
Abstract
Background
CRTC1 (CREB regulated transcription coactivator 1) gene plays a role in synaptic plasticity, learning and long-term memory formation in the hippocampus. Recently, CRTC1 has been shown to be downregulated in Alzheimer’s disease (AD). Nevertheless, the mechanisms underlying CRTC1 dysregulation in AD remain unclear.
Methods
To understand better the epigenetic mechanisms regulating CRTC1 expression that may be altered in AD, we profiled DNA methylation at CpG site resolution by bisulfite cloning sequencing in two promoter regions (referred to as Prom1 and Prom2) of the CRTC1 gene in human hippocampus from controls and AD cases. Next, we correlated DNA methylation levels with AD-related pathology, i.e., β-amyloid and phosphorylated-tau (p-tau) burden and also measured CRTC1 mRNA levels by RT-qPCR.
Results
Methylation levels were lower in AD cases as compared to controls within both promoter regions (Prom1: 0.95 % vs. 5 %, p-value < 0.01 and Prom2: 2.80 % vs. 17.80 %, p-value < 0.001). Interestingly, CRTC1 methylation levels inversely correlated with AD-related neuropathological changes, particularly with p-tau deposition (rSpearman = -0.903, p < 0.001). Moreover, a 1.54-fold decrease in CRTC1 mRNA levels was observed in hippocampus of AD cases compared to controls (p < 0.05) supporting the notion that CRTC1 is downregulated in the AD hippocampus.
Conclusions
DNA methylation levels within two distinct promoter regions of the CRTC1 gene were decreased in human hippocampus affected by AD compared with controls and methylation within Prom1 showed a strong inverse correlation with p-tau deposition. Further studies are guaranteed to elucidate the precise role that CRTC1 methylation plays in AD pathophysiology.
Similar content being viewed by others


Background
CRTC1 (CREB regulated transcription coactivator 1) [EMBL: BC017075] plays a critical role as coactivator of the CREB (cAMP-responsive element binding)-dependent gene transcription pathway [1], which in turn is essential to synaptic plasticity, learning and long-term memory formation in the hippocampus [2–7]. CRTC1 is highly expressed in brain, particularly in hippocampal neurons [8, 9], where it translocates from synapses and dendrites to the nucleus upon the concomitant activation of calcium and cAMP signaling pathways [6, 10, 11]. Moreover, it has been shown that CRTC1 is involved in dendritic growth of developing cortical neurons [12]. The relevance of this gene to brain function is additionally supported by experiments performed on knockout mouse models. Mice lacking Crtc1 develop psychomotor retardation along with neurobehavioral endophenotypes. Interestingly, these mice show reduced dopamine and serotonin turnover in the prefrontal cortex and decreased expression of several downstream genes involved in neuroplasticity [13]. In addition, CRCT1 was recently identified as a modulator of mitochondrial metabolism and longevity [14, 15].
All this evidence makes CRTC1 an intriguing candidate to be examined in neurodegenerative disorders at the molecular level [16]. Indeed, the role of CRCT1 in Alzheimer’s disease (AD) has been previously studied in mouse models. Gene transcription mediated by CRTC1 is impaired in neurons and brain from APP (Sw, Ind) transgenic mice, an AD mouse model overexpressing the human β-amyloid precursor protein (APP) [PDB: E9PG40] carrying human mutations [17, 18]. Remarkably, β-amyloid-induced spatial learning and memory deficits in APP (Sw, Ind) transgenic mice are reversed by restoring a specific subset of CRTC1 target genes [19]. However, studies performed on postmortem human brain samples are still very scarce. In this regard, a recent report shows that CRTC1-dependent genes and CRTC1 protein [PDB: Q6UUV9] levels are significantly reduced in human hippocampus at intermediate and advanced (IV-VI) Braak pathological stages [19]. Nevertheless, the specific mechanisms underlying CRTC1-dependent gene transcriptional disruption in AD still remain unclear.
DNA methylation is a widely studied epigenetic mechanism that contributes to regulate gene expression. The addition of a methyl group to the 5-carbon position of the cytosine-phosphate-guanine (CpG) sites, mostly located at regulatory elements in the genome, modulates the transcription of nearby or related genes. Interestingly, DNA methylation patterns change during the ageing process [20–22] and may have an impact on gene expression in the human brain [23]. In addition, aberrant DNA methylation patterns are involved in an increasing number of common diseases [24, 25], including neurodegenerative conditions. Based on that evidence, we hypothesized that certain regulatory elements of the CRTC1 gene may be differentially methylated in the hippocampus of AD patients. Hence, we aimed to profile DNA methylation levels by bisulfite cloning sequencing in two distinct promoter regions of CRTC1 in hippocampal samples from a cohort of neuropathologically defined “pure” AD cases and controls. We also explored the relationship between DNA methylation levels and AD-related neuropathological changes. Next, CRTC1 mRNA expression was assessed in AD and control hippocampus.
Methods
Human brain samples
We conducted an observational, case-control study comparing postmortem hippocampal samples from AD patients and control hippocampus. Frozen postmortem hippocampal samples from 30 Alzheimer’s disease (AD) cases and 12 controls were provided by the Navarrabiomed Brain Bank. After death, half brain specimens from donors were cryopreserved at −80 °C.
Immunostaining protocol
Formalin-fixed (4 %), paraffin-embedded tissue from mid-hippocampus region was cut (5-mm sections) and placed on StarFrost Microscope Slides. After deparaffinizing, endogenous peroxidase activity was quenched by incubation with 0.3 % (vol/vol) H2O2 in methanol for 30 minutes at room temperature. Antigen retrieval was performed by soaking the sections in 10-mM citrate buffer pH 6.0, heated and boiled for 10 minutes in a microwave oven.
Assessment of β-amyloid deposition was carried out with a mouse monoclonal (S6F/3D) anti β-amyloid antibody (Leica Biosystems Newcastle Ltd, Newcastle upon Tyne, UK) which identified the presence of amyloid pathology.
Evaluation of neurofibrillary pathology was performed with a mouse monoclonal antibody anti-human PHF-TAU, clone AT-8, (Tau AT8) (Innogenetics, Ghent, Belgium), which identified hyperphosphorylated tau (p-tau) [26] in the shape of dystrophic neurites and neurofibrillary tangles. The reaction product was visualized using an automated slide immunostainer (Leica Bond Max) with Bond Polymer Refine Detection (Leica Biosystems, Newcastle Ltd). Omission of the primary antibody was used as a negative control for all stainings.
Neuropathological assessment
Neuropathological examination was completed following the usual recommendations [27] and assessment of AD was performed according to the updated National Institute on Aging-Alzheimer's Association guidelines [26]. Samples were further classified, based on the ABC score, into four groups: control (n = 12), initial AD (including A1B1C1, A1B2C1, A1B2C2, and A1B2C3 scores) (n = 9), intermediate AD (including A2B2C2, A2B2C3, and A2B3C3 scores) (n = 7) and advanced AD (including A3B2C1, A3B2C3, A3B3C2, and A3B3C3 scores) (n = 14) (Additional file 1: Figure S1). Importantly, to avoid spurious associations, those individuals showing coexisting protein deposits different from p-tau or β-amyloid were not eligible for the study. This approach maximizes chances of finding true associations with AD, even though reducing the final sample size. Neuropathological and demographic features of subjects, including age, gender, ABC score and postmortem interval are listed in Additional file 2: Table S1.
Quantitative assessment of β-amyloid and p-tau deposits in brain tissues
In order to quantitatively assess the β-amyloid and p-tau burden for further statistical analysis we applied a method to quantify protein deposits. This method generates a numeric measurement that reflects the extent of β-amyloid and p-tau deposition. Sections of the mid hippocampus were examined after performing immunostaining with anti β-amyloid and anti p-tau antibodies as described above. Three pictures were obtained for each immunostained section using an Olympus BX51 microscope at 10x magnification power. Morphological deposits of β-amyloid, as described by Braak and Braak (neuritic, immature and compact plaque), were manually determined and those areas were further edited and analyzed with ImageJ software [28] (Additional file 1: Figure S2). Next, β-amyloid plaque count referred to as amyloid plaque score (APS) and total area of β-amyloid deposition were automatically measured by ImageJ and averaged for each section. Regarding p-tau deposit, pictures were also analyzed with ImageJ software, by adjusting in the threshold color hue (maximum range: 45–197), saturation (maximum range: 7–243) and brightness (maximum range: 50–195), in order to obtain an averaged quantitative measure of the global p-tau deposit for each section (Additional file 1: Figure S2).
CRTC1 methylation profiling by bisulfite cloning sequencing
Genomic DNA was isolated from hippocampal tissue by standard methods [29]. Next, 500 ng of genomic DNA was bisulfite converted using the EpiTect Bisulfite Kit (QIAGEN, Redwood City, CA, USA) according to the manufacturer’s instructions. Two promoter regions (Prom1 and Prom2) within the CRTC1 gene were amplified by PCR (Additional file 1: Figure S3). Genomic coordinates were obtained from GRCh37/Hg19 assembly. Primer pair sequences were designed by MethPrimer [30] and are listed in Additional file 2: Table S2. PCR products were cloned using the TopoTA Cloning System (Invitrogen, Carlsbad, CA, USA) and a minimum of 12 independent clones were sequenced for each examined subject and region. Methylation graphs were obtained with the QUMA software [31].
CRTC1 mRNA expression analysis
Total RNA was isolated from the 42 hippocampus homogenates using RNeasy Lipid Tissue Mini kit (QIAGEN, Redwood City, CA, USA), following the manufacturer’s instructions. Genomic DNA was removed with recombinant DNase (TURBO DNA-free™ Kit, Ambion, Inc., Austin, TX, USA). RNA integrity was checked by 1.25 % agarose gel electrophoresis under denaturing conditions. Concentration and purity of RNA were both evaluated with a NanoDrop spectrophotometer. Only RNA samples showing a minimum quality index (260 nm/280 nm absorbance ratios between 1.8 and 2.2 and 260 nm/230 nm absorbance ratios higher than 1.8) were included in the study. Complementary DNA (cDNA) was reverse transcribed from 1500 ng total RNA with SuperScript® III First-Strand Synthesis Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) after priming with oligo-d (T) and random primers. RT-qPCR reactions were performed in triplicate with Power SYBR Green PCR Master Mix (Invitrogen, Carlsbad, CA, USA) in an Applied 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Sequences of primer pair were designed using the Real Time PCR tool (IDT, Coralville, IA, USA) and are listed in Additional file 2: Table S2. Relative expression level of CRTC1 mRNA in a particular sample was calculated as previously described [32] and the geometric mean of GAPDH and ACTB genes were used to normalize expression values.
Statistical data analysis
Statistical analysis was performed with SPSS 21.0 (IBM, Inc., USA). Data represent the mean ± SEM or median (range), depending on the type of variable. Differences with p-value < 0.05 were considered significant. Statistical significance for expression and bisulfite intergroup differences were assessed by the Mann-Whitney U test and the Kruskal–Wallis test. Spearman’s rank correlation coefficient was used to determine correlation between AD-related pathology and methylation levels. When multiple comparisons were performed, as was the case of correlation between methylation levels and AD-related pathological burden, a Bonferroni correction was applied and the significance threshold was set at p-corrected value = 0.008. GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla, CA, USA) was used to draw the graphs except for methylation figures that were obtained by QUMA software.
Ethics, consent and permissions
Our study was carried out in accordance with the Declaration of Helsinki and handling of human brain samples was performed according to the current Spanish national legislation (Law 14/2007 and Royal Decree RD1716/2011). The Ethics Committee of the “Complejo Hospitalario de Navarra” approved the use of human subjects for this study (90/2014). Written informed consent was obtained from all subjects or next of kin, previous to brain donation, to perform research projects related to neurodegenerative conditions. The consent form is held by the authors’ institution and is available for review by the Editor-in-Chief.
Results
CRTC1 methylation is decreased in hippocampus of AD cases compared to controls
To begin to ask whether DNA methylation within CRTC1 is altered in AD hippocampus, two different promoters of the CRTC1 gene were examined by bisulfite cloning sequencing. Promoters were identified by using chromatin immunoprecipitation sequencing (ChIP-seq) data from the track Chromatin State Segmentation by HMM (Hidden Markov Model) from ENCODE project and Broad Institute, shown at the UCSC Genome Browser [33]. This track displays chromatin state segmentation data for each of nine human cell types (Fig. 1a, Additional file 1: Figure S3). We designed primers to amplify two independent amplicons. Promoter 1 (Prom1) amplicon was placed at the 5’ end of CRTC1, partially overlapping exon 1 and the beginning of intron 1, and contained 37 CpG sites within the southern shore of a 628 bp-CpG island (chr19:18794192–18794819). Whereas promoter 2 (Prom2) amplicon was located within intron 1 and encompassed 15 CpG sites at the southern shore of a 230 bp-CpG island (chr19:18811562–18811791) (Fig. 1a, Additional file 1: Figure S3). To sum up, a total of 52 individual CpG sites, representing 63.8 % and 88.2 % of each CpG island, respectively, were analyzed in a subset of five control and eight AD cases. To calculate the average methylation level, DNA methylation percentage was measured at CpG site resolution and further averaged across all the CpG sites for each promoter region and subject. Interestingly, we found that average methylation level at Prom1 was decreased in AD cases compared to controls (0.95 % vs. 5 %, p-value < 0.01) (Fig. 1b). Similarly, average methylation level was significantly decreased within Prom2 in AD cases compared to controls (2.80 % vs. 17.80 %, p-value < 0.001) (Fig. 1c).

DNA methylation levels within two promoter regions of CRTC1 in Alzheimer’s disease (AD) and control hippocampus. a The graph shows genomic position of the two amplicons (Prom1 and Prom2) within the two promoter regions of the CRTC1 gene that were examined by bisulfite cloning sequencing. At the bottom of the graph, predicted functional elements are shown for each of nine human cell lines explored by chromatin immunoprecipitation (ChIP) combined with massively parallel DNA sequencing. Boxes represent promoter regions (red), enhancers (yellow), transcriptional transition and elongation (dark green) and weak transcribed regions (light green). CpG islands are also represented by green boxes. The track was obtained from Chromatin State Segmentation by HMM from ENCODE/Broad track shown at the UCSC Genome Browser. Next, dot plot charts and representative examples of the methylation graphs for Prom1 (b) and Prom2 (c), respectively, are shown. A decrease in methylation was observed within both promoters in AD hippocampus compared to controls. Horizontal lines within dot plots represent median methylation values for each group. Boxes on the right represent individual patients. Black and white circles denote methylated and unmethylated cytosines, respectively. Each column symbolizes a unique CpG site in the examined amplicon and each line represents an individual DNA clone. *p-value < 0.05; ** p-value < 0.005. CpG cytosine-phosphate-guanine
CRTC1 methylation correlates with p-tau burden
Next, we aimed to correlate DNA methylation levels with AD-related neuropathological changes recorded from the hippocampus sections. In brief, p-tau and β-amyloid burden were measured and averaged by using the ImageJ software (Additional file 1: Figure S2). The amyloid plaque score (APS) was also recorded (Additional file 2: Table S1). Notably, the average methylation level within Prom 1 showed a strong inverse correlation with p-tau deposition (rSpearman = −0.903, p < 0.001). Although we found a correlation between Prom1 average methylation level and β-amyloid burden (Table 1), the p-value for this correlation did not reach the Bonferroni-corrected significance threshold (corrected p-value = 0.008). Likewise, average methylation level within Prom2 was no longer correlated with p-tau deposition and β-amyloid burden after applying the Bonferroni correction (Table 1).
CRTC1 mRNA levels are downregulated in Alzheimer’s disease hippocampus
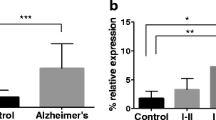
To test if CRTC1 was differentially expressed in our cohort, we measured CRTC1 mRNA expression levels by RT-qPCR in hippocampal samples from Alzheimer’s disease (AD) cases and controls. Four samples did not pass the RNA quality threshold and so were not included in the experiments (see CRTC1 mRNA expression analysis in the Methods section). Eventually, 26 AD cases were compared to 12 controls. RT-qPCR reactions were performed in triplicate for each sample and repeated twice within independent cDNA sets. Moreover, two different sets of primers (CRTC1-qPCR1 and CRTC1-qPCR2) were used to increase the reliability of the experiment (Additional file 2: Table S2). A 1.54-fold decrease in CRTC1 mRNA levels was observed in the hippocampus of AD cases compared to controls (p < 0.05) (Fig. 2a). Next, a disease-staging analysis was conducted to investigate changes of CRTC1 mRNA levels across AD stages. We found that CRTC1 mRNA levels significantly decreased across AD stages (p < 0.05) and post-hoc analysis showed that CRTC1 expression was significantly reduced at advanced stages compared to controls (p < 0.05) (Fig. 2b).

CRTC1 mRNA expression is decreased in human hippocampus in Alzheimer’s disease (AD). a The graph shows a significant 1.54-fold decrease in CRTC1 mRNA levels in AD hippocampal samples compared to control hippocampal samples. b CRTC1 mRNA expression decreases across AD stages, as shown when CRTC1 expression levels are sorted based on ABC score. Boxes represent percentage of CRTC1 expression relative to the geometric mean of GAPDH and ACTB housekeeping genes expression. Bars represent the standard error of the mean. *p-value < 0.05; ** p-value < 0.005. c Two different CRTC1 transcript variants are shown. CRTC1 variant 1 has 14 exons and CRTC1 variant 3 shares all the exons of variant 1 plus an additional exon 3. Black boxes represent exons, arrows represent transcription start sites and white boxes denote CpG islands. CpG cytosine-phosphate-guanine
We further tested if differences in CRTC1 mRNA expression resulted from a decrease in a particular type of CRTC1 transcript variant. Two different CRTC1 transcripts were identified in the Reference Sequence (RefSeq) database, i.e., CRTC1 transcript variant 1 (NM_015321) and transcript variant 3 (NM_001098482), sharing all the exons but for an additional exon (exon 3) belonging to CRTC1 variant 3 (Fig. 2c). Due to the extra exon 3, we were able to design a primer pair to selectively amplify CRTC1 variant 3 (Additional file 2: Table S2), whereas a primer pair to amplify only CRTC1 variant 1 was not able to be designed since all the exons of this variant are shared with CRTC1 variant 3. We found that levels of CRTC1 variant 3 mRNA were not different between AD cases and controls (p = 0.206), suggesting that significant differences in global CRTC1 mRNA levels between AD cases and controls might result from a reduction in CRTC1 variant 1 mRNA.
Finally, we studied the relationship between promoter methylation and mRNA expression for the CRTC1 gene. We tested the correlation between DNA methylation levels and mRNA expression levels. In order to avoid potential bias from disease status, we tested the potential correlation separately in two groups, controls and AD patients. Interestingly, CRTC1 mRNA expression showed a trend to be inversely correlated with Prom1 methylation levels (rSpearman = −0.949, p = 0.051) in the control group; meanwhile, no significant correlation was found between methylation and expression among AD patients (rSpearman = −0.287, p = 0.490).
mRNA levels of CRTC1 downstream target genes are altered in Alzheimer’s disease hippocampus
We also wanted to test whether CRTC1-dependent genes were also dysregulated in our set of AD hippocampal samples. To this end, we measured mRNA levels of BDNF, c-fos, EGR4, and EGR1 by RT-qPCR in our set of samples. Primer pairs and specific transcripts that were amplified by RT-qPCR are shown in Additional file 2: Table S2.
Interestingly, we found significantly decreased mRNA levels of BDNF, EGR4, and EGR1 in AD hippocampus compared to control hippocampus (p-value < 0.05) (Additional file 1: Figure S4). These results are in accordance with previous reports based on AD in vitro models, murine models and human brain. On the contrary, c-fos mRNA expression was significantly increased in AD cases compared to controls in our set of samples (p-value < 0.005) (Additional file 1: Figure S4).
Discussion
Here we report altered patterns of DNA methylation within two distinct promoter regions of CRTC1 in the human hippocampus affected by Alzheimer’s disease (AD). Interestingly, DNA methylation levels are decreased in AD and inversely correlate with p-tau burden in the hippocampus.
Our results indicate that important regulatory regions for the CRTC1 gene are differentially methylated in the AD hippocampus compared to controls. It is well known that CRTC1 is mainly expressed in the CA1 region of the hippocampus [8] and is essential to synaptic plasticity and memory consolidation [2–7]. Moreover, studies conducted on AD mouse models highlight its relevance to AD pathogenesis [3, 17–19] and CRTC1 protein has been found to be downregulated in the human hippocampus affected by AD [19]. Thus, studying regulatory mechanisms of CRTC1 gene expression in the human hippocampus, one of the more vulnerable regions to AD pathology, is of utmost interest. Our finding suggests that altered patterns of methylation at regulatory regions might contribute to disruption of the CRTC1 gene function that was previously described in AD [19], as DNA methylation is one of the most important epigenetic mechanisms that regulate gene expression. Most interestingly, methylation levels correlated to AD-related pathology changes in our cohort. In particular, a strong inverse correlation was found between DNA methylation levels and p-tau burden in the hippocampus (rSpearman = −0.903, p < 0.001). It is of note that neurofibrillary tangles, which are aggregates of p-tau protein that accumulate within neurons, are found in the first stages of AD restricted to the hippocampus and entorhinal cortex, while the other hallmark of AD, β-amyloid deposition, begins to accumulate in the neocortex. From our data showing such a strong correlation, we may hypothesize that altered methylation within CRTC1 may be part of the molecular signature of p-tau pathology in the hippocampus. Anyhow, the relationship between CRTC1 gene and p-tau pathology merits further investigations.
We also found a mild but otherwise significant decrease in CRTC1 mRNA expression in AD hippocampus compared to controls (Fig. 2). This finding is in concordance with a previous report that showed both total and phosphorylated CRTC1 protein downregulation in human hippocampus affected by AD [19]. Moreover, CRTC1 mRNA expression has been found to be decreased in other neurodegenerative conditions and brain diseases, such as frontotemporal lobar degeneration with progranulin mutations [34] and Down syndrome [35] (Additional file 1: Figure S5). Down syndrome is considered a natural model for AD, since most people affected by Down syndrome will develop early AD [36]. Interestingly, a significant decrease in CRTC1 mRNA levels was shown for CRTC1 transcript variant 1 (NM_015321) but not for CRTC1 transcript variant 3 (NM_001098482) in Down syndrome adult brain [35] (Additional file 1: Figure S5), which is in line with our present work.
To understand better the relationship between methylation and expression in our study we tested the correlation between CRTC1 methylation and mRNA levels. Among control hippocampus samples, we found an inverse relationship between methylation and expression at the Prom1 region. In other words, lower levels of DNA methylation within Prom1 showed a trend to be correlated with higher levels of CRTC1 mRNA (rSpearman = −0.949, p = 0.051). This correlation is consistent with the classical model of epigenetic regulation where loss of methylation in the promoter region results in an increase in gene expression, indicating that CRTC1 is probably regulated by DNA methylation under normal circumstances. However, no correlation between methylation and expression was identified among AD patients in our analysis, suggesting that proper functioning of DNA methylation in regulating CRTC1 expression may be disturbed in the AD context.
Finally, we show significantly decreased mRNA levels of several CRTC1-dependent genes, such as BDNF, EGR4, and EGR1 in AD hippocampus compared to control hippocampus (p-value < 0.05) (Additional file 1: Figure S4), which adds consistency to the present study. These results are in accordance with previous reports performed on AD in vitro models, murine models and human brain. For instance, oligomeric Aβ peptide downregulates BDNF levels via Aβ-induced CREB transcriptional downregulation in human neuroblastoma (SH-SY5Y) cells [34]. In murine models, it has been largely demonstrated that several CRTC1-dependent genes are downregulated in the brain tissue affected by AD pathology, including BDNF and EGR4 [17, 18, 35]. Most interestingly, not only are BDNF mRNA levels decreased in AD human frontal cortex but also BDNF mRNA levels correlate with cognitive performance of patients, assessed by Mini-Mental State Examination scores, as was shown in a recent report [36].
On the other hand, c-fos mRNA expression was significantly increased in AD cases compared to controls in our set of samples (p-value < 0.005) (Additional file 1: Figure S4). Remarkably, this is in good agreement with the results obtained from earlier studies that used AD human hippocampus to assess c-fos expression [37-39] but, in turn, differs from several reports that have shown c-fos mRNA levels to be decreased in AD murine models [17, 18, 35]. This discrepancy among the results obtained from murine and human studies regarding c-fos mRNA expression in the context of AD pathology will need further research.
Conclusions
In conclusion, this study provides evidence that CRTC1 methylation levels are altered in the human hippocampus affected by AD. It also suggests that the functional relationship between DNA methylation and gene expression for CRTC1 may be altered in the AD context. Further studies are needed to better understand the precise role that CRTC1 methylation plays in AD pathophysiology.
Abbreviations
- AD:
-
Alzheimer’s disease
- APP:
-
amyloid precursor protein
- APS:
-
amyloid plaque score
- BDNF :
-
Brain-derived Neurotrophic Factor
- bp:
-
base pair
- cDNA:
-
complementary DNA
- ChIP-seq:
-
chromatin immunoprecipitation sequencing
- CpG:
-
cytosine-phosphate-guanine dinucleotide
- CREB :
-
cAMP-responsive element binding
- CRTC1 :
-
CREB regulated transcription coactivator 1
- EGR1 :
-
Early growth response protein 1
- EGR4 :
-
Early growth response protein 4
- ENCODE:
-
Encyclopedia of DNA Elements
- GEO:
-
Gene Expression Omnibus
- HMM:
-
Hidden Markov Model
- mRNA:
-
messenger RNA
- PCR:
-
polymerase chain reaction
- Prom 1:
-
promoter number 1
- Prom2:
-
promoter number 2
- p-tau:
-
hyperphosphorylated tau
- RT-qPCR:
-
real time-qPCR
- UCSC:
-
University of California, Santa Cruz
References
Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, et al. TORCs: transducers of regulated CREB activity. Mol Cell. 2003;12(2):413–23.
Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79(1):59–68.
Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1(7):595–601. doi:10.1038/2830.
Guzowski JF, McGaugh JL. Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Acad Sci U S A. 1997;94(6):2693–8.
Benito E, Valor LM, Jimenez-Minchan M, Huber W, Barco A. cAMP response element-binding protein is a primary hub of activity-driven neuronal gene expression. J Neurosci. 2011;31(50):18237–50. doi:10.1523/jneurosci.4554-11.2011.
Kovacs KA, Steullet P, Steinmann M, Do KQ, Magistretti PJ, Halfon O, et al. TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104(11):4700–5. doi:10.1073/pnas.0607524104.
Sekeres MJ, Mercaldo V, Richards B, Sargin D, Mahadevan V, Woodin MA, et al. Increasing CRTC1 function in the dentate gyrus during memory formation or reactivation increases memory strength without compromising memory quality. J Neurosci. 2012;32(49):17857–68. doi:10.1523/jneurosci.1419-12.2012.
Kang MG, Byun K, Kim JH, Park NH, Heinsen H, Ravid R, et al. Proteogenomics of the human hippocampus: the road ahead. Biochim Biophys Acta. 2015;1854(7):788–97. doi:10.1016/j.bbapap.2015.02.010.
Watts AG, Sanchez-Watts G, Liu Y, Aguilera G. The distribution of messenger RNAs encoding the three isoforms of the transducer of regulated cAMP responsive element binding protein activity in the rat forebrain. J Neuroendocrinol. 2011;23(8):754–66. doi:10.1111/j.1365-2826.2011.02178.x.
Zhou Y, Wu H, Li S, Chen Q, Cheng XW, Zheng J, et al. Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS One. 2006;1:e16. doi:10.1371/journal.pone.0000016.
Ch'ng TH, Uzgil B, Lin P, Avliyakulov NK, O'Dell TJ, Martin KC. Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell. 2012;150(1):207–21. doi:10.1016/j.cell.2012.05.027.
Li S, Zhang C, Takemori H, Zhou Y, Xiong ZQ. TORC1 regulates activity-dependent CREB-target gene transcription and dendritic growth of developing cortical neurons. J Neurosci. 2009;29(8):2334–43. doi:10.1523/jneurosci.2296-08.2009.
Breuillaud L, Rossetti C, Meylan EM, Merinat C, Halfon O, Magistretti PJ, et al. Deletion of CREB-regulated transcription coactivator 1 induces pathological aggression, depression-related behaviors, and neuroplasticity genes dysregulation in mice. Biol Psychiatry. 2012;72(7):528–36. doi:10.1016/j.biopsych.2012.04.011.
Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, et al. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15(5):713–24. doi:10.1016/j.cmet.2012.04.007.
Burkewitz K, Morantte I, Weir HJ, Yeo R, Zhang Y, Huynh FK, et al. Neuronal CRTC-1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell. 2015;160(5):842–55. doi:10.1016/j.cell.2015.02.004.
Xue ZC, Wang C, Wang QW, Zhang JF. CREB-regulated transcription coactivator 1: important roles in neurodegenerative disorders. Sheng Li Xue Bao. 2015;67(2):155–62.
Espana J, Valero J, Minano-Molina AJ, Masgrau R, Martin E, Guardia-Laguarta C, et al. beta-Amyloid disrupts activity-dependent gene transcription required for memory through the CREB coactivator CRTC1. J Neurosci. 2010;30(28):9402–10. doi:10.1523/jneurosci.2154-10.2010.
Saura CA. CREB-regulated transcription coactivator 1-dependent transcription in Alzheimer's disease mice. Neurodegener Dis. 2012;10(1-4):250–2. doi:10.1159/000333341.
Parra-Damas A, Valero J, Chen M, Espana J, Martin E, Ferrer I, et al. Crtc1 activates a transcriptional program deregulated at early Alzheimer's disease-related stages. J Neurosci. 2014;34(17):5776–87. doi:10.1523/jneurosci.5288-13.2014.
Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A. 2012;109(26):10522–7. doi:10.1073/pnas.1120658109.
Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13(10):R97. doi:10.1186/gb-2012-13-10-r97.
Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15(2):R24. doi:10.1186/gb-2014-15-2-r24.
Hernandez DG, Nalls MA, Gibbs JR, Arepalli S, van der Brug M, Chong S, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20(6):1164–72. doi:10.1093/hmg/ddq561.
Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012. doi:10.1038/nrg3270.
Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol. 2013;20(3):274–81. doi:10.1038/nsmb.2518.
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. doi:10.1007/s00401-011-0910-3.
Bell JE, Alafuzoff I, Al-Sarraj S, Arzberger T, Bogdanovic N, Budka H, et al. Management of a twenty-first century brain bank: experience in the BrainNet Europe consortium. Acta Neuropathol. 2008;115(5):497–507. doi:10.1007/s00401-008-0360-8.
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–31.
Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 2008;36(Web Server issue):170–5. doi:10.1093/nar/gkn294.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi:10.1006/meth.2001.1262.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi:10.1101/gr.229102.
Chen-Plotkin AS, Geser F, Plotkin JB, Clark CM, Kwong LK, Yuan W, et al. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum Mol Genet. 2008;17(10):1349–62. doi:10.1093/hmg/ddn023.
Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90(6):647–60. doi:10.1016/j.ygeno.2007.08.005.
Hartley D, Blumenthal T, Carrillo M, DiPaolo G, Esralew L, Gardiner K, et al. Down syndrome and Alzheimer's disease: common pathways, common goals. Alzheimers Dement. 2015;11(6):700–9. doi:10.1016/j.jalz.2014.10.007.
Acknowledgements
This work was supported by the Spanish Government through a grant from the Institute of Health Carlos III (FIS PI13/02730), jointly funded by European Regional Development Fund (ERDF), European Union, “A way of shaping Europe”; the Regional Basque Government through a grant from The Basque Foundation for Health Innovation and Research (BIOEF) (BIO12/ALZ/007), a grant from Fundación Caja-Navarra and the Trans-Pyrenean Biomedical Research Network (REFBIO). NC was also supported by REFBIO funding. Sponsors did not play any role in design, in the collection, analysis, and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
We want to kindly thank Teresa Tuñón M.D., Ph.D. (Department of Pathology, Complejo Hospitalario de Navarra, technical support), Federico García-Bragado M.D., Ph.D. (Department of Pathology, Complejo Hospitalario de Navarra, technical support) and Isabel Gil M.D. (Navarrabiomed BrainBank, technical support) for their help. Finally, we are very grateful to the patients and relatives that generously donor the brain tissue to the Navarrabiomed Brain Bank.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MM conceived the study, participated in its design and coordination, obtained funds, analyzed data and contributed to writing/revising the manuscript. NC participated in designing parts of the study and contributed to drafting/revising the manuscript for content. MA participated in acquisition of data, revised subject diagnosis and contributed to drafting/revising the manuscript for content. JSR contributed to drafting/revising the manuscript for content, analysis and interpretation of data (p-tau and amyloid deposits), sorting of patients into different stages and acquisition and analysis of image data. MVZ participated in the study design, acquired data, revised subject diagnosis, classified patients, and contributed to drafting/revising the manuscript for content. MR contributed to design gene maps, performed bisulfite experiments, was involved in the interpretation of data, and revised the methods section of the manuscript. IR contributed to drafting/revising the manuscript for content and acquisition of data. RL contributed to acquisition of data, revised subject diagnosis and revised the draft. EE made substantial contribution to drafting/revising the manuscript for content with significant contribution to patients’ staging and p-tau analysis. IM contributed to the study design, to obtaining funding and drafting/revising the manuscript for content. CE contributed to the study design, acquisition of data, sorting of samples, diagnosis of subjects and drafting/revising the manuscript for content. All authors read and approved the final manuscript.
Additional files
Additional file 1: Figure S1.
Pictures obtained at 10x from the most representative cases showing different degrees of protein deposit. Figure S2. Beta-amyloid and tau protein measurement in hippocampal sections. Figure S3. Maps of the two CRTC1 regions analyzed by bisulfite sequencing cloning in human hippocampus. Figure S4. mRNA expression levels of CRTC1 downstream genes in human AD hippocampus compared to controls. Figure S5. Gene Expression Omnibus (GEO) data analysis for CRTC1 mRNA expression levels in Down syndrome and Frontotemporal Lobar Degeneration with progranulin mutations. (PDF 974 kb)
Additional file 2:
Supplemental Tables. Table S1. Brain sample set characteristics. Table S2. Bisulfite and quantitative-PCR primers. (PDF 308 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mendioroz, M., Celarain, N., Altuna, M. et al. CRTC1 gene is differentially methylated in the human hippocampus in Alzheimer’s disease. Alz Res Therapy 8, 15 (2016). https://doi.org/10.1186/s13195-016-0183-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-016-0183-0