
Abstract
Background
The phenocopy syndrome of behavioral variant of frontotemporal dementia (phFTD) refers to patients presenting with neuropsychiatric symptoms mimicking the behavioral variant frontotemporal dementia (bvFTD), but lacking frontotemporal atrophy/hypometabolism on neuroimaging and not evolving to dementia during the follow-up. It is important to recognize phFTD for clinical and research purposes.
Objective
The aim of this study was to perform a systematic review of the available literature on phFTD taking into account its clinical, cognitive, imaging, genetic, and pathological features.
Methods and results
We searched for the following terms in two electronic databases (PubMed and Scopus): “frontotemporal dementia and slowly progressive,” “frontotemporal dementia and phenocopy,” “frontotemporal dementia and non-progressive,” “frontotemporal dementia and benign progression,” and “frontotemporal dementia and benign.” We did not include review articles. Papers had to be written in English, French, Portuguese, or Spanish. Overall, 235 studies were retrieved in the initial search. A total of 31 studies composed the final selection, comprising 292 patients. Patients with phFTD are predominantly male and have no major cognitive deficits, with globally preserved executive functions and episodic memory. Some cases (n = 7) of slowly progressive FTD have been associated with C9orf72 genetic expansion. There are only four reports of phFTD neuropathological data, with two patients with no neurodegenerative findings and two with frontotemporal lobar degeneration with ubiquitin-positive inclusions.
Conclusion
The neurobiological underpinnings of phFTD remain unknown. It is controversial whether phFTD belongs to the FTD spectrum. Studies with biomarkers and pathological data are needed to solve the phFTD conundrum.
Similar content being viewed by others

Background
Frontotemporal dementia (FTD) is a neurodegenerative disorder characterized by progressive deterioration of behavior and/or language associated with marked atrophy of frontal and/or temporal lobes [1]. FTD is the second most frequent cause of early-onset dementia, also affecting older subjects [1]. FTD comprises three distinct clinical phenotypes: behavioral variant, semantic variant of primary progressive aphasia (PPA), and non-fluent/agrammatic PPA. The behavioral variant of FTD (bvFTD) is the most frequent subtype [1].
Patients with bvFTD have progressive changes in personality and social conduct. According to consensual diagnostic criteria for bvFTD [2], the diagnosis of possible bvFTD requires at least three of six characteristics: disinhibition, apathy/inertia, loss of empathy and/or sympathy, perseveration/compulsive behaviors, hiperorality, and neuropsychological profile of executive dysfunction with relative sparing of episodic memory and visuospatial skills. Diagnosis of probable bvFTD additionally requires functional impairment and prominent signs of focal frontotemporal involvement in either structural or functional neuroimaging exams [2]. Definite bvFTD is reserved for patients with known pathogenic genetic mutation or with histopathological evidence of frontotemporal lobar degeneration (FTLD) [2].
The estimated mean survival for patients with bvFTD ranges from 6 to 8 years since symptom onset [3]. However, over the past few years, some studies have identified a group of patients clinically indistinguishable from typical FTD who does not progress to frank dementia on follow-up. These patients fulfill criteria for possible bvFTD and have limited or no imaging abnormalities, such as focal prefrontal atrophy on magnetic resonance imaging (MRI) or frontal hypometabolism on fluorodeoxyglucose-positron emission tomography (FDG-PET) [4]. Since their condition remains stable over many years, such group has been called “phenocopy” of FTD (phFTD), FTD “phenocopy syndrome,” “non-progressive” FTD, “benign” FTD, or slowly progressive FTD [4,5,6,7]. It is worth emphasizing that patients with phFTD do not satisfy the diagnostic criteria for probable bvFTD since neuroimaging is unremarkable or almost normal.
Despite efforts to characterize phFTD, results have been controversial. For instance, while some studies reported preserved global cognitive efficiency in phFTD [8,9,10], others described that phFTD patients perform worse than controls in general measures of cognition [5, 11].
From a neuropathological point of view, the question also remains unclear as there are only a few histopathological studies of these patients. As some patients with slowly progressive bvFTD have been diagnosed with chromosome 9 open reading frame 72 (C9orf72) mutation [6, 12], it has been suggested that phFTD could be due to FTLD, the so-called indolent variant of FTD [13]. The hypothesis of an “indolent” FTD places the phenocopy group in the FTD spectrum and raises the issue whether phFTD represents a slow neurodegenerative process. Conversely, phFTD has been conceptualized as late-onset forms of psychiatric disorders including late-onset bipolar disorder and personality disorders [8, 14, 15].
In practical terms, phFTD has been regarded as a clinical entity similar to bvFTD but with relatively normal cognitive performance, intact activities of daily living, no neuroimaging features of bvFTD, and without clinical progression over three or more years of follow-up [7]. However, several questions remain unanswered. How is it possible to explain the behavioral overlap between phFTD and typical bvFTD in the absence of structural or functional brain abnormalities in the former? Are there any other clinical and cognitive features distinguishing phFTD from bvFTD? The identification of phFTD is highly relevant for clinical purposes in order to provide good clinical care, family support and to establish realistic outcomes for these patients. The aim of this study was to perform a systematic review of the available literature about phFTD taking into account its clinical, cognitive, imaging, biological, genetic, and pathological aspects.
Methods
We conducted a systematic review according to the guidelines proposed by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) [16]. This search was independently performed by two investigators (ESV and LCS) in July 2018. We searched for the following terms in two electronic databases (PubMed and Scopus): “frontotemporal dementia and slowly progressive,” “frontotemporal dementia and phenocopy,” “frontotemporal dementia and non-progressive,” “frontotemporal dementia and benign progression,” and “frontotemporal dementia and benign.” We adopted the following filters: clinical articles, comparative studies, historical articles, journal articles, letter, classical articles, case report, comments, and clinical trials. We did not include review articles or abstracts of scientific meetings. They had to be written in English, French, Portuguese, or Spanish. No chronological limits were adopted. Disagreement of eligibility (n = 1) was resolved through a consensual agreement between authors (ESV and LCS).
We carried out the following procedure: (1) titles and abstracts were screened and non-pertinent studies were excluded; (2) after this initial screen, selected articles were subsequently read in full-text and non-pertinent ones were excluded.
This systematic review was registered in the PROSPERO international platform under the number CRD42018107060.
Results
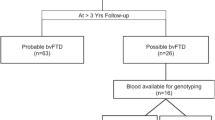
A total of 235 studies were retrieved in the initial search. A total of 31 studies composed the final selection (see Fig. 1), comprising 292 patients. Table 1 presents the main findings from the selected studies. Table 1 also provides detailed information on how each study defined phFTD.

PRISMA flow diagram for studies of phenocopy syndrome of frontotemporal dementia
Results are presented in five parts: part I, epidemiological aspects; part II, cognitive and functional profiles; part III, behavioral and psychiatric profiles; part IV, neuroimaging; and part V, biomarkers, genetic and neuropathological findings in phFTD.
Part I: Epidemiological aspects
The frequency of phFTD among bvFTD series was variable [3,4,5, 8, 17, 18], ranging from 0 [18] to 52% [4] in the selected studies. Most studies reported a higher percentage of men among phFTD patients [3,4,5, 8, 11, 17, 19,20,21,22,23,24,25]. Considering all the reported cases (n = 292), there is a clear male predominance (male = 255, female = 25, missing data = 12), with a male to female ratio of 10:1. Some studies reported that patients with phFTD were younger (45–65 years) than bvFTD (50–75 years) [3, 17], but this was not observed in other series [4, 5, 21, 22]. Non-progressive FTD usually does not present with neurological signs (e.g., primitive reflexes) on physical examination [20], and family history for dementia is typically absent [3, 20].
Part II: Cognitive and functional profiles
Most studies did not find differences between phFTD patients and healthy controls on measures of global cognitive efficiency, such as the Mini-Mental State Examination (MMSE) and Addenbrooke’s Cognitive Examination-Revised (ACE-R) [10, 19, 22]. However, Steketee et al. reported that phFTD patients had significantly lower MMSE scores than controls [11]. Similarly, patients with non-progressive FTD performed worse than controls on ACE-R in another study [5].
Most studies reported that phFTD patients had better global cognitive performance than bvFTD, as measured by the MMSE and the ACE-R [3, 5, 19, 21].
One study assessed structural (MRI) and functional neuroimaging (FDG-PET) in a group of 24 patients with bvFTD [26]. bvFTD patients were classified according to brain MRI in a subgroup with atrophy pattern suggestive of bvFTD (n = 15) and a subgroup without abnormalities (n = 9). bvFTD patients with abnormal MRI performed worse than the bvFTD group with normal MRI on the ACE-R, but almost a third of bvFTD with abnormal MRI had ACE-R scores overlapping with the normal MRI subgroup [26]. This finding suggests that global cognitive efficiency may not be a good measure to differentiate progressive from non-progressive FTD (phFTD).
There is evidence indicating that episodic memory performance may distinguish progressive from non-progressive patients presenting with FTD-related behavioral disorders. Patients with phFTD syndrome had normal performance on episodic memory tests, performing better than typical bvFTD patients [10, 19, 27]. Moreover, memory scores seemed to be very sensitive to detect progressive bvFTD cases at initial presentation [20, 27]. Source memory tasks may also distinguish bvFTD from phFTD at initial presentation. Patients with progressive bvFTD had impairment on temporal and spatial source retrieval, while phFTD patients displayed only temporal source deficits [22].
Executive tasks did not seem to provide a distinction between bvFTD and phFTD, as some bvFTD patients had normal executive performance [5]. phFTD patients performed better than bvFTD in executive tests, such as Digit Span, Letter Fluency, Trail Making, and Hayling [5]. However, up to 20% of bvFTD patients had normal performance on these same tests [5]. Of note, the frequency of dysexecutive syndrome at presentation did not differ phFTD from bvFTD in a cohort of 91 subjects [3].
A longitudinal study investigated emotion recognition and sarcasm detection in bvFTD [28], suggesting that social cognition tasks may be a useful tool to differentiate progressive from non-progressive bvFTD. However, in this study, bvFTD patients had a functional decline at baseline and the follow-up was inferior to 3 years, preventing their classification as phFTD [28].
The functional profile of phFTD has also been investigated. One study compared performance on activities of daily living (ADL) in phFTD and bvFTD [25], according to two ADL measures: a caregiver-based scale, the Disability Assessment of Dementia (DAD), and a patient-based scale, the Assessment of Motor and Process Skills (AMPS). The minimal follow-up period for the phFTD group in this study was 5 years. There was no difference between phFTD and bvFTD in DAD scale, but there was a clear distinction on the performance-based measure (AMPS), with bvFTD patients exhibiting worse performance than phFTD [25]. Another study [24] evaluated the rate of change in ADLs in phFTD and bvFTD. Although both groups had similar levels of functional skills at baseline, bvFTD patients deteriorated in ADLs over 12 months, while phFTD patients did not. Taken together, these data support that there is no evidence of functional impairment in phFTD, suggesting that the assessment of daily activities may be useful to differentiate phFTD from bvFTD.
Part III: Behavioral and psychiatric profiles
Some studies compared behavioral features between bvFTD and phFTD. One study [21] compared progressor and non-progressor patients regarding their profile on the Cambridge Behavioral Inventory (CBI) at initial presentation. There was no difference on bvFTD core diagnostic features between progressors and non-progressors. However, distractibility and stereotypic speech were more common in progressors, while current depression was more frequent in non-progressors [21].
Stereotypical and compulsive behaviors have also been associated with clinical and functional decline in a large series of patients followed up to 5 years [20]. Confabulation was reported in one phFTD patient [29]. On the contrary, other studies showed that bvFTD and phFTD were indistinguishable on behavioral features at presentation [4, 5, 25].
One retrospective study [17] investigated psychiatric and psychological features in patients with phFTD, reporting a higher frequency of recent life events, relationship problems, and cluster C personality traits in this group when compared with bvFTD patients. Bipolar disorder seemed to be more frequent in phFTD patients than in bvFTD group, and one phFTD patient was considered to have autism spectrum disorder [17].
Dols et al. reported four patients with bipolar disorder slowly developing a clinical syndrome marked by apathy, disinhibition, loss of empathy, stereotypical behavior, and compulsiveness, similar to bvFTD [14]. Patients had modest cognitive impairment and did not progress over 3 to 7 years of follow-up. Neuroimaging was unrevealing and C9orf72 screening was negative in all cases. These authors hypothesized that end-stage bipolar disorder could be the underlying cause of the phenocopy syndrome in these patients [14].
In sum, only one study systematically assessed psychiatric antecedents among phFTD patients [17]. There is some evidence suggesting a clinical overlap between phFTD and bipolar disorder in the elderly.
Part IV: Neuroimaging
Patients with phFTD do not exhibit evident frontotemporal atrophy in brain MRI or focal hypometabolism/hypoperfusion in functional neuroimaging methods. It is worth emphasizing that focal frontotemporal atrophy is a marker of clinical and functional decline during the follow-up, ruling out phFTD [4, 23].
Steketee et al. compared bvFTD, phFTD, and healthy controls with quantitative methods in functional and structural MRI [11]. The phFTD group (n = 7) showed cortical atrophy, most prominently in the right temporal lobe, whereas bvFTD group (n = 11) had extensive frontotemporal atrophy [11]. Compared to bvFTD and controls (n = 20), cerebral perfusion was increased in phFTD patients, with higher perfusion in the left prefrontal cortex [11].
Functional connectivity and white matter (WM) microstructure were investigated in bvFTD and phFTD [9]. Compared to controls (n = 17), phFTD patients (n = 7) showed higher connectivity on the default mode network (DMN) than bvFTD patients (n = 12). There were frontotemporal WM abnormalities in both bvFTD and phFTD groups, but they more pronounced in bvFTD [9]. Increased DMN connectivity was also reported in slowly progressive patients with C9orf72 expansion and no characteristic atrophy on structural MRI [30].
Brain metabolism on FDG-PET may be abnormal in cases of normal brain MRI [23, 26]. A typical pattern of frontotemporal hypometabolism is usually associated with a functional decline over the years, but patients with clinical behavioral features of bvFTD and abnormal metabolism on FDG-PET may also remain stable over the years [23, 26, 31]. Normal MRI has a high negative predictive value of normal FDG-PET [26], while FDG-PET increases the sensitivity of the diagnosis of bvFTD [23]. As FDG-PET is a sensitive marker of neurodegeneration, the results showing the absence of typical frontotemporal metabolism in most phFTD patients reinforce the absence of an underlying neurodegenerative process in this condition. Taken together, while data with quantitative methods suggest that phFTD patients share some structural and functional abnormalities with bvFTD [9, 30], other findings are not indicative of an underlying neurodegenerative process [26].
Table 2 shows a synthesis of the comparison of clinical, cognitive, behavioral, and imaging features between phFTD and bvFTD.
Part V: Biomarkers, genetics, and neuropathological data
C9orf72 expansion has been identified in patients with slowly progressive FTD [6, 12, 32, 33]. For instance, three cases of slowly progressive FTD associated with C9orf72 expansion were reported in the same family [12].
The R406W microtubule-associated protein tau (MAPT) mutation is typically associated with a slowly progressive memory decline with symmetrical frontotemporal atrophy on MRI. A novel phenotype associated with the R406W mutation has been identified, marked by a slowly progressive behavioral disorder related to predominant right temporal lobe atrophy [34].
So far, only four patients with phFTD underwent autopsy [13, 35]. Two phFTD cases with behavioral disorders, mild dysexecutive function, and unchanged neuropsychological testing during follow-up (5 and 10 years) did not have FTLD pathology on postmortem pathological exam [35].
On the other hand, spongiosis and gliosis associated with ubiquitin-positive inclusions were reported in one patient featuring typical FTD behavioral symptoms, but no abnormalities on both structural and functional neuroimaging after 3 years of follow-up [36]. In the same study, the profile of peptides in the cerebrospinal fluid (CSF) differed between patients with rapidly progressive FTD (n = 13) and slowly progressive FTD (n = 11), indicating that these may be valuable markers of establishing FTD prognosis [36].
FTLD with ubiquitin pathology was also found in a patient with a 20-year history of behavioral disorders with slow functional decline [13]. Staining for fused-in-sarcoma (FUS) and TAR DNA-binding protein 43 (TDP-43) proteins was negative, and no amyloid plaques were observed; tau pathology was scarce [13].
Discussion
For many years, bvFTD has been considered a clinically homogeneous condition marked by stereotypical behaviors, typical neuropsychological profile (severe executive dysfunction and relative sparing of episodic memory), and shorter survival than Alzheimer’s disease. Recent data from longitudinal studies with bvFTD patients with cognitive, molecular, and neuroimaging tools have challenged this classic clinical profile, highlighting the phenotypical heterogeneity of FTD. More specifically, a subgroup of slowly progressive patients with no evident neuroimaging features of FTD has been recognized. FTD patients with no or slow decline over at least 3 years after symptom onset have been referred as phenocopies of bvFTD (phFTD). In other terms, phFTD is characterized by changes in behavior but with normal neuroimaging, thus fulfilling criteria for possible bvFTD. Moreover, phFTD patients have no or very slow cognitive and functional decline on follow-up.
The phenocopy syndrome of FTD is a clinical and scientific challenge. From a clinical perspective, distinguishing bvFTD from phFTD is crucial for prognosis purpose, clinical care as well as for patient and family counseling and support. From a scientific perspective, the inclusion of phFTD patients in cohorts of bvFTD patients may hinder the development of disease-modifying strategies against FTD. Researchers in the field of FTD should be aware of phFTD for optimal cognitive and behavioral characterization of these patients.
There is a variable frequency of phFTD patients among FTD series [18, 20]. Methodological issues, such as different diagnostic definitions of phFTD and distinct periods of follow-up, hamper establishing a precise prevalence of phFTD.
There is some evidence that cognitive measures may help to differentiate bvFTD from phFTD. Some studies found that bvFTD and phFTD differ in terms of performance in episodic memory tests [10, 19, 27]. Consensual diagnostic criteria for bvFTD state that episodic memory is relatively spared in bvFTD [2]. However, there is increasing evidence that episodic memory impairment occurs in bvFTD [37, 38] in a similar degree as observed in Alzheimer’s disease [19, 27, 39,40,41]. Actually, amnesia in bvFTD is associated with the involvement of medial temporal structures, such as hippocampal and perihippocampal regions [37, 39,40,41]. phFTD patients seem to have normal performance on episodic memory tests, suggesting preservation of the Papez’s circuit. These findings suggest that episodic memory impairment may be a marker of progressive FTD, distinguishing bvFTD from phFTD [19, 21, 27].
Executive functions seem to be more impaired in bvFTD than in phFTD [5, 21], but dysexecutive syndrome at presentation does not seem to be a prognostic factor for bvFTD patients [3]. Moreover, a subset of bvFTD patients may not manifest prominent executive dysfunction at presentation, performing within normal values in executive tests [5, 42, 43]. Therefore, the absence of executive dysfunction in a patient with behavioral features of bvFTD should not be considered as a marker of non-progression. A word of caution is needed here as studies in the field did not always include healthy controls, therefore limiting the interpretation of the prognostic value of cognitive parameters.
The overall preservation of cognitive functions including episodic memory, executive functions and social cognition in phFTD is in line with the lack of significant neuroimaging abnormalities in these patients. By definition, phFTD patients do not exhibit clear frontotemporal involvement in brain imaging. However, a recent study reported functional connectivity changes and microstructural WM abnormalities in phFTD [9]. Compared to phFTD, patients with bvFTD had a similar topographical pattern of alterations, but with more intense abnormalities [9]. Compared to healthy controls, patients with phFTD had a mild increase in DMN connectivity, while bvFTD had a lower increase in the same measure. These authors proposed that DMN increased connectivity would be a compensatory mechanism to early impairment in neuronal functioning [9]. They also suggested that those findings supported the hypothesis that phFTD may belong to the FTD spectrum or might constitute a prodromal phase of bvFTD [9]. More studies are necessary to test this hypothesis.
A fundamental question is whether there is an underlying neurodegenerative process in phFTD. There are only four phFTD reports with postmortem neuropathological assessment. No FTLD pathology was found in two cases [35], while FTLD pathology was documented in two patients [13, 36].
Neurodegeneration biomarkers could shed some light into the question whether phFTD belongs to FTD spectrum. To the best of our knowledge, there is no study investigating phFTD patients with molecular neuroimaging markers such as flortaucipir, which has already been used in FTD patients [44, 45]. So far, there is only one study investigating CSF markers in phFTD patients [36]. In the next future, CSF biomarkers and/or molecular neuroimaging with pathophysiological markers may contribute to the in vivo distinction between phFTD and bvFTD.
To further complicate the scenario, recent studies have reported slowly progressive bvFTD in carriers of C9orf72 expansion [6, 12, 20, 32, 33]. The C9orf72 mutation has important diagnostic implications. The presence of pathogenic mutation in bvFTD patients, regardless of neuroimaging findings, establishes the diagnosis of “definite” bvFTD [2]. One half of C9orf72 carriers initially met criteria for possible bvFTD in a large longitudinal series of patients [20]. Thus, some patients with phenocopy syndrome may have a neurodegenerative pathology and a definite FTD diagnostic when a screening for the C9orf72 mutation is applied to them. Genetic investigation for C9orf72 must be considered in cases of suspected phFTD. Taking into account that R406W MAPT mutation has also been associated with slowly progressive behavioral disorder [34], the genetic screening for this mutation must also be considered in selected cases.
The issue becomes more complex with the finding of the C9orf72 expansion in psychiatric disorders like late-onset psychosis and bipolar disorder [9]. Some authors argue that phFTD actually represents late-onset psychiatric disorders, including late-onset schizophrenia and bipolar disorder, and/or autism spectrum disorders and personality traits decompensated in old age [4, 5, 21, 31]. Indeed, there is evidence of higher frequency of psychiatric or psychological syndromes in phFTD in comparison to bvFTD [17].
The predominance of men among phFTD patients led some authors [5, 8, 10, 20, 21, 27] to hypothesize that phFTD could be a late outcome of Asperger’s disorder, which is more frequent in male than female [46, 47]. Neurodevelopmental disorders are generally more common in men [46, 47]. However, the clinical characteristics of autism spectrum disorder are evident from early childhood [31, 47, 48]. Interestingly, Gossink et al. found only one case of autism spectrum disorder among 33 patients with phFTD [17]. Late-onset schizophrenia is more common in women and its course is characterized by psychotic symptoms with relative preservation of affect.
The end-stage of bipolar disorder was proposed to be the underlying cause of phFTD [14]. In this context, it is important to discuss the “neuroprogression phenomenon” [49] to explain long-term outcomes in brain structure, cognition, functionality, and response to treatments in patients with bipolar disorder. The neuroprogression model, defined by the pathological reorganization and changes of the central nervous system along the course of severe mental disorders, would occur due to successive insults, involving inflammation and oxidative stress, directly related to repeated acute episodes. This theoretical model may include the diagnosis of phFTD syndrome as a possible outcome for the natural course of bipolar disorder and other severe psychiatric disorders.
Cluster C personality traits seem to be more frequent among phFTD patients compared to the bvFTD group [17]. It is controversial whether personality traits change in old age. The question whether phenocopy syndrome represents a late-onset primary psychiatric disorder or a slowly neurodegenerative process remains open. phFTD does not exhibit the unequivocal features of bvFTD, such as a progressive course and neuroimaging abnormalities, neither fulfilling the typical symptoms of a primary psychiatric disorder. phFTD patients may fit the criteria for “mild behavioral impairment” (MBI). MBI is defined by late-life behavioral changes, with no functional decline and no cognitive deficits [50, 51]. Importantly, MBI patients do not fulfill the criteria for psychiatric disorders. Despite the variable outcome, it has been proposed that MBI patients have a higher risk to develop FTD [52]. MBI is a very controversial construct, with an unclear neurobiological basis, and possibly refers to a heterogeneous group of clinical conditions.
The nomenclature “phenocopy syndrome of bvFTD” also deserves critical considerations. The term “phenocopy” is usually employed to refer to “a non-genetically produced phenotype that mimics or resembles the genetically produced one” [13]. It has been used, for instance, to refer to patients with Huntington’s disease phenotype but who lack the typical genetic mutation [53]. Most cases of bvFTD are not monogenic as Huntington’s disease [1], and most studies on phFTD did not test for known pathogenic mutations related to FTD such as C9orf72. Therefore, the term “phenocopy” may not be the most appropriate one to refer to this complex presentation.
Some important caveats about the literature on phFTD must be highlighted: (1) Studies are limited by a small number of patients. (2) Most studies were carried out at few research centers, and it is highly possible that the same patients were enrolled in different studies from the same research group. (3) The length of follow-up is variable, with some studies including phFTD patients with a clinical follow-up as short as 1 year. It is possible that a longer follow-up would detect patients with clinical progression or imaging changes. (4) Most studies did not perform genetic investigation for C9orf72 expansion. (5) The dearth of neuropathological data must be noticed as well.
Conclusions
In conclusion, phFTD represents a clinical condition with the same behavioral features of typical bvFTD, but without neuroimaging abnormalities and no functional decline. Whether these cases belong to the FTD spectrum is still controversial. The next advances on biomarkers and molecular neuroimaging may provide valuable tools for the diagnosis and follow-up of these patients and also clarify the pathophysiological pathways underlying phFTD and bvFTD.
Abbreviations
- ACE-R:
-
Addenbrooke’s Cognitive Examination-Revised
- ADL:
-
Activities of daily living
- AMPS:
-
Assessment of Motor and Process Skills
- bvFTD:
-
Behavioral variant frontotemporal dementia
- C9orf72 :
-
Chromosome 9 open reading frame 72
- CBI:
-
Cambridge Behavioral Inventory
- DAD:
-
Disability Assessment of Dementia
- DMN:
-
Default mode network
- FDG-PET:
-
Fluorodeoxyglucose-positron emission tomography
- FTD:
-
Frontotemporal dementia
- FTLD:
-
Frontotemporal lobar degeneration
- FUS :
-
Fused-in-sarcoma
- MAPT :
-
Microtubule-associated protein tau
- MBI:
-
Mild behavioral impairment
- MMSE:
-
Mini-Mental Status Examination
- MRI:
-
Magnetic resonance imaging
- phFTD:
-
Phenocopy syndrome of behavioral variant of frontotemporal dementia
- PRISMA:
-
Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- TDP-43 :
-
TAR DNA-binding protein 43
- WM:
-
White matter
References
Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):1672–82.
Rascovsky K, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–77.
Garcin B, et al. Determinants of survival in behavioral variant frontotemporal dementia. Neurology. 2009;73(20):1656–61.
Davies RR, et al. Progression in frontotemporal dementia: identifying a benign behavioral variant by magnetic resonance imaging. Arch Neurol. 2006;63(11):1627–31.
Hornberger M, et al. Executive function in progressive and nonprogressive behavioral variant frontotemporal dementia. Neurology. 2008;71(19):1481–8.
Khan BK, et al. Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry. 2012;83(4):358–64.
Kipps CM, Hodges JR, Hornberger M. Nonprogressive behavioural frontotemporal dementia: recent developments and clinical implications of the ‘bvFTD phenocopy syndrome’. Curr Opin Neurol. 2010;23(6):628–32.
Devenney E, et al. The behavioural variant frontotemporal dementia phenocopy syndrome is a distinct entity - evidence from a longitudinal study. BMC Neurol. 2018;18(1):56.
Meijboom R, et al. Functional connectivity and microstructural white matter changes in phenocopy frontotemporal dementia. Eur Radiol. 2017;27(4):1352–60.
Pennington C, Hodges JR, Hornberger M. Neural correlates of episodic memory in behavioral variant frontotemporal dementia. J Alzheimers Dis. 2011;24(2):261–8.
Steketee RM, et al. Structural and functional brain abnormalities place phenocopy frontotemporal dementia (FTD) in the FTD spectrum. Neuroimage Clin. 2016;11:595–605.
Gomez-Tortosa E, et al. Familial benign frontotemporal deterioration with C9ORF72 hexanucleotide expansion. Alzheimers Dement. 2014;10(5 Suppl):S284–9.
Brodtmann A, et al. Phenocopy or variant: a longitudinal study of very slowly progressive frontotemporal dementia. BMJ Case Rep. 2013;2013:bcr2012008077. https://doi.org/10.1136/bcr-2012-008077.
Dols A, et al. Late life bipolar disorder evolving into frontotemporal dementia mimic. Neuropsychiatr Dis Treat. 2016;12:2207–12.
Gambogi LB, et al. Long-term severe mental disorders preceding behavioral variant frontotemporal dementia: frequency and clinical correlates in an outpatient sample. J Alzheimers Dis. 2018;66(4):1577–1585. https://doi.org/10.3233/JAD-180528.
Moher D, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097.
Gossink FT, et al. Psychiatric diagnoses underlying the phenocopy syndrome of behavioural variant frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2016;87(1):64–8.
Nunnemann S, et al. Survival in a German population with frontotemporal lobar degeneration. Neuroepidemiology. 2011;37(3–4):160–5.
Bertoux M, et al. Two distinct amnesic profiles in behavioral variant frontotemporal dementia. Biol Psychiatry. 2014;75(7):582–8.
Devenney E, et al. Progression in behavioral variant frontotemporal dementia: a longitudinal study. JAMA Neurol. 2015;72(12):1501–9.
Hornberger M, et al. Can progressive and non-progressive behavioural variant frontotemporal dementia be distinguished at presentation? J Neurol Neurosurg Psychiatry. 2009;80(6):591–3.
Irish M, et al. Differential impairment of source memory in progressive versus non-progressive behavioral variant frontotemporal dementia. Arch Clin Neuropsychol. 2012;27(3):338–47.
Kerklaan BJ, et al. The added value of 18-fluorodeoxyglucose-positron emission tomography in the diagnosis of the behavioral variant of frontotemporal dementia. Am J Alzheimers Dis Other Dement. 2014;29(7):607–13.
Mioshi E, Hodges JR. Rate of change of functional abilities in frontotemporal dementia. Dement Geriatr Cogn Disord. 2009;28(5):419–26.
Mioshi E, Kipps CM, Hodges JR. Activities of daily living in behavioral variant frontotemporal dementia: differences in caregiver and performance-based assessments. Alzheimer Dis Assoc Disord. 2009;23(1):70–6.
Kipps CM, et al. Combined magnetic resonance imaging and positron emission tomography brain imaging in behavioural variant frontotemporal degeneration: refining the clinical phenotype. Brain. 2009;132(Pt 9):2566–78.
Hornberger M, et al. How preserved is episodic memory in behavioral variant frontotemporal dementia? Neurology. 2010;74(6):472–9.
Kumfor F, et al. Tracking the progression of social cognition in neurodegenerative disorders. J Neurol Neurosurg Psychiatry. 2014;85(10):1076–83.
Poletti M, Borelli P, Bonuccelli U. The neuropsychological correlates of pathological lying: evidence from behavioral variant frontotemporal dementia. J Neurol. 2011;258(11):2009–13.
Lee SE, et al. Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain. 2014;137(Pt 11):3047–60.
Kipps CM, et al. Behavioural variant frontotemporal dementia: not all it seems? Neurocase. 2007;13(4):237–47.
Llamas-Velasco S, et al. Slowly progressive behavioral frontotemporal dementia with C9orf72 mutation. Case report and review of the literature. Neurocase. 2018;24(1):68–71.
Suhonen NM, et al. Slowly progressive frontotemporal lobar degeneration caused by the C9ORF72 repeat expansion: a 20-year follow-up study. Neurocase. 2015;21(1):85–9.
Wood R, et al. Slowly progressive behavioural presentation in two UK cases with the R406W MAPT mutation. Neuropathol Appl Neurobiol. 2016;42(3):291–5.
Devenney E, et al. The bvFTD phenocopy syndrome: a clinicopathological report. J Neurol Neurosurg Psychiatry. 2016;87(10):1155–6.
Mattsson N, et al. Novel cerebrospinal fluid biomarkers of axonal degeneration in frontotemporal dementia. Mol Med Rep. 2008;1(5):757–61.
Hornberger M, Piguet O. Episodic memory in frontotemporal dementia: a critical review. Brain. 2012;135(Pt 3):678–92.
Poos JM, et al. Meta-analytic review of memory impairment in behavioral variant frontotemporal dementia. J Int Neuropsychol Soc. 2018;24(6):593–605.
Bertoux M, et al. Structural anatomical investigation of long-term memory deficit in behavioral frontotemporal dementia. J Alzheimers Dis. 2018;62(4):1887–900.
Fernandez-Matarrubia M, et al. Episodic memory dysfunction in behavioral variant frontotemporal dementia: a clinical and FDG-PET study. J Alzheimers Dis. 2017;57(4):1251–64.
Hornberger M, et al. In vivo and post-mortem memory circuit integrity in frontotemporal dementia and Alzheimer’s disease. Brain. 2012;135(Pt 10):3015–25.
Castiglioni S, et al. The frontal assessment battery does not differentiate frontotemporal dementia from Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;22(2):125–31.
Torralva T, et al. A neuropsychological battery to detect specific executive and social cognitive impairments in early frontotemporal dementia. Brain. 2009;132(Pt 5):1299–309.
Makaretz SJ, et al. Flortaucipir tau PET imaging in semantic variant primary progressive aphasia. J Neurol Neurosurg Psychiatry. 2018;89(10):1024–1031. https://doi.org/10.1136/jnnp-2017-316409.
Spina S, et al. Frontotemporal dementia with the V337M MAPT mutation: Tau-PET and pathology correlations. Neurology. 2017;88(8):758–66.
Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383(9920):896–910.
Masi A, et al. An overview of autism spectrum disorder, heterogeneity and treatment options. Neurosci Bull. 2017;33(2):183–93.
Midorikawa A, Kawamura M. The relationship between subclinical Asperger’s syndrome and frontotemporal lobar degeneration. Dement Geriatr Cogn Dis Extra. 2012;2:180–6.
Gama CS, et al. Staging and neuroprogression in bipolar disorder: a systematic review of the literature. Rev Bras Psiquiatr. 2013;35(1):70–4.
Stella F, et al. Neuropsychiatric symptoms in the prodromal stages of dementia. Curr Opin Psychiatry. 2014;27(3):230–5.
Taragano FE, Allegri RF, Lyketsos C. Mild behavioral impairment: a prodromal stage of dementia. Dement Neuropsychol. 2008;2(4):256–60.
Taragano FE, et al. Risk of conversion to dementia in a mild behavioral impairment group compared to a psychiatric group and to a mild cognitive impairment group. J Alzheimers Dis. 2018;62(1):227–38.
Hensman Moss DJ, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology. 2014;82(4):292–9.
Acknowledgements
Not applicable
Funding
ALT, LCS and PC are supported by Brazilian National Council for Scientific and Technological Development (CNPq – bolsa de produtividade em pesquisa). This publication was partially funded by UFMG (Grant PRPq 02/2019).
Availability of data and materials
This review was based on published literature, all of which is fully listed.
Author information
Authors and Affiliations
Contributions
ESV and LCS performed the systematic review and drafted the first version of the manuscript. PC, HCG, LBG, LIM, and ALT critically reviewed the manuscript for intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Valente, E.S., Caramelli, P., Gambogi, L.B. et al. Phenocopy syndrome of behavioral variant frontotemporal dementia: a systematic review. Alz Res Therapy 11, 30 (2019). https://doi.org/10.1186/s13195-019-0483-2
Published:
DOI: https://doi.org/10.1186/s13195-019-0483-2