AD patients with IL-33 expression did not have lower levels of Aβ and tau. However, we found that the IL-33-expressing patients had higher levels of IL-1β. IL-33 and IL-1β belong to the IL-1 family, and IL-1β has been shown to reduce amyloid plaque pathology in AD mouse models [31, 32]. Moreover, we found a significant and positive association between IL-33 expression and cognitive preservation, and the levels of IL-33 were positively associated with the MMSE scores. These findings indirectly support the role of IL-33 in cognitive preservation in patients with aMCI or AD. Future longitudinal studies are needed to warrant the link between Aβ and IL-33 in human studies.
Our data with follow-up were consistent with a human study addressing the baseline differences between MCI with subsequent AD conversion and MCI without subsequent AD conversion [20]. Compared with AD converters, AD non-converters had increased baseline levels of IL-33+ cell that was also positively correlated with baseline bilateral hippocampus volumes [20]. These findings indirectly supported the association between IL-33 deficiency and neurodegeneration. Indeed, in our study, the AD converters had higher proportion of IL-33 non-expression than the AD non-converters.
Although our study found a positive linear trend of IL-33 expression among heathy controls (3.8%), aMCI (25.7%), and AD (43.6%), the longitudinal changes of IL-33 expression remain unclear in aMCI and AD. Several lines of evidence suggest that AD-related neurodegeneration begins 20 years or more before the affected individual experiences noticeable symptoms [1]. The IL-33 non-expression in aMCI and AD may be a condition of deficiency (insufficient production) or a consequence of depletion (excessive consumption). A previous study found lower baseline levels of IL-33+ cell in MCI patients with AD conversion [20], which was consistent with our study showing a higher peripheral IL-33 non-expression in aMCI patients with AD conversion than non-AD converters. Another human genetic study also reported lower levels of IL-33 expression in the brain of AD cases than controls [21]. Taking these findings together, insufficient production of IL-33, rather than IL-33 depletion, might be associated with the risk of AD conversion and rapid cognitive decline. However, future studies need to prospectively examine the levels of IL-33 in the preclinical stage of aMCI and AD.
Limitations
First, the bioactivity of IL-33 is limited in blood [33]. A study measured the serum levels of IL-33 in 30 healthy controls and found that all of the samples were undetectable (lowest limit of detection, 75 pg/mL) [33]. Future study addressing the role of IL-33 in AD can simultaneously measure IL-33 and its receptor ST2. Second, we used a multiplex bead-based assay method to measure the plasma levels of IL-33; however, this method does not specify which IL-33 isoforms (full-length or cleaved) were measured. Several studies have reported that the size of the IL-33 molecules strongly influences their bioactivity [34,35,36]. For example, the mature forms of IL-33 (cleaved) are 30-fold more potent than full-length IL-33 for activation of innate lymphoid cells [35]. Therefore, future studies should consider a Western blot analysis to identify different forms of IL-33 in association with aMCI and AD. Third, our sample size was small; therefore, our findings need to be validated by future large-scale studies. Fourth, several biomarkers were only measured once. Future studies should assess the longitudinal changes of IL-33, Aβ, and tau in association with the cognitive decline. Finally, the correlation between the CSF levels of IL-33 and the peripheral levels of IL-33 remains to be determined.
Conclusions
This is an early foray into the association of IL-33 in human AD research, indicating an association between IL-33 expression and cognitive preservation in aMCI and AD patients. Unanswered key questions include the underlying mechanism of IL-33 deficiency in mediating cognition decline in aMCI and AD and the trajectory of IL-33 expression from preclinical AD stage to full-blown AD. In conclusion, our findings suggest that IL-33 or its associated signaling pathways may represent a new treatment paradigm for AD.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Association. As: 2019 Alzheimer’s disease facts and figures. Alzheimers Dement 2019, 15(3):321–387.
Startin CM, Ashton NJ, Hamburg S, Hithersay R, Wiseman FK, Mok KY, Hardy J, Lleo A, Lovestone S, Parnetti L, et al. Plasma biomarkers for amyloid, tau, and cytokines in Down syndrome and sporadic Alzheimer’s disease. Alzheimers Res Ther. 2019;11(1):26.
Shin WS, Di J, Cao Q, Li B, Seidler PM, Murray KA, Bitan G, Jiang L. Amyloid beta-protein oligomers promote the uptake of tau fibril seeds potentiating intracellular tau aggregation. Alzheimers Res Ther. 2019;11(1):86.
Halawa OA, Gatchel JR, Amariglio RE, Rentz DM, Sperling RA, Johnson KA, Marshall GA, Alzheimer’s Disease Neuroimaging I. Inferior and medial temporal tau and cortical amyloid are associated with daily functional impairment in Alzheimer’s disease. Alzheimers Res Ther. 2019;11(1):14.
Degerman Gunnarsson M, Ingelsson M, Blennow K, Basun H, Lannfelt L, Kilander L. High tau levels in cerebrospinal fluid predict nursing home placement and rapid progression in Alzheimer’s disease. Alzheimers Res Ther. 2016;8(1):22.
Abu-Rumeileh S, Steinacker P, Polischi B, Mammana A, Bartoletti-Stella A, Oeckl P, Baiardi S, Zenesini C, Huss A, Cortelli P, et al. CSF biomarkers of neuroinflammation in distinct forms and subtypes of neurodegenerative dementia. Alzheimers Res Ther. 2019;12(1):2.
McManus RM, Heneka MT. Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther. 2017;9(1):14.
Theriault P, ElAli A, Rivest S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res Ther. 2015;7(1):41.
Nagga K, Wattmo C, Zhang Y, Wahlund LO, Palmqvist S. Cerebral inflammation is an underlying mechanism of early death in Alzheimer’s disease: a 13-year cause-specific multivariate mortality study. Alzheimers Res Ther. 2014;6(4):41.
Lane RF, Shineman DW, Fillit HM. Beyond amyloid: a diverse portfolio of novel drug discovery programs for Alzheimer’s disease and related dementias. Alzheimers Res Ther. 2011;3(6):36.
Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–72.
Rosenberg RN, Fu M, Lambracht-Washington D. Active full-length DNA Abeta42 immunization in 3xTg-AD mice reduces not only amyloid deposition but also tau pathology. Alzheimers Res Ther. 2018;10(1):115.
Wang JH, Lei X, Cheng XR, Zhang XR, Liu G, Cheng JP, Xu YR, Zeng J, Zhou WX, Zhang YX. LW-AFC, a new formula derived from Liuwei Dihuang decoction, ameliorates behavioral and pathological deterioration via modulating the neuroendocrine-immune system in PrP-hAbetaPPswe/PS1(DeltaE9) transgenic mice. Alzheimers Res Ther. 2016;8(1):57.
Wang S, Yu Y, Geng S, Wang D, Zhang L, Xie X, Wu B, Li C, Xu H, Li X, et al. A coimmunization vaccine of Abeta42 ameliorates cognitive deficits without brain inflammation in an Alzheimer’s disease model. Alzheimers Res Ther. 2014;6(3):26.
Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16(11):676–89.
Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J. The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron. 2015;85(4):703–9.
Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-Inoue H, Dorman LC, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359(6381):1269–73.
Fu AK, Hung KW, Yuen MY, Zhou X, Mak DS, Chan IC, Cheung TH, Zhang B, Fu WY, Liew FY, et al. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc Natl Acad Sci U S A. 2016;113(19):E2705–13.
Carlock C, Wu J, Shim J, Moreno-Gonzalez I, Pitcher MR, Hicks J, Suzuki A, Iwata J, Quevado J, Lou Y. Interleukin33 deficiency causes tau abnormality and neurodegeneration with Alzheimer-like symptoms in aged mice. Transl Psychiatry. 2017;7(8):e1191.
La Rosa F, Saresella M, Baglio F, Piancone F, Marventano I, Calabrese E, Nemni R, Ripamonti E, Cabinio M, Clerici M. Immune and imaging correlates of mild cognitive impairment conversion to Alzheimer’s disease. Sci Rep. 2017;7(1):16760.
Chapuis J, Hot D, Hansmannel F, Kerdraon O, Ferreira S, Hubans C, Maurage CA, Huot L, Bensemain F, Laumet G, et al. Transcriptomic and genetic studies identify IL-33 as a candidate gene for Alzheimer’s disease. Mol Psychiatry. 2009;14(11):1004–16.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–9.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–9.
Hogervorst E, Xin X, Rahardjo T, Shifu X. The Hopkins Verbal Learning Test and detection of MCI and mild dementia: a literature review. J Alzheimers Dis Parkinsonism. 2014;4(166):2161–0460.1000166.
Tsai CL, Liang CS, Lee JT, Su MW, Lin CC, Chu HT, Tsai CK, Lin GY, Lin YK, Yang FC: Associations between plasma biomarkers and cognition in patients with Alzheimer’s disease and amnestic mild cognitive impairment: a cross-sectional and longitudinal study. J Clin Med. 2019;8(11):1893.
Ho PS, Yeh YW, Huang SY, Liang CS. A shift toward T helper 2 responses and an increase in modulators of innate immunity in depressed patients treated with escitalopram. Psychoneuroendocrinology. 2015;53:246–55.
Ho PS, Yen CH, Chen CY, Huang SY, Liang CS. Changes in cytokine and chemokine expression distinguish dysthymic disorder from major depression and healthy controls. Psychiatry Res. 2017;248:20–7.
Pomeshchik Y, Kidin I, Korhonen P, Savchenko E, Jaronen M, Lehtonen S, Wojciechowski S, Kanninen K, Koistinaho J, Malm T. Interleukin-33 treatment reduces secondary injury and improves functional recovery after contusion spinal cord injury. Brain Behav Immun. 2015;44:68–81.
Korhonen P, Kanninen KM, Lehtonen S, Lemarchant S, Puttonen KA, Oksanen M, Dhungana H, Loppi S, Pollari E, Wojciechowski S, et al. Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav Immun. 2015;49:322–36.
Augustine J, Pavlou S, Ali I, Harkin K, Ozaki E, Campbell M, Stitt AW, Xu H, Chen M. IL-33 deficiency causes persistent inflammation and severe neurodegeneration in retinal detachment. J Neuroinflammation. 2019;16(1):251.
Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O'Banion MK. Chronic interleukin-1beta expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosci. 2007;27(35):9301–9.
Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O'Banion MK. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013;33(11):5053–64.
Dreis C, Ottenlinger FM, Putyrski M, Ernst A, Huhn M, Schmidt KG, Pfeilschifter JM, Radeke HH. Tissue cytokine IL-33 modulates the cytotoxic CD8 T lymphocyte activity during nutrient deprivation by regulation of lineage-specific differentiation programs. Front Immunol. 2019;10:1698.
Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, Martin MU. The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription. J Immunol. 2011;187(4):1609–16.
Lefrancais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, Girard JP. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci U S A. 2014;111(43):15502–7.
Morita H, Nakae S, Saito H, Matsumoto K. IL-33 in clinical practice: size matters? J Allergy Clin Immunol. 2017;140(2):381–3.
Acknowledgements
We thank Shieh-Yueh Yang at MagQu Co., Ltd. and MagQu LLC for assistance with the assays for plasma biomarkers and Ruei-Yi Chao for performing neuropsychological assessments during the study period. The study was conducted using Taiwan Biobank resources.
Funding
This study was supported by grants from the Ministry of Science and Technology of Taiwan (MOST 106-2314-B-016-007-MY2, MOST 108-2314-B-016-023-), Tri-Service General Hospital (TSGH-C106-068, TSGH-C108-100, TSGH-C108-216, TSGH-D-109-101, TSGH-D-109-185), and Beitou Branch, Tri-Service General Hospital (TSGH-BT-108-01).
Author information
Authors and Affiliations
Contributions
CSL managed the literature review, conducted the statistical analyses, interpreted the results, and wrote the first draft of the manuscript. KPS designed the study, directed the data collection, provided the conceptualization and theory used to integrate the findings, and edited the manuscript. CLT played a major role in the acquisition of data and revised the manuscript for intellectual content. JTL designed the study, provided the conceptualization and theory used to integrate the findings, and edited the manuscript. CSC interpreted the data and revised the manuscript for intellectual content. TCY interpreted the data and revised the manuscript for intellectual content. MWS interpreted the data and revised the manuscript for intellectual content. GYL interpreted the data and revised the manuscript for intellectual content. YKL interpreted the results and provided feedback and comments on the various versions of the manuscript. HTC interpreted the data and revised the manuscript for intellectual content. CKT interpreted the results and provided feedback and comments on the various versions of the manuscript. FCY designed the study, directed the data collection, provided the overall scientific supervision, interpreted the results, and edited the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocol was approved by the Institutional Review Board for the Protection of Human Subjects at the Tri-Service General Hospital (TSGHIRB 1-107-05-111). Written informed consent was obtained from all participants.
Consent for publication
All authors have approved of the manuscript and agree with its submission.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liang, CS., Su, KP., Tsai, CL. et al. The role of interleukin-33 in patients with mild cognitive impairment and Alzheimer’s disease. Alz Res Therapy 12, 86 (2020). https://doi.org/10.1186/s13195-020-00652-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-020-00652-z