
Abstract
Alzheimer’s disease (AD) prevalence is twice as high in non-Hispanic Blacks (NHBs) as in non-Hispanic Whites (NHWs). The objective of this study was to determine whether aberrant methylation at imprint control regions (ICRs) is associated with AD. Differentially methylated regions (DMRs) were bioinformatically identified from whole-genome bisulfite sequenced DNA derived from brain tissue of 9 AD (5 NHBs and 4 NHWs) and 8 controls (4 NHBs and 4 NHWs). We identified DMRs located within 120 regions defined as candidate ICRs in the human imprintome (https://genome.ucsc.edu/s/imprintome/hg38.AD.Brain_track). Eighty-one ICRs were differentially methylated in NHB-AD, and 27 ICRs were differentially methylated in NHW-AD, with two regions common to both populations that are proximal to the inflammasome gene, NLRP1, and a known imprinted gene, MEST/MESTIT1. These findings indicate that early developmental alterations in DNA methylation of regions regulating genomic imprinting may contribute to AD risk and that this epigenetic risk differs between NHBs and NHWs.
Similar content being viewed by others


Introduction
More than six million Americans are affected by Alzheimer’s disease (AD) [1], which is the most common form of dementia (60% to 80% of cases) [2, 3], and it is now the sixth leading cause of death in the USA [3]. Additionally, AD places a tremendous burden not only on the patients, but also on the caregivers and the healthcare system. AD is a disease of cognitive changes, but also increases susceptibility to multiple comorbidities, including pneumonia, femur fractures, and increased mortality risk [3]. This neurodegenerative disease is associated with cell death and atrophy involving various brain regions, which progress along anatomically connected networks, starting in the entorhinal cortex and medial temporal lobes, and extending into the neocortex over time. Although it is now accepted that this neuropathology is characterized by the aggregation of extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) composed of the hyperphosphorylated tau protein [4], the etiologic factors contributing to AD are still largely unknown. Established risk factors for AD, including advanced age, familial history, genetics, history of head trauma, and cardiovascular diseases do not fully explain the formation of Aβ plaques and NFTs [1].
A modest number of genetic variants derived from hypothesis-driven and agnostic approaches have been associated with AD. For familial AD, which affects ~ 5% of cases, genetic risk factors include mutations in genes such as apolipoprotein E (APOE; Chr19) [5, 6], amyloid precursor protein (APP; Chr21) [7, 8], presenilin 1 (PSEN1; Chr14) [7, 9], presenilin 2 (PSEN2; Chr1) [7, 10,11,12], and beta-site APP-cleaving enzyme 1 (BACE1; Chr11) [13, 14]. APOE contributes to both familial and sporadic AD, and has three variants, ε2, ε3, and ε4 [5, 15] of which ε4 is the most prevalent isoform found in AD cases [16]. These genetic risk factors are often associated with abnormal protein function (i.e., proteinopathies), which is believed to play a significant role in familial AD [16].
However, up to 95% of the disease is estimated to be sporadic [16, 17]. Such cases share common neuropathological endpoints with familial AD, including Aβ plaque accumulation, NFTs, synaptic loss, excess inflammation, oxidative damage, and neuronal death [18]. While genetic factors, such as APOE variants, appear to influence sporadic AD through intricate interplay with each other and environmental influences, it is important to note that they are neither necessary nor sufficient for the development of AD [17]. This has led to the emerging hypothesis that environmental stressors accumulated over the life course contribute to the later development and progression of AD [19].
Globally, there is a slight geographic variation in AD prevalence [20]. Among Americans aged 65 years and older, the risk of AD is twofold higher in non-Hispanic Blacks (NHBs) and 1.5-fold higher in Hispanics compared to non-Hispanic Whites (NHWs) [1]. The causes for this disparate outcome are presently unclear. Although a single nucleotide polymorphism (rs115550680) of ABCA7, which regulates lipid transport [21], was recently associated with late-onset AD in a NHB population but not NHW or Hispanics [22], this known genetic variant does not fully explain the higher disease burden in NHBs. A plausible hypothesis to account for the elevated rates of disease in NHB and Hispanic populations is that environmental or life course stressors, such as migration and segregation [23], inadequate medical surveillance, and living in polluted environments, are more common in these populations and result in functional and enduring alterations in the epigenome [24, 25]. Thus, profiling epigenetic marks that link established risk factors to AD holds promise for early detection, and for identifying novel mechanistic pathways contributing to AD.
Epigenetic dysregulation, which can cause alterations in gene expression in response to environmental stressors, may cause long-term changes in molecular pathways contributing to AD. Indeed, it was recently documented that the average 5-methylcytosine level is decreased in the entorhinal cortex of individuals with AD compared to that in controls [26]. DNMT1, a critical factor in the maintenance of DNA methylation, and MeCP1/MBD2, components of the methylation complex, are also significantly decreased in the entorhinal cortex of AD individuals compared to controls [27]. Furthermore, brain-derived neurotrophic factor (BDNF), which functions in cortical neuron maintenance, has increased promoter CpG methylation in both AD brain tissue and blood [28], in support of similarities between methylation patterns in the blood and brain tissues of AD and other dementia patients [29, 30]. Nevertheless, the interpretation of these data is complicated by lack of replication, and the possibility that methylation levels may change between tissue/cell type and throughout life.
In epidemiological studies, DNA methylation is frequently identified in accessible peripheral blood [31,32,33,34], but because these epigenetic marks can differ between tissues and cell types, they do not always correlate with those from inaccessible cells of affected brain regions. Moreover, because epigenetic marks respond to various environmental cues throughout life, causality is difficult to discern. One exception to these issues is the repertoire of methylation marks controlling genomic imprinting—the human imprintome—which epigenetically regulates the expression of imprinted genes crucial to tissue development during the intrauterine period [35]. After fertilization, DNA methylation of imprint control regions (ICRs) in the primordial germ cells (PGCs) undergoes complete erasure, and after sex determination, these regions are remethylated in a sex-dependent manner. This time frame is a window of high susceptibility to epigenetic perturbations due to environmental exposures and stressors that can alter the methylation of these ICRs in PGCs [36, 37]. PGCs with aberrant methylation can then transfer altered gene expression to the next generation and because of mitotic heritability, this aberrant methylation is conserved in all cell types and tissues in the offspring resulting in altered health effects over the life course [38], and increased susceptibility in adulthood to diseases, such as AD.
Thus, the complete mapping of the human imprintome that is susceptible to environmentally influenced alterations is key to understanding the non-genetic factors in complex diseases [35]. It is also important to distinguish between the non-imprinted epigenetic-controlled regulatory sites, and the ICRs involved in regulating the parental-dependent expression of imprinted genes. DNA methylation patterns at non-imprinted sites are cell type-specific and can be responsive to environmental cues throughout life. In contrast, the inherited ICRs, or the somatic ICRs that occur at the stem cell stage of embryonic development, should have the same stable methylation status across all tissues throughout life, including peripheral blood cells and the brain [39]. As changes in the brain are likely to start decades before clinical symptoms of AD appear [40, 41], the purpose of this study was to use genome-wide approaches to comprehensively identify dysregulated ICRs [35] associated with AD that trace their origin to adverse events in early development. The consistency of imprinted methylation marks across tissues and cell types makes them attractive as early epigenetic biomarkers for AD obtainable from accessible tissues.
Results
Patient and sample characteristics
Characteristics of the 17 NHW and NHB individuals who donated the brain samples used in this study are shown in Additional file 1: Table S1. AD cadavers ranged in age from 63 years to > 89 years (median 84 years), and controls ranged in age from 56 to 87 years (median 74 years). All nine AD samples were obtained from the temporal cortex. Five of eight control brain samples were obtained from the temporal cortex while three were obtained from the cerebellum. The diagnosis of AD was made postmortem through a comprehensive neuropathologic evaluation (Additional file 1: Table S1). The brain tissues used in this study were obtained from the Joseph and Kathleen Bryan Brain Bank at Duke University, which has historical significance, as it contained brain samples that were instrumental in the discovery of the association between APOE-ε4 and late onset and sporadic Alzheimer's disease [5, 6].
Association of ICRs with Alzheimer’s disease
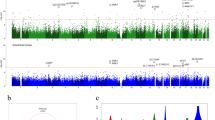
Bisulfite conversion rate of AD and control samples taken from individuals of NHBs and NHWs showed a > 97% bisulfite conversion in all sample groups. Quality controls revealed no sequence duplication bias (Additional file 1: Fig. S1a), and sequence coverage between 15X and 36X (Additional file 1: Fig. S1b, c). We identified the CpG methylation ratio from replicate bam files for AD samples and controls. Later, differentially methylated regions (DMRs) were called using model-based analysis of bisulfite sequencing data criteria (MOABS, version 1.3.8.7) [42]. We performed three different analyses to identify AD-associated DMRs (Additional file 2: Table S2) using a 10% differential methylation threshold, a minimum read depth \(\ge\) 7, and a maximum distance between consecutive CpG’s \(\le\) 300, consistent with MOABS [42]. The resulting set of AD-related DMRs were analyzed against the 1488 candidate ICRs reported by our group [43], resulting in the identification of 120 candidate ICRs, including four of the 25 confirmed ICRs, that exhibit differential methylation in AD patients compared to controls (Fig. 1a, Table 1). Stratified by group, I. 40 (33.3%) differentially methylated ICRs are observed between all AD samples (n = 9) and controls (n = 8), II. 81 (67.5%) are observed between NHB-AD cases (n = 5) and controls (n = 4), and III. 27 (22.5%) are observed between NHW-AD cases (n = 4) and controls (n = 4). Interestingly, our results indicate that NHBs exhibit a threefold increase relative to NHWs in AD-associated differential methylation of regions postulated to be ICRs (Fig. 1a, Table 1). Alignment of AD-related DMRs and candidate ICRs can be accessed at https://genome.ucsc.edu/s/imprintome/hg38.AD.Brain_track.

AD-associated candidate ICRs in NHBs and NHWs. a DMRs that differed in DNA methylation (≥ 10%) between AD cases and controls in NHBs and NHWs were determined by WGBS. b Venn diagram of ICRs from ALL [40], NHB [81] and NHW [27] when DNA methylation differed by ≥ 10% between AD cases and controls. c Venn diagram of ICRs from ALL [10], NHB [32] and NHW [10] when DNA methylation differed by ≥ 15% between AD cases and controls. Created with BioRender.com
Notably, the AD-associated ICRs we identified are plausible targets for AD pathogenesis. For example, we found that ICR_20, near CASZ1 (Fig. 2a) and ICR_1027, near RBFOX3 (Fig. 2b) were differentially methylated in only NHBs. Methylation of DMRs overlapping ICR_20 (CDIF: 0.227, p-value: 1.07E-33) and ICR_1027 (CDIF: 0.208, p-value: 1.68E-25; CDIF: 0.166, p-value: 1.85E-16) were increased more than 10% in AD cases when compared to controls (Table 2). The CASZ1 gene encodes the Castor zinc finger 1 protein involved in neuronal differentiation [44]. An in vitro study demonstrated a gain of 5mC in the CASZ1 region (chr1:10732049–10732050) in AD neurons compared to wildtype cells [45]. However, our results showed hypermethylation in AD brain samples compared to controls, in NHBs (chr1:10682586–10683160) and in ALL group (chr1:10682972–10683160) overlapping the CASZ1 ICR (Table 1, Additional file 2: Table S2). RBFOX3 encodes NeuN [46, 47], and is expressed in approximately 68% of cells in the gray matter of the cerebral cortex [47, 48]. Human brains affected by AD have decreased RBFOX3 expression in the hippocampus when compared to non-AD brains [47]. Additionally, in a mouse study, RBFOX3 was found to be developmentally regulated, and its expression is reported to coincide with 4R-tau expression resulting from alternative splicing of tau exon 10 [49].

Race/ethnicity dependent ICRs in AD. a ICR_20 (CASZ1) and b ICR_1027 (RBFOX3) differed by ≥ 10% in DNA methylation between AD cases and controls only in NHBs. Candidate ICRs (horizontal red boxes) are delineated by vertical dashed red lines. The candidate ICRs were previously defined by having 5 or more consecutive CpGs with methylation levels of 50% ± 15% (green dots) for tissues in all three germ layers (i.e. brain, kidney, and liver); methylation levels for sperm and oocytes are also shown (i.e. ≥ 90% methylation—yellow dots and ≤ 10% methylation—blue dots) [35]
Only two ICRs (ICR_481, chr7:130490640-130494200 and ICR_987, chr17:5771207-5771575), proximal to MEST/MESTIT1 and NLRP1, respectively, were differentially methylated in both NHBs and NHWs (Fig. 1b and Fig. 3a, b). Methylation of ICR_481 (CDIFs: -0.159, -0.155, -0.137, and -0.118, p-values: 1.08E-09, 2.02E-14, 2.16E-09, and 7.50E-09) is decreased, whereas methylation of ICR_987 (CDIFs: 0.138, 0.218, and 0.257, p-values: 4.64E-21, 1.26E-09, and 2.41E-50) is increased more than 10% in AD cases when compared to controls (Table 2). MEST is a paternally expressed imprinted gene and highly expressed in mesoderm and adult brain [50]. It is linked to intrauterine growth retardation and abnormal maternal behavior in adult mice [51]. In humans, the maternal uniparental disomy related to imprinting at the PEG1/MEST region located at 7q32 causes Silver-Russell syndrome [52]. The NLR family pyrin domain-containing 1 (NLRP1) inflammasome is widely expressed in humans [53]. In the central nervous system, it is primarily expressed by pyramidal neurons and oligodendrocytes where overexpression triggers caspase 1 and 6 activation, eventually leading to axonal degeneration and neuronal death by pyroptosis, an inflammatory form of programmed cell death [53, 54]. Repeating these analyses using a more stringent cutoff of 15% for a methylation difference reduced the total number of AD-associated ICRs from 120 to 45. Furthermore, it is noteworthy that ICR_987 maintained its differential methylation status in both NHBs and NHWs under the more stringent conditions. On the other hand, with a 15% cutoff ICR_481 was differentially methylated in only NHWs (Fig. 1c and Table 1).

Race/ethnicity independent ICRs in AD. a ICR_481 (MEST/MESTIT1) and b ICR_987 (NLRP1) differed by ≥ 10% in DNA methylation between AD cases and controls in both NHBs and NHWs. Candidate ICR (horizontal red box) and a known ICR (horizontal yellow box) is delineated by vertical dashed red lines. The candidate ICR was previously defined by having 5 or more consecutive CpGs with methylation levels of 50% ± 15% (green dots) for tissues in all three germ layers (i.e., brain, kidney, and liver); methylation levels for sperm and oocytes are also shown (i.e., ≥ 90% methylation—yellow dots and ≤ 10% methylation—blue dots) [35]
When we constrained the analysis of the 1488 ICRs, specifically to the 332 for which gametic methylation patterns were available [35], we did not observe differences that would affect the proportional differences between AD and controls in both NHBs and NHWs. For example, we identified 37 of 332 candidate ICRs associated with AD, with 31 ICRs found in NHBs and two ICRs found in NHWs (Additional file 1: Table S3). Notably, we again observed a common ICR, ICR_481 (MEST/MESTIT1), which exhibited differential methylation in AD brains compared to controls. Despite observing a reduced number of DMRs associated with AD when constraining the analysis to the 332 ICRs for which there is gametic data, the demonstration of parental origin of methylation strengthens our confidence in the 37 identified differentially methylated regions as robust candidates for regulatory regions in the development and progression of AD.
We identified 106 genes in closest proximity to the 120 AD-associated ICRs (i.e., between 0 and 186,698 bp; average 11,270 bp) (Table 1). This is well within the range of known imprinted domains. For example, the H19/IGF2 and KCNQ1 imprinted domain is about 1.4 Mb long; that of MEST/MESTIT1 is around 4.0 Mb; and that of the NPP5F_v2 is nearly 8.6 Mb DNA [55].
When stratified by race/ethnicity, there were 85 NHB-AD associated ICRs and 26 were linked to NHW-AD associated ICRs. Network analysis conducted separately on the 85 ICRs in NHBs and 26 ICR in NHWs using ingenuity pathway analysis (IPA) unveiled shared functions such as cell signaling, cellular development, embryonic development, and organ development in both NHBs and NHWs (Additional file 1: Table S4, S5). Interestingly, two pathways, namely white adipose tissue browning and gap junction signaling, were also identified as common features in both NHBs and NHWs. On the other hand, the netrin signaling pathway, known to regulate axonal growth, was found only in NHBs.
The 106 genes linked to the 120 AD-associated candidate ICRs contained 16 previously known imprinted genes [56] (http://www.geneimprint.com) in close proximity to 13 AD-associated candidate ICRs. Of these imprinted genes, nine (MEST [57, 58], MESTIT1 [58], PEG13 [59], SNRPN [60], SNURF [61], ZIM2 [62, 63], PEG3 [62, 63], MIMT1 [63], NNAT [64]) are paternally expressed, five (SVOPL [65], H19 [66], MEG3 [67], ZNF597 [68], NAA60 [68]) are maternally expressed, and two (i.e., BLCAP [69], GNAS [70]) have isoform-dependent expression (71). This indicates that monoallelic parent-of-origin expression can present in some gene transcripts (i.e., isoforms), but not in others.
lncRNAs and microRNAs analyses
Many known epigenetic regulators of gene expression include lncRNAs and microRNAs (miRNAs). Thus, we examined the lncRNAs associated with the 120 ICRs linked to AD using LncExpDB [72]. We found 9 lncRNAs (BABAM2-AS1, LINC01101, HTR5A-AS1, PEG13, FAM27E3, H19, PCCA-DT, SNHG1, and GNAS-AS1) within or near differentially methylated AD-associated ICRs in NHBs and only one lncRNA (GNAS-AS1) in NHWs, all of which are linked to brain development. To identify microRNAs overlapping the 120 ICRs, we used hsa.gff3 data provided by miRbase [73], and found two microRNAs (i.e., miR1260b, miR1587) in NHBs among the annotated genes. miR1260b is reported to regulate two tumor suppressor genes, sFRP1 and SMAD4, in prostate cancer through epigenetic mechanisms [74, 75], and to also extensively participate in arthritis, osteogenic differentiation, and Alzheimer's disease [76].
Discussion
Although it is established that mutations in APOE, APP, PSEN1/2, and BACE1 contribute to AD risk [77, 78], and that the APOE-ε4 allele affects cognitive function [77, 79], known genetic variation alone explains only a small proportion of AD. Evidence in the last decade supports that epigenetics may contribute substantially to altered gene function and disease development. The highly conserved and stable methylation pattern of ICRs makes them particularly valuable in the study of diseases like AD, which do not manifest until adulthood or advanced age. After fertilization and remodeling of methylation status during the intrauterine period, ICRs normally maintain the same methylation status in all cells and tissues, including in blood and brain tissue, throughout life. For this reason, early changes in the methylation of ICRs could potentially serve as susceptibility biomarkers for disease risk, and they could be measured at any time during an individual's life. To elucidate the AD associated candidate ICRs in brain tissue that have potential regulatory functions, we determined the methylation pattern of the genome by WGBS and identified the differentially methylated regions in AD brain samples compared to that in controls. We identified 120 candidate ICRs with altered methylation levels in patients with AD.
We next determined whether the patterns of AD-related methylation in candidate ICRs differ by racial/ethnic group. A threefold difference in the number of AD-related ICRs was found in the brain samples of NHBs (67.5%) when compared to NHWs (22.5%). This finding is consistent with the postulate that environmentally responsive epigenetic differences in the methylation of ICRs could contribute to the racial/ethnic disparities observed in AD between NHBs and NHWs [1].
Remarkably, one of the two common ICRs identified in NHBs and NHWs was ICR_481 (MEST/MESTIT1). MEST/PEG1, a paternally expressed imprinted gene, was shown to regulate neuronal migration in development of neocortex [80, 81], block neuron differentiation when knocked out, and inhibit Wnt signaling when expressed [50]. This is functionally significant, as Wnt signalling has been reported to be associated with age related neurodegenerative diseases including AD [80, 82]. As MEST expression is reduced by promoter hypermethylation, this would result in activation of Wnt signaling in brain tissues of AD patients, potentially facilitating the progression of AD [80].
The second ICR identified in NHBs and NHWs was ICR_987. It is closest to the gene NLRP1, a component of the inflammasome complex that triggers an immunostimulatory form of cell death called pyroptosis, which is activated in neuronal cells in response to amyloid-β (Aβ) aggregates [53, 83]. Inflammasomes are multiprotein complexes that are assembled in response to a cellular stressor, including infection, and lead to caspase activation [84]. They also are associated with neurodegenerative diseases such as AD [53], consistent with the hypothesis that neuroinflammation contributes substantially to neurodegeneration [53]. Studies in murine AD models indicate that the Nlrp1 inflammasome is indeed upregulated, and neuronal death is observed, leading to cognitive decline [53]. Kaushal et al. [54] reported a 25- to 30-fold higher number of NLRP1-expressing neurons in AD brains compared to control brains. The existence of significantly increased methylation of the candidate ICR_987 (NLRP1) in both NHBs and NHWs supports the neuroinflammation hypothesis of AD formation, one of the most studied mechanisms in AD pathogenesis.
Furthermore, there are four single nucleotide polymorphisms (SNPs) (rs2137722, rs11657747, rs34733791 and rs11651595) in the NLRP1 region that have been reported as significantly associated with AD [85]. The major alleles for SNPs rs2137722 and rs11657747 are Gs in CpG sites, such that the minor alleles abolish potential methylation sites. Interestingly, the minor A allele for rs2137722, which would block methylation, appears to provide a protective effect against AD [85]. This functional importance of NLRP1 in the development of AD further supports the potential regulatory importance of the proximal differentially methylated ICR_987.
We conducted a comparative analysis of data from three previous epigenome studies: Zhang et al. (2020) in prefrontal cortex [86], Smith et al. (2021) in prefrontal cortex, temporal gyrus, and entorhinal cortex [87], and Breen et al. (2023) in blood [88]. We aimed to identify overlaps between previously reported associations and DMRs identified in this study (Additional file 2: Table S2). The first two studies (Smith et al. (2021) [87] and Zhang et al. (2020) [86]) utilized methylation data from the Illumina HumanMethylation 450 k beadchip, from which we identified commonalities with our AD-associated DMRs (Additional file 3: Table S6). However, none of the 120 candidate ICRs overlapped the differentially methylated positions (DMPs) reported in these two studies. A limiting factor in using the Illumina HumanMethylation 450 k beadchip array is that coverage is for approximately 450,000 methylation sites, constituting only 3% of the total 28,084,558 CpGs in the human genome. Comparison of the 450 K manifest with the coordinates of the AD-ICRs identified 102 CpG sites in common, in 45 ICRs. The lack of common regions between these different approaches is at least partially attributable to this low coverage.
The study by Breen et al. (2023) employed whole-genome methyl-sequencing in blood and looked at differentially methylated positions (DMPs) in AD patients compared to controls [88]. The comparisons between the DMRs identified in this recent study and our AD associated DMRs listed in Table 1 revealed 102 overlapping DMRs in NHBs, including DMR chr7:76149513–76150704, which overlaps with candidate ICR_473, two DMRs in NHWs, and 22 DMRs in ALL group that are common in both studies (Additional file 3: Table S6).
Additionally, an epigenome-wide association study (EWAS) conducted by Piras et al. [89] revealed differential methylation patterns in the genes of AD brains as compared to non-demented control brains. The study identified 832 DMRs, out of which five DMRs were found associated with CASZ1, MAD1L1, MRPL23, RIMBP2, SLC9A3R2, ZCCHC14, and NFIC. Interestingly, these genes were each in proximity to an ICR identified in our study (ICR_20, ICR_434, ICR_716/ICR_719, ICR_805, ICR_922, ICR_976, and ICR_1071, respectively; see Table 1). There are other epigenome-wide studies that showed DMRs associated with various regions of the brain [87, 90]. However, more studies with a focus on ICRs and higher coverage are needed to understand the association between ICRs and AD development.
The AD-associated ICRs described here also overlap other known early-established methylation-dependent gene regulatory regions. A previous study determined regions of systemic interindividual variations (SIVs), characterized by consistent methylation across tissues within individuals, but with significant variation among individuals [91]. Like ICRs, SIV methylation is established before tissue specification, hence the consistency within individuals, but unlike ICRs, methylation is not restricted to parent-of-origin status. The SIV control regions comprise approximately 0.1% of the human genome and regulate the expression of metastable epialleles. The most widely known example of a metastable epiallele is the agouti viable yellow (Avy) locus in mice used to demonstrate that environmentally induced epigenetic modifications during early development can produce a range of phenotypes and alter disease susceptibility in adulthood [24]. SIVs are defined as regions “conserved across diverse human ethnic groups, sensitive to periconceptional environmental exposures, and associated with genes implicated in a broad range of human disorders and phenotypes” [91, 92]. Interestingly, 15 of the AD-associated candidate ICRs overlap with previously described SIVs (Table 1) [91], including ICR_987 (chr17:5771207–5771575, NLRP1), common to both NHB-AD and NHW-AD case control comparisons.
Obesity is known as one of the modifiable risk factors for dementia [93]. According to a study by Nianogo et al. [2] one third of the AD related dementia cases were linked to a combination of modifiable risk factors, including midlife obesity, physical inactivity, and educational attainment [2]. In support of this, our analysis revealed the white adipose tissue browning pathway as a common enriched pathway associated with AD for both NHB and NHW populations. Changes in adipose tissue are part of the normal aging process [94], as adipose tissue is highly dynamic and has a role in homeostatic processes. White adipose tissue (WAT) may actively change into beige or brown adipose tissue (BAT) with environmental factors [95] through the white adipose tissue browning pathway. WAT is known to function as an endocrine organ secreting various types of adipokines including TNFα, which increases with aging. High WAT mass is related to metabolic disorders and linked to insulin resistance [94], and high BMI is associated with a reduction in brain volume [96, 97]. It is suggested that impairment in adipose tissue-derived adipokines may cause problems in brain homeostasis [94], which may eventually lead to neurodegenerative diseases.
Additionally, we identified the gap junction signaling pathway in both NHB and NHW populations as an enriched pathway, which is potentially dysregulated in AD. GNAS, an imprinted gene that is involved in multiple signaling pathways associated with G protein-coupled receptors, is also one of the molecules involved in the gap junction signaling pathway. Transcription of connexins, involved in the gap junction signaling pathway, are reported to be regulated by epigenetic modifications [98]. GJA3, in close proximity to ICR_814 (chr13:20142811–20142911), is a member of the gap junction signaling pathway and was found to contain a differentially methylated CpG (chr13: 20736075) in AD hippocampus samples compared to controls [99]. Future studies on the possible epigenetic regulation of these pathways may elucidate their mechanisms in the development of AD.
Interestingly, our network analysis identified netrin signaling in NHBs only. Netrins are axon guidance molecules associated with the regulation of axonal growth and play roles in neuroinflammation. Netrin 1 is reported to inhibit Aβ production [100]. UNC5B, a molecule belonging to the netrin signaling pathway, was the closest gene to candidate ICR_664 (chr10:71266448–71266685), which was hypomethylated in AD brain samples compared to controls (Table 1, Additional file 2: Table S2). The UNC5B receptor is activated by the Netrin 1 molecule in the netrin signaling pathway. It is reported to have inhibitory roles in the inflammatory response in nervous system and may have a protective role in neurodegeneration [100, 101].
A major strength of our study is that it is one the largest investigations, with deeply phenotyped participants with WGBS data, that uses DNA derived from brain samples not only from NHWs, but also NHBs, who are rarely included in the study cohorts, despite their higher risk of developing AD. Nevertheless, the data should be interpreted in the context of its limitations. Firstly, the relatively small sample size (n = 17) could diminish the statistical power required to identify some ICRs associated with AD. Despite the sample size, our use of agnostic WGBS revealed novel changes in ICRs that should be detectable in accessible tissues. Moreover, WGBS has revealed racial/ethnicity-dependent differences in ICR methylation that may lead to a better understanding of disparities in AD. Further analysis of this phenomenon may improve the treatment of AD by providing a mechanism for determining at-risk individuals. While the inclusive nature of our study aimed to encompass a diverse cohort, the small sample size also introduces an inevitable limitation, preventing us from drawing definitive conclusions regarding sex-specific ICR methylation patterns in the context of AD. However, further analyses with a larger sample size to address this issue in future studies are necessary.
Secondly, brain structural changes in AD start in the entorhinal cortex and medial temporal lobes and extend into the neocortex [4] and cerebellum [102] over time. We cannot exclude the possibility that the threefold difference in the number of differentially methylated ICRs associated with AD is due, in part, to epigenetic differences given the heterogeneity in NHB control tissues, which were a combination of temporal cortex and cerebellum. However, ICR methylation should be similar across tissue and cell types and should not be affected by tissue/cell heterogeneity. For that reason, we are confident with our definition of differentially methylation in candidate ICRs.
We also repeated our initial analyses using the more stringent DMR cutoff of 15% to reduce false positives and restricted our analysis to a subset of the 1488 ICRs previously reported, specifically the 332 ICRs with gametic methylation data. This more stringent analysis did not alter the findings that aberrant ICRs methylation is over-represented among AD cases, more so in NHBs than NHWs. Although differential ICR methylation holds promise in surveillance to identify AD, replicating these findings in high-powered studies with DNA derived from various brain regions and accessible tissues such as blood or central nervous system fluids, in diverse populations, are necessary.
Finally, while we acknowledge the existence of epigenetic drift associated with aging, we lack information regarding the methylation status of ICRs throughout the aging process. ICRs represent specific regulatory regions, and the DNA methylation patterns established during the intrauterine stage remain conserved. Furthermore, a study conducted by Mancino et al. (2023) on the hippocampus of mice, highlights a notable age-related increase in DNA methylation—an established transcriptional indicator of aging [103]. Interestingly, the authors observed that genomic imprinting, specifically parent-of-origin-specific DNA methylation, remained largely unaffected by the aging process. This observed stability extended across various brain regions, including the cerebellum, nucleus accumbens, hypothalamus, and prefrontal cortex. Transcriptomic analysis further substantiated these findings, confirming the preservation of imprinted expression in the aged hippocampus [103]. Nevertheless, our current dataset does not provide insights into age-related changes. With the anticipation of acquiring more comprehensive data through larger sample sizes and long-term follow-up studies, we aim to unravel the methylation dynamics of ICRs in the future, drawing from an extended population.
Conclusion
Using unbiased WGBS, we provide the first evidence that DNA methylation in 120 ICRs varies markedly between AD cases and controls. The number of ICRs with altered methylation is three times higher in NHBs with AD than in NHWs with AD, which may contribute to the higher prevalence of AD in NHBs compared to NHWs [1]. Our findings are also consistent with the developmental origins of health and disease (DOHaD) hypothesis that increased susceptibility to adult-onset chronic diseases such as AD frequently have their origins in early development [104], and support the findings that AD is characterized by changes in the brain that likely start decades before the clinical symptoms appear [40, 41]. Thus, alteration in ICR methylation may serve as an early detection tool of AD risk that is essential for slowing the progression of this disease.
Methods
Human brain specimens
To identify differentially methylated ICRs associated with AD, we obtained frozen autopsy brain specimens from the Joseph and Kathleen Bryan Brain Bank of the Duke University/University of North Carolina at Chapel Hill Alzheimer’s Disease Research Center (Duke/UNC ADRC); informed consent documentation is in IRB ID# 00016278. The AD and control brain tissues were selected according to their neuropathologic diagnosis of AD. Nine brain samples from AD cadavers (five NHBs and four NHWs) and eight brain samples from control cadavers (four NHBs and four NHWs) were collected. For all individuals, the time elapsed between patient death and collection and snap-freezing of samples was less than 24 h (Additional file 1: Table S1).
Library preparation of specimens, WGBS, and identification of AD-associated DMRs
We performed WGBS to identify DMRs associated with AD in all brain samples (n = 17). Libraries were prepared from extracted DNA using EpiGnome™ Methyl-Seq reagents (Illumina, Inc, San Diego, CA), index-tagged for multiplexing, and sequenced on an Illumina NextSeq platform (Illumina, Inc, San Diego, CA). Reads were assigned back to individuals by index reads, and aligned in silico to a bisulfite-converted reference genome (i.e., hg38 version 87). Reads without unique alignments, due to either repetitive genomic sequence or loss of specificity from bisulfite conversion of cytosines, and duplicate reads, indicative of clonal amplification of original random DNA fragments, were eliminated. The quality of each paired-end sequence file was inspected using fastqc, and adapter trimming and quality control were performed using the Trim Galore wrapper script that calls the cutadapt [105] script internally. Paired-end reads with a trimmed adapter sequence were aligned to the human reference genome (i.e., hg38 version 87) downloaded from Ensembl using bsmap aligner [106]. The following options were passed to the aligner: -p 8 -L 135 -w 100 -v 10 -q 10 -R -V 1. The aligned bam files were sorted and indexed using samtools [107], and duplicate sequence reads were removed using the Picard [108] application.
We analyzed three groups from 17 study participants whose clinic and demographic characteristics are summarized in Additional file 1: Table S1. These included: I) All AD samples vs. all controls, II) NHB-AD vs. NHB-control samples, III) NHW-AD vs. NHW-control samples (Fig. 1a). Briefly, a methylation ratio for CpG dinucleotides was generated from replicate bam files for each AD samples and controls using mcall, and DMRs were called between AD samples and controls using mcomp. The following options were utilized for mcomp inputs: –doStrandSpecifiMeth = 1, –doDmrScan = 1, –doDmcScan = 1, –dmrMethods = 2 –minDmcsInDmr = 4, –minCredibleDif = 0.1 or 0.15, –maxDistConsDmcs = 300, –minDepthForComp = 7 –pFetDmc = 0.05. DMRs were called using Model-based analysis of bisulfite sequencing data (MOABS, version 1.3.8.7), which relies on Credible Methylation Difference (CDIF) as a single metric for both statistics and biological significance of differential methylation, i.e., significant DMRs were generated using MOABS, and confirmed with known DMRs as positive controls [42]. The following criteria were used in calling a DMR: minimum differentially methylated C’s \(\ge\) 4, credible cis-acting methylation difference \(\ge\) 10% or \(\ge\) 15%, minimum read depth \(\ge\) 7, and max distance between consecutive CpG’s \(\le\) 300 (Fig. 4). After merging the bam files, the total coverage from certain CpGs divided by total coverage for all CpG’s (wig sum percentages) were plotted versus increasing read depth using mmint (https://github.com/lijiacd985/Mmint) (Additional file 1: Fig. S1a). This plot helped us check if there were high duplication levels or sequence bias. There was no sequence bias observed for all AD and control samples. We have generated the coverage plot using the plot coverage function from deepTools [109] using the merged bam files per group (Additional file 1: Fig. S1b, c).

Experimental workflow. The steps used to identify DMRs in AD cases vs controls, overlapping ICRs, and closest genes. Created with BioRender.com
Identification of AD-related ICRs in NHBs and NHWs
The DMRs associated with AD were intersected with the recently defined ICRs [35] using bedtools. A description of the process for defining human ICRs was recently published [35]. Briefly, puticr, a custom tool implemented in Python (Version \(\ge\) 2.7), was used to identify 1488 ICRs in tissue from multiple germ layers [35]. We used Bedtools [110] to identify ICRs that intersected with AD-associated DMRs, and then used two-sided Fisher’s exact test to identity enrichment of ICRs in the set of AD DMRs (Fig. 4).
To explore the functional significance of these AD-associated ICRs, genes closest to either side of the 120 AD-associated ICRs were identified from the UCSC genome browser. To explore molecular and cellular functions, potentially enriched pathways, and associations with disease, we analyzed our gene set using online tools, including Ingenuity Pathway Analysis (IPA) [111].
Microsoft Excel v16.46 (21021202) and Bedtools v2.30.0 were used to compare ICR coordinates with published data. Using the Expression Database of Human Long noncoding RNAs (LncExpDB)[72], we compared lncRNAs associated with brain development genes, and the genes in close proximity to ICRs. Furthermore, using hsa.gff3 data provided by miRbase [73], we identified microRNAs among the annotated genes that overlap with AD-associated ICRs.
Availability of data and materials
The raw sequencing data obtained by our study are in the process of being submitted to dbGaP.
References
Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimers Dement. 2022;18(4):700–89.
Nianogo RA, Rosenwohl-Mack A, Yaffe K, Carrasco A, Hoffmann CM, Barnes DE. Risk factors associated with alzheimer disease and related dementias by sex and race and ethnicity in the US. JAMA Neurol. 2022;79(6):584–91.
Heun R, Schoepf D, Potluri R, Natalwala A. Alzheimer’s disease and co-morbidity: increased prevalence and possible risk factors of excess mortality in a naturalistic 7-year follow-up. Eur Psychiatry. 2013;28(1):40–8.
Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312–39.
Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43(8):1467–72.
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977–81.
Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23(4):213–27.
Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 1992;357(6378):500–3.
Theuns J, Del-Favero J, Dermaut B, van Duijn CM, Backhovens H, Van den Broeck MV, et al. Genetic variability in the regulatory region of presenilin 1 associated with risk for Alzheimer’s disease and variable expression. Hum Mol Genet. 2000;9(3):325–31.
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376(6543):775–8.
Sherrington R, Froelich S, Sorbi S, Campion D, Chi H, Rogaeva EA, et al. Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet. 1996;5(7):985–8.
Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, et al. A familial Alzheimer’s disease locus on chromosome 1. Science. 1995;269(5226):970–3.
Ou-Yang MH, Kurz JE, Nomura T, Popovic J, Rajapaksha TW, Dong H, et al. Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci Transl Med. 2018;10(459).
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–41.
Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Chakrabarti S, Khemka VK, Banerjee A, Chatterjee G, Ganguly A, Biswas A. Metabolic risk factors of sporadic Alzheimer’s disease: implications in the pathology, pathogenesis and treatment. Aging Dis. 2015;6(4):282–99.
van der Flier WM, Scheltens P. Epidemiology and risk factors of dementia. J Neurol Neurosurg Psychiatry. 2005;76(5):v2-7.
LaFerla FM, Green KN. Animal models of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(11).
Sharma VK, Mehta V, Singh TG. Alzheimer’s disorder: epigenetic connection and associated risk factors. Curr Neuropharmacol. 2020;18(8):740–53.
Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11(2):111–28.
Aikawa T, Holm ML, Kanekiyo T. ABCA7 and pathogenic pathways of Alzheimer's disease. Brain Sci. 2018;8(2).
Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ϵ4, and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013;309(14):1483–92.
Glymour MM, Manly JJ. Lifecourse social conditions and racial and ethnic patterns of cognitive aging. Neuropsychol Rev. 2008;18(3):223–54.
Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23(15):5293–300.
Dolinoy DC. Epigenetic gene regulation: early environmental exposures. Pharmacogenomics. 2007;8(1):5–10.
Tecalco-Cruz AC, Ramirez-Jarquin JO, Alvarez-Sanchez ME, Zepeda-Cervantes J. Epigenetic basis of Alzheimer disease. World J Biol Chem. 2020;11(2):62–75.
Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging. 2010;31(12):2025–37.
Chang L, Wang Y, Ji H, Dai D, Xu X, Jiang D, et al. Elevation of peripheral BDNF promoter methylation links to the risk of Alzheimer’s disease. PLoS ONE. 2014;9(11): e110773.
Declerck K, Vanden BW. Characterization of blood surrogate immune-methylation biomarkers for immune cell infiltration in chronic inflammaging disorders. Front Genet. 2019;10:1229.
Karlsson IK, Ploner A, Wang Y, Gatz M, Pedersen NL, Hagg S. Apolipoprotein E DNA methylation and late-life disease. Int J Epidemiol. 2018;47(3):899–907.
Madrid A, Hogan KJ, Papale LA, Clark LR, Asthana S, Johnson SC, et al. DNA hypomethylation in blood links B3GALT4 and ZADH2 to Alzheimer’s disease. J Alzheimers Dis. 2018;66(3):927–34.
Wang E, Wang M, Guo L, Fullard JF, Micallef C, Bendl J, et al. Genome-wide methylomic regulation of multiscale gene networks in Alzheimer’s disease. Alzheimers Dement. 2023;19(8):3472–95.
Kim BH, Vasanthakumar A, Li QS, Nudelman KNH, Risacher SL, Davis JW, et al. Integrative analysis of DNA methylation and gene expression identifies genes associated with biological aging in Alzheimer’s disease. Alzheimers Dement (Amst). 2022;14(1): e12354.
Peng X, Zhang W, Cui W, Ding B, Lyu Q, Wang J. ADmeth: a manually curated database for the differential methylation in Alzheimer’s disease. IEEE/ACM Trans Comput Biol Bioinform. 2023;20(2):843–51.
Jima DD, Skaar DA, Planchart A, Motsinger-Reif A, Cevik SE, Park SS, et al. Genomic map of candidate human imprint control regions: the imprintome. Epigenetics. 2022;17(13):1920–43.
Smallwood SA, Kelsey G. De novo DNA methylation: a germ cell perspective. Trends Genet. 2012;28(1):33–42.
Cowley M, Oakey RJ. Resetting for the next generation. Mol Cell. 2012;48(6):819–21.
Mandy M, Nyirenda M. Developmental Origins of Health and Disease: the relevance to developing nations. Int Health. 2018;10(2):66–70.
Lorgen-Ritchie M, Murray AD, Staff R, Ferguson-Smith AC, Richards M, Horgan GW, et al. Imprinting methylation predicts hippocampal volumes and hyperintensities and the change with age in later life. Sci Rep. 2021;11(1):943.
Beason-Held LL, Goh JO, An Y, Kraut MA, O’Brien RJ, Ferrucci L, et al. Changes in brain function occur years before the onset of cognitive impairment. J Neurosci. 2013;33(46):18008–14.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–92.
Sun D, Xi Y, Rodriguez B, Park HJ, Tong P, Meong M, et al. MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 2014;15(2):R38.
Jima DD, Skaar DA, Planchart A, Motsinger-Reif A, Cevik SE, Park SS, et al. Genomic map of candidate human imprint control regions: the imprintome. Epigenetics. 2022:1–24.
Wang C, Liu Z, Woo CW, Li Z, Wang L, Wei JS, et al. EZH2 mediates epigenetic silencing of neuroblastoma suppressor genes CASZ1, CLU, RUNX3, and NGFR. Cancer Res. 2012;72(1):315–24.
Fetahu IS, Ma D, Rabidou K, Argueta C, Smith M, Liu H, et al. Epigenetic signatures of methylated DNA cytosine in Alzheimer’s disease. Sci Adv. 2019;5(8):eaaw2880.
Dredge BK, Jensen KB. NeuN/Rbfox3 nuclear and cytoplasmic isoforms differentially regulate alternative splicing and nonsense-mediated decay of Rbfox2. PLoS ONE. 2011;6(6): e21585.
Hokama M, Oka S, Leon J, Ninomiya T, Honda H, Sasaki K, et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb Cortex. 2014;24(9):2476–88.
Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol. 2009;513(5):532–41.
Gu J, Chen F, Chu D, Lu Y, Iqbal K, Gong CX, et al. Rbfox3/NeuN regulates alternative splicing of tau exon 10. J Alzheimers Dis. 2018;66(4):1695–704.
Jung H, Lee SK, Jho EH. Mest/Peg1 inhibits Wnt signalling through regulation of LRP6 glycosylation. Biochem J. 2011;436(2):263–9.
Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, Surani MA. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet. 1998;20(2):163–9.
Eggermann T, Spengler S, Begemann M, Binder G, Buiting K, Albrecht B, et al. Deletion of the paternal allele of the imprinted MEST/PEG1 region in a patient with Silver-Russell syndrome features. Clin Genet. 2012;81(3):298–300.
Yap JKY, Pickard BS, Chan EWL, Gan SY. The role of neuronal NLRP1 inflammasome in Alzheimer’s disease: bringing neurons into the neuroinflammation game. Mol Neurobiol. 2019;56(11):7741–53.
Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, et al. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015;22(10):1676–86.
Bina M. Discovering candidate imprinted genes and imprinting control regions in the human genome. BMC Genomics. 2020;21(1):378.
Morison IM, Paton CJ, Cleverley SD. The imprinted gene and parent-of-origin effect database. Nucleic Acids Res. 2001;29(1):275–6.
Riesewijk AM, Hu L, Schulz U, Tariverdian G, Höglund P, Kere J, et al. Monoallelic expression of human PEG1/MEST is paralleled by parent-specific methylation in fetuses. Genomics. 1997;42(2):236–44.
Li T, Vu TH, Lee KO, Yang Y, Nguyen CV, Bui HQ, et al. An imprinted PEG1/MEST antisense expressed predominantly in human testis and in mature spermatozoa. J Biol Chem. 2002;277(16):13518–27.
Court F, Camprubi C, Garcia CV, Guillaumet-Adkins A, Sparago A, Seruggia D, et al. The PEG13-DMR and brain-specific enhancers dictate imprinted expression within the 8q24 intellectual disability risk locus. Epigenetics Chromatin. 2014;7(1):5.
Lorgen-Ritchie M, Murray AD, Ferguson-Smith AC, Richards M, Horgan GW, Phillips LH, et al. Imprinting methylation in SNRPN and MEST1 in adult blood predicts cognitive ability. PLoS ONE. 2019;14(2): e0211799.
Cao Y, AlHumaidi SS, Faqeih EA, Pitel BA, Lundquist P, Aypar U. A novel deletion of SNURF/SNRPN exon 1 in a patient with Prader-Willi-like phenotype. Eur J Med Genet. 2017;60(8):416–20.
Kim J, Bergmann A, Stubbs L. Exon sharing of a novel human zinc-finger gene, ZIM2, and paternally expressed gene 3 (PEG3). Genomics. 2000;64(1):114–8.
Yeung KS, Ho MSP, Lee SL, Kan ASY, Chan KYK, Tang MHY, et al. Paternal uniparental disomy of chromosome 19 in a pair of monochorionic diamniotic twins with dysmorphic features and developmental delay. J Med Genet. 2018;55(12):847–52.
Evans HK, Wylie AA, Murphy SK, Jirtle RL. The neuronatin gene resides in a “micro-imprinted” domain on human chromosome 20q112. Genomics. 2001;77(1–2):99–104.
Hannula-Jouppi K, Muurinen M, Lipsanen-Nyman M, Reinius LE, Ezer S, Greco D, et al. Differentially methylated regions in maternal and paternal uniparental disomy for chromosome 7. Epigenetics. 2014;9(3):351–65.
Zhang Y, Tycko B. Monoallelic expression of the human H19 gene. Nat Genet. 1992;1(1):40–4.
Wylie AA, Murphy SK, Orton TC, Jirtle RL. Novel imprinted DLK1/GTL2 domain on human chromosome 14 contains motifs that mimic those implicated in IGF2/H19 regulation. Genome Res. 2000;10(11):1711–8.
Nakabayashi K, Trujillo AM, Tayama C, Camprubi C, Yoshida W, Lapunzina P, et al. Methylation screening of reciprocal genome-wide UPDs identifies novel human-specific imprinted genes. Hum Mol Genet. 2011;20(16):3188–97.
Schulz R, McCole RB, Woodfine K, Wood AJ, Chahal M, Monk D, et al. Transcript- and tissue-specific imprinting of a tumour suppressor gene. Hum Mol Genet. 2009;18(1):118–27.
Liu J, Yu S, Litman D, Chen W, Weinstein LS. Identification of a methylation imprint mark within the mouse Gnas locus. Mol Cell Biol. 2000;20(16):5808–17.
Skaar DA, Li Y, Bernal AJ, Hoyo C, Murphy SK, Jirtle RL. The human imprintome: regulatory mechanisms, methods of ascertainment, and roles in disease susceptibility. ILAR J. 2012;53(3–4):341–58.
Li Z, Liu L, Jiang S, Li Q, Feng C, Du Q, et al. LncExpDB: an expression database of human long non-coding RNAs. Nucleic Acids Res. 2021;49(D1):D962–8.
Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–62.
Hirata H, Hinoda Y, Shahryari V, Deng G, Tanaka Y, Tabatabai ZL, et al. Correction: Genistein downregulates onco-miR-1260b and upregulates sFRP1 and Smad4 via demethylation and histone modification in prostate cancer cells. Br J Cancer. 2018;119:388.
Hirata H, Hinoda Y, Shahryari V, Deng G, Tanaka Y, Tabatabai ZL, et al. Genistein downregulates onco-miR-1260b and upregulates sFRP1 and Smad4 via demethylation and histone modification in prostate cancer cells. Br J Cancer. 2014;110(6):1645–54.
Shen S, Huang J, Xu C, Shen Y, Jiang S, Li Y, et al. ERK modulates macrophage polarization and alters exosome miRNA expression in diabetic nephropathy. Clin Lab. 2021;67(12).
Armstrong AR. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019;57(2):87–105.
Hoenicka J. Genes in Alzheimer’s disease. Rev Neurol. 2006;42(5):302–5.
Liu G, Yao L, Liu J, Jiang Y, Ma G, et al. Cardiovascular disease contributes to Alzheimer’s disease: evidence from large-scale genome-wide association studies. Neurobiol Aging. 2014;35(4):786–92.
Prasad R, Jung H, Tan A, Song Y, Moon S, Shaker MR, et al. Hypermethylation of Mest promoter causes aberrant Wnt signaling in patients with Alzheimer’s disease. Sci Rep. 2021;11(1):20075.
Ji L, Bishayee K, Sadra A, Choi S, Choi W, Moon S, et al. Defective neuronal migration and inhibition of bipolar to multipolar transition of migrating neural cells by Mesoderm-Specific Transcript, Mest, in the developing mouse neocortex. Neuroscience. 2017;355:126–40.
Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810.
Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, et al. Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 2014;5(8): e1382.
de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol. 2014;6(12): a016287.
Pontillo A, Catamo E, Arosio B, Mari D, Crovella S. NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis Assoc Disord. 2012;26(3):277–81.
Zhang L, Silva TC, Young JI, Gomez L, Schmidt MA, Hamilton-Nelson KL, et al. Epigenome-wide meta-analysis of DNA methylation differences in prefrontal cortex implicates the immune processes in Alzheimer’s disease. Nat Commun. 2020;11(1):6114.
Smith RG, Pishva E, Shireby G, Smith AR, Roubroeks JAY, Hannon E, et al. A meta-analysis of epigenome-wide association studies in Alzheimer’s disease highlights novel differentially methylated loci across cortex. Nat Commun. 2021;12(1):3517.
Breen C, Papale LA, Clark LR, Bergmann PE, Madrid A, Asthana S, et al. Whole genome methylation sequencing in blood identifies extensive differential DNA methylation in late-onset dementia due to Alzheimer's disease. Alzheimers Dement. 2023.
Piras IS, Brokaw D, Kong Y, Weisenberger DJ, Krate J, Delvaux E, et al. Integrated DNA methylation/RNA profiling in middle temporal gyrus of Alzheimer’s disease. Cell Mol Neurobiol. 2023;43(5):2289–307.
Li QS, Sun Y, Wang T. Epigenome-wide association study of Alzheimer’s disease replicates 22 differentially methylated positions and 30 differentially methylated regions. Clin Epigenetics. 2020;12(1):149.
Gunasekara CJ, Scott CA, Laritsky E, Baker MS, MacKay H, Duryea JD, et al. A genomic atlas of systemic interindividual epigenetic variation in humans. Genome Biol. 2019;20(1):105.
Gunasekara CJ, Hannon E, MacKay H, Coarfa C, McQuillin A, Clair DS, et al. A machine learning case-control classifier for schizophrenia based on DNA methylation in blood. Transl Psychiatry. 2021;11(1):412.
Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396(10248):413–46.
Parimisetty A, Dorsemans AC, Awada R, Ravanan P, Diotel N, Lefebvre DC. Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research. J Neuroinflammation. 2016;13(1):67.
Zoico E, Rubele S, De Caro A, Nori N, Mazzali G, Fantin F, et al. Brown and beige adipose tissue and aging. Front Endocrinol (Lausanne). 2019;10:368.
Ward MA, Carlsson CM, Trivedi MA, Sager MA, Johnson SC. The effect of body mass index on global brain volume in middle-aged adults: a cross sectional study. BMC Neurol. 2005;5:23.
Debette S, Beiser A, Hoffmann U, Decarli C, O’Donnell CJ, Massaro JM, et al. Visceral fat is associated with lower brain volume in healthy middle-aged adults. Ann Neurol. 2010;68(2):136–44.
Sanchez OF, Rodriguez AV, Velasco-Espana JM, Murillo LC, Sutachan JJ, Albarracin SL. Role of Connexins 30, 36, and 43 in brain tumors, neurodegenerative diseases, and neuroprotection. Cells. 2020;9(4):846.
Altuna M, Urdánoz-Casado A, Sánchez-Ruiz de Gordoa J, Zelaya MV, Labarga A, Lepesant JMJ, et al. DNA methylation signature of human hippocampus in Alzheimer’s disease is linked to neurogenesis. Clin Epigenetics. 2019;11(1):91.
Lee WS, Lee WH, Bae YC, Suk K. Axon guidance molecules guiding neuroinflammation. Exp Neurobiol. 2019;28(3):311–9.
Li Q, Wang BL, Sun FR, Li JQ, Cao XP, Tan L. The role of UNC5C in Alzheimer’s disease. Ann Transl Med. 2018;6(10):178.
Hoxha E, Lippiello P, Zurlo F, Balbo I, Santamaria R, Tempia F, et al. The emerging role of altered cerebellar synaptic processing in Alzheimer’s disease. Front Aging Neurosci. 2018;10:396.
Mancino S, Seneviratne J, Mupo A, Krueger F, Oxley D, Eckersley-Maslin MA, et al. Stability of genomic imprinting and X-chromosome inactivation in the aging brain. BioRxiv. 2023:2023.09.29.560184.
Gauvrit T, Benderradji H, Buee L, Blum D, Vieau D. Early-life environment influence on late-onset Alzheimer’s disease. Front Cell Dev Biol. 2022;10: 834661.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetJournal. 2011. https://doi.org/10.14806/ej.17.1.200.
Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinf. 2009;10:232.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Institute B. Picard Toolkit. Broad Institue. 2019.
Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42(Web Server issue):W187–91.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2.
Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30(4):523–30.
Acknowledgements
This work was supported in part by National Institutes of Health grants R01HD098857 (D.A.S., C.H., R.L.J.), and R01MD017696, R01MD011746, P30ES025128, R01ES032462 (C.H., D.A.S., D.D.J., R.L.J.), and Office of Extramural Research, National Institutes of Health.
Author information
Authors and Affiliations
Contributions
C.H., and R.L.J. conceived the idea and obtained funding, A.P., S.E.C., and D.A.S. contributed mechanistic expertise to advancing the thesis, and D.D.J. performed bioinformatics, A.L provided expertise in the clinical aspects of AD, T. Ø. and H.E.W. provided expertise in AD epidemiology. All authors contributed to drafting and editing the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Brain samples were obtained from Joseph and Kathleen Bryan Brain Bank of the Duke University/University of North Carolina at Chapel Hill Alzheimer’s Disease Research Center, and all informed consent documentation and protocols are in IRB ID# 00016278.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
This table provides demographic and clinical characteristics of both Alzheimer's disease (AD) cases and controls from which the brain samples were obtained. Table S3. This table presents a compilation of AD-associated differentially methylated regions (DMRs) that overlap with 332 candidate inherited ICRs, which have been validated through parental allele confirmation. Table S4 and S5. These tables contain results from the functional and pathway analysis of genes that are in close proximity to AD-associated ICRs. Table S4 pertains to non-Hispanic Black (NHB) populations, while Table S5 focuses on non-Hispanic White (NHW) populations. Figure S1. This figure shows the results of the quality control for the WGBS data, ensuring the reliability and accuracy of the obtained results.
Additional file 2: Table S2.
The supplementary materials comprise additional details of the DMRs identified across three distinct comparison groups, the overall group (ALL), NHBs, and NHWs, including: Chromosome number: Indicating the chromosome on which the DMR is located. DMR coordinates: Providing the precise coordinates of the DMR on the chromosome. Mean ratio: The average methylation ratio across the DMR region. Cytosine number: Specifying the count of cytosines within the DMR. Number of CpG sites: Enumerating the quantity of CpG sites present within the DMR for both controls and AD patients. Methylation difference: Representing the difference in methylation levels between AD patients and controls. p-value: the statistical significance of the observed methylation differences. Hyper/hypomethylation status: Indicating whether the region is hypermethylated or hypomethylated in AD patients relative to controls.
Additional file 3: Table S6.
The supplementary materials comprise additional details of the DMRs shared between AD-associated DMRs identified in our study in brain stratified by race/ethnicity and the study by Breen et al. (2023) in blood [88].
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cevik, S.E., Skaar, D.A., Jima, D.D. et al. DNA methylation of imprint control regions associated with Alzheimer’s disease in non-Hispanic Blacks and non-Hispanic Whites. Clin Epigenet 16, 58 (2024). https://doi.org/10.1186/s13148-024-01672-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-024-01672-4