
Abstract
Background
Previous studies have traditionally attributed the initiation of cancer cells to genetic mutations, considering them as the fundamental drivers of carcinogenesis. However, recent research has shed light on the crucial role of epigenomic alterations in various cell types present within the tumor microenvironment, suggesting their potential contribution to tumor formation and progression. Despite these significant findings, the progress in understanding the epigenetic mechanisms regulating tumor heterogeneity has been impeded over the past few years due to the lack of appropriate technical tools and methodologies.
Results
The emergence of single-cell sequencing has enhanced our understanding of the epigenetic mechanisms governing tumor heterogeneity by revealing the distinct epigenetic layers of individual cells (chromatin accessibility, DNA/RNA methylation, histone modifications, nucleosome localization) and the diverse omics (transcriptomics, genomics, multi-omics) at the single-cell level. These technologies provide us with new insights into the molecular basis of intratumoral heterogeneity and help uncover key molecular events and driving mechanisms in tumor development.
Conclusion
This paper provides a comprehensive review of the emerging analytical and experimental approaches of single-cell sequencing in various omics, focusing specifically on epigenomics. These approaches have the potential to capture and integrate multiple dimensions of individual cancer cells, thereby revealing tumor heterogeneity and epigenetic features. Additionally, this paper outlines the future trends of these technologies and their current technical limitations.
Similar content being viewed by others
Background
The term “epigenetics” was introduced by Conrad Waddington in 1942 [1] to describe the phenomenon wherein alterations in gene phenotype occur without any corresponding changes in the DNA sequence [2]. At the molecular level, numerous specific modifying proteins are involved in such modifications, including "writers" that catalyze the deposition of specific modifications, "erasers" that catalyze the removal of specific modifications, and "readers" that recognize and bind to modification sites catalyzed by the "writers" and form complexes [3].
The prevailing genetic theory posits that the accumulation of somatic mutations is responsible for the formation of tumors, indicating that cancer cells may emerge from mutations in specific genes that drive oncogenesis [4, 5]. Nevertheless, the lack of mutations with potent oncogenic drivers in various tumor types has prompted researchers to redirect their attention toward non-genetic determinants, such as epigenetic modifications, in the investigation of tumor heterogeneity. Research has demonstrated that changes in epigenetic modifications are significant contributors to malignant biological outcomes, including tumor proliferation, self-renewal, differentiation [6], treatment resistance [7], and tumor metastasis [8].
Despite the advancements brought about by Bulk sequencing in elucidating the mechanisms of epigenetic regulation of tumor heterogeneity, numerous inquiries remain unresolved. Notably, in the investigation of the intricate mechanism of HCC pathogenesis, a handful of studies have highlighted the involvement of ALKBH5 and METTL4 in the pathogenesis of HCC; however, conflicting outcomes have been reported [9,10,11]. Similarly, in hepatocellular carcinoma, YTHDF2 and METTL3, among other m6A-related proteins, have also yielded conflicting findings [12,13,14,15,16]. The aforementioned phenomenon could potentially be attributed to the intricate cellular heterogeneity of tumors. Regrettably, the comprehensive gene expression data acquired through conventional sequencing techniques may obscure the infrequent cellular subtypes that play a pivotal role in tumor advancement, and the heterogeneity of tumor cells is concealed by the average information of a substantial cell population.
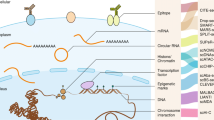
Currently, the field of cancer medicine has transitioned into a phase of precision, whereby the advent of single-cell sequencing technology has provided a promising avenue for investigating the heterogeneity of tumors and the intricate epigenetic regulatory mechanisms that underlie them [17, 18]. In contrast to conventional bulk sequencing, single-cell sequencing offers a notable benefit in its ability to evaluate tumor heterogeneity at the level of individual cells. This overcomes the limitation of traditional sequencing, which can only furnish aggregated data and potentially obscure valid information. Consequently, single-cell sequencing has emerged as a potent instrument for uncovering the epigenetic mechanisms that govern tumor heterogeneity (Fig. 1).

Comparison of the characteristics of single-cell sequencing technology and Bulk sequencing technology
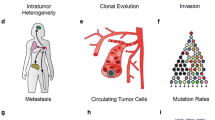
Tumors are intricate and heterogeneous biological systems, and it has been recognized that epigenetics plays a crucial role in cancer initiation and progression. Bulk sequencing alone is insufficient to fully elucidate the intricate epigenetic regulatory mechanisms underlying tumors. Single-cell epigenetic analysis has emerged as a valuable tool for exploring previously uncharted aspects of epigenetic heterogeneity associated with tumor biology, including clonal heterogeneity, the tumor microenvironment, cancer stem cells, circulating tumor cells, treatment resistance, intercellular communication, and tumor metastasis (Fig. 2).

Use of single-cell sequencing allows assessment of epigenetic regulation of tumor heterogeneity not accurately assessed by previous bulk methodologies. Due to the complex cellular heterogeneity of many types of tumors, valid information about individual cells is often masked by the average data of bulk sequencing when using bulk sequencing. However, the emergence of single-cell sequencing has enabled the investigation of epigenetic regulation of tumor heterogeneity, including clonal heterogeneity, cellular crosstalk, tumor stem cells, tumor metastasis, circulating tumor cells, treatment resistance, spatial organization, and tumor microenvironment (TME) mechanisms, which was previously unattainable. A Clonal heterogeneity: single-cell sequencing can monitor novel tumor cell subtypes adapted to the tumor microenvironment in the context of epigenetic alterations, revealing the impact of tumor heterogeneity in cancer patients by epigenetic modifications. B Single-cell sequencing can infer cellular interactions by correlating the expression of known ligands and receptors, and unravel the epigenomic alterations regulated by this interaction set that lead to tumor development. C Cancer stem cells: single-cell sequencing can study the epigenetic background of tumor stem cell differentiation trajectory to predict tumor progression and reveal the heterogeneity of tumor cells. D Tumor metastasis: single-cell sequencing can monitor rare cellular mutations that acquire invasiveness, metastasis, immune escape and EMT during tumor development and investigate the mechanisms underlying the epigenetic regulation of this process. E Circulating tumor cells: single-cell sequencing can be used to investigate the epigenetic regulation of metastasis by comparing the expression differences between CTCs in blood with metastatic dissemination of tumors and the primary tumor. F Treatment resistance: single-cell sequencing allows monitoring rare cellular mutations that acquire treatment resistance in tumors (e.g., intrinsic drug resistance and acquired drug resistance) and investigating the mechanisms of epigenetic regulation of this process. G Spatial organization: single-cell sequencing can obtain temporal and spatial information on gene expression, and in situ measurement of the epigenome can better reveal the association between cellular spatial distribution and gene regulation in tumor tissues. H Tumor microenvironment (TME): single-cell sequencing can investigate the phenotypic and functional heterogeneity of various cell types caused by epigenomic alterations in the tumor microenvironment (TME) during tumorigenesis and progression
However, there are extensive interactions between epigenomes and other omics, and examining epigenomics exclusively at the single-cell resolution can limit a comprehensive comprehension of epigenetic heterogeneity in tumors. By integrating single-cell epigenomic, transcriptomic, genomic analyses, we anticipate expanding the current understanding of how epigenetic domains influence tumor heterogeneity through a multi-dimensional single-cell epigenetic multi-omics approach. This approach is often unachievable with bulk or single-cell epigenetic sequencing alone.
Single-cell sequencing technology applied to omics to explore the impact of epigenetic modifications on tumor heterogeneity
Since the first appearance of single-cell transcriptomics technology in 2009 [19], a plethora of such techniques have emerged. With continuous improvements in these technologies, we are now able to perform individualized analyses of specific cellular features, moving from the multi-cellular level of tissues to the single-cell realm, thereby obtaining a more detailed understanding of tumor cell heterogeneity.
In recent years, the advent of numerous bulk and single-cell level epigenetic sequencing methods has provided powerful tools for mapping various epigenetic modifications, such as chromatin accessibility, histone modifications, DNA methylation, and single-cell nuclear organization [20]. These tools have proven instrumental in unraveling the epigenetic mechanisms underlying malignant tumor cells [21].
However, studying only the epigenome of tumors is insufficient to fully reveal the complexity of tumor heterogeneity [22]. To address this issue, researchers have proposed integrating epigenomic data with transcriptomic, and genomic data from other omics levels at the single-cell resolution. This comprehensive analysis enables a holistic understanding of the relationship between epigenetic regulation and cancer heterogeneity [23] (Fig. 3).

Single-cell mono-omics and single-cell multi-omics sequencing approaches to research the epigenetic mechanisms of cancer
Transcriptomics
Transcriptome sequencing is a method used to understand the gene expression profile by sequencing all the expressed genes in a cell. Traditional transcriptome sequencing methods often require a large amount of cell samples, which obscures subtle differences between individual cells within a cell population, especially in highly heterogeneous tumor cells.
Single-cell transcriptome sequencing technology is a high-resolution approach that enables sequencing of mRNA in individual cells. Common techniques for single-cell transcriptome sequencing include single-cell RNA sequencing (scRNA-seq) [24] and single-cell nuclear sequencing (scNuc-seq). These techniques utilize microfluidic chips, microarrays, droplet-based or partition-based approaches to isolate and capture individual cells, perform cDNA synthesis, DNA amplification, and high-throughput sequencing, to obtain transcriptomic information from individual cells and deeply study the heterogeneity and characteristics of tumor cells [25].
Single-cell transcriptome sequencing has broad applications in tumor research. Through this technology, we can identify and define different subgroups and cell types within tumor cells, gaining insights into the cellular heterogeneity within tumors [26]. Additionally, the utilization of scRNA-seq assays to track temporal genealogy at various stages of tumor development can facilitate the identification of key gene mutations that initiate tumor progression, while also providing insights into the interplay between heterogeneity and temporal dynamics [27]. In the study of tumor immune microenvironment, this technology helps to identify and characterize tumor-infiltrating immune cells and immune suppressive cells, predict patient responses to immunotherapy, and provide foundations for the development of immunotherapy strategies [28]. Moreover, single-cell transcriptome sequencing can also predict patient responses to specific treatments, enabling personalized treatment selection, and provide insights into the mechanisms of treatment resistance for theoretical research [29]. Finally, this technology contributes to the discovery and validation of novel tumor-related genes and signaling pathways, providing potential targets and theoretical basis for targeted therapy and new drug development [17, 30, 31]. In summary, single-cell transcriptome sequencing provides a powerful and comprehensive tool for tumor research, greatly advancing our understanding of tumor development and treatment, and providing important scientific foundations for personalized treatment strategies and new drug development.
A study utilized single-cell RNA sequencing to analyze individual cells from pancreatic ductal adenocarcinoma (PDAC) tumors and control tissues, revealing the cellular heterogeneity and progression mechanisms within PDAC. The study identified a high degree of heterogeneity in PDAC, encompassing various malignant and stromal cell types. The malignant subtypes were found to consist of multiple subpopulations with distinct proliferation and migration capabilities. Additionally, the study observed a correlation between a subset of ductal cells and the inactivated state of tumor-infiltrating T cells. These findings provide valuable resources and insights for understanding the intra-tumoral heterogeneity in PDAC and identifying potential biomarkers for anti-cancer therapies [26].
There have also been studies through single-cell RNA sequencing (scRNA-seq) to analyze metastatic lung cancer before and during targeted therapy. They found a diverse and dynamic tumor ecosystem consisting of cancer cells and the tumor microenvironment (TME). The scRNA-seq analysis identified targetable oncogenes in cancer cells and revealed different gene expression patterns in residual disease (RD) and progressive disease (PD) under therapy. RD cells showed a transition to a primitive cell state, while PD cells exhibited upregulated pathways related to kynurenine, plasminogen, and gap junctions. This study also observed the presence of active T-lymphocytes and reduced macrophages in RD, while immunosuppressive cell states characterized PD. These biological features identified through scRNA-seq analysis served as biomarkers for clinical outcomes in independent patient cohorts. This research highlights the impact of therapy-induced adaptations in the multi-cellular ecosystem of metastatic lung cancer on treatment response and patient outcomes [32].
Another study analyzed cancer-associated fibroblasts (CAFs) in gastric cancer using single-cell RNA sequencing. The researchers discovered the heterogeneity of CAFs and their dynamic communication with components of the tumor microenvironment. Four distinct subsets of CAFs were identified, and two subsets, namely inflammatory CAFs (iCAFs) and extracellular matrix CAFs (eCAFs), exhibited enhanced pro-invasive activities and interactions with immune cell subsets. eCAFs were also associated with poorer overall survival in patients. Therefore, inhibiting the activation of these CAF subsets holds promise as a therapeutic strategy for improving the treatment of gastric cancer [33].
After the first single-cell transcriptome sequencing paper was published in 2009 [19], there have been significant improvements in the capacity and resolution of single-cell sequencing libraries. These advancements have enabled us to simultaneously explore whole transcriptome profiles of gene expression in thousands of cells (Table 1).
Single-cell transcriptomics has also been integrated with various novel technologies to provide a more comprehensive understanding of tumor heterogeneity. CITE-seq is an example of such technology that combines transcriptome analysis with phenotypic characterization, allowing simultaneous sequencing of the single-cell transcriptome and indexing of cell surface proteins [47]. CROP-seq, by integrating CRISPR screening with single-cell transcriptome resolution, facilitates comprehensive analyses of complex regulatory mechanisms and different cell populations, enabling high-throughput functional profiling [48, 49]. The LINNAEUS technique enables simultaneous lineage tracing and transcriptomic analysis of numerous single cells, providing a systematic approach for tracing the origins of novel cell types or identifying known cell types under different conditions [50]. These advantages make single-cell transcriptome sequencing technology particularly suitable for studying the heterogeneity of tumor cells.
Table 1 presents the unique advantages and limitations of single-cell transcriptomics in terms of characteristics, throughput, and applicability.
Epigenomes
The significance of epigenetic regulation in the progression of development and disease is widely recognized. The introduction of single-cell sequencing technologies has facilitated the development of various methods for analyzing epigenetic regulation at different levels, including chromatin accessibility, DNA methylation, histone modifications, and nucleosome positioning, among others. These technologies have enabled researchers to investigate various aspects of epigenetics at the single-cell level [20, 51]. Such techniques have emerged as powerful tools for uncovering the distinctive epigenomic characteristics of rare cellular subtypes and the epigenetic diversity present within cellular populations.
Chromosome accessibility
Nucleosomes serve as the fundamental structural units of eukaryotic chromatin which consist of DNA wrapped around a core of histone proteins. The chromatin structure is further organized into a three-dimensional arrangement, which includes densely packed regions and more open, accessible regions. These distinct regions of chromatin structure play a crucial role in regulating gene expression. During replication and transcription processes, the tightly packed chromatin structure needs to be opened up to expose specific DNA sequences for regulatory factors to bind and carry out their functions. This opening up of the chromatin structure to allow regulatory factor binding is referred to as chromosome accessibility [52]. It involves the dynamic modulation of the chromatin structure, allowing access to the DNA. Studies have demonstrated that the accessibility of DNA sequences within the chromatin structure can influence gene transcription activity. Changes in chromatin accessibility can either promote or repress gene expression [52, 53]. This process is mediated by various mechanisms, including histone modifications, chromatin remodeling complexes, and interactions with transcription factors.
Chromatin accessibility is a field of active research and understanding its impact on gene expression has important implications in various cellular processes and diseases. By studying the accessibility of DNA within chromatin, scientists can gain insights into the mechanisms underlying gene regulation and potentially discover new targets for therapeutic interventions. The field of single-cell chromatin accessibility sequencing has made significant progress, enabling the detailed exploration of chromatin accessibility in individual cells shown in Fig. 4. Among the techniques developed for this purpose, single-cell ATAC-seq (scATAC-seq) has emerged as a powerful method. scATAC-seq leverages the Tn5 transposase’s sensitivity to identify open and accessible chromatin regions [54]. It allows for the investigation of chromatin accessibility landscapes at a single-cell resolution, providing insights into the epigenetic regulation and heterogeneity within tumors.

Insights into the dynamic regulation of chromatin accessibility and methods for single-cell chromatin accessibility sequencing
In addition to scATAC-seq, other techniques have been employed to assess chromatin accessibility at the single-cell level. For example, single-cell DNase-seq (scDNase-seq) utilizes DNase I digestion to identify accessible DNA regions in individual cells [55]. This method enables the detection of regions of open chromatin and provides valuable information about transcription factor binding and regulatory elements. Other new technologies are shown in Table 2.
These single-cell chromatin accessibility methods have been widely applied in tumor research, offering insights into the heterogeneity and regulatory dynamics within tumor cell populations. For instance, a study utilized single-cell ATAC-seq technology to analyze the cellular composition and state changes that occur during the transformation from healthy colon to precancerous adenomas to colorectal cancer (CRC). It revealed the cellular composition and state changes that occur during the transformation from healthy colon to adenomas and subsequently to CRC. In the cancerous state, the study observed T cell exhaustion, RUNX1-regulated cancer-associated fibroblasts, and increased accessibility associated with HNF4A motifs in epithelial cells. Furthermore, in sporadic CRC, the DNA methylation changes were strongly anti-correlated with the accessibility changes along this continuum, thus providing additional regulatory markers for molecular staging of polyps [55].
The process of tumor metastasis is a prominent contributor to mortality among individuals diagnosed with cancer [62, 63], and the utilization of scATAC-seq has significantly enhanced our comprehension of the underlying mechanisms involved in tumor metastasis. A study identified novel cell subpopulations with abnormally high CXCL14 expression levels in patients with breast cancer PL by transposase accessible chromatin (ATAC) sequencing (scATAC-seq) of breast cancer negative (NL) and positive lymph nodes (PL), and also identified potential regulators that may be associated with breast cancer lymph node metastasis, improving our understanding of the mechanism of lymph node metastasis of lymph node metastasis and provide a new prognostic marker for breast cancer lymphatic metastasis [64]. Another study confirmed that TCF7 promotes epithelial-mesenchymal transition (EMT) and that activation of EMT is a key process in cancer metastasis by performing single-cell RNA sequencing and scATAC-seq on tumors from patients with low risk of recurrence, high risk of recurrence, and recurrent bladder cancer [65].
The tumor microenvironment encompasses diverse cellular and structural constituents that exert a pivotal influence on tumor advancement and resistance to therapeutic interventions. Moreover, the examination of single-cell chromatin accessibility can be extended to the analysis of the tumor microenvironment. In one study, by performing scRNA-seq and scATAC-seq on ccRCC primary tumor tissues, investigators identified key regulatory molecules in the tumor microenvironment that mediate tumor progression and manipulate immune cell function, and further experimentally validated their role in tumor growth [66]. Another study identified regulatory mechanisms associated with CD8 T-cell depletion by analyzing scATAC-seq profiles of serial tumor biopsies before and after programmed cell death protein 1 blockade [67].Twenty-two human IDH mutant gliomas were analyzed by scATAC-seq, which explained how different subtypes of IDH mutant gliomas maintain different phenotypes and tumor microenvironments despite being derived from a common spectral hierarchy, and found that ATRX regulates glial identity and tumor microenvironment in IDH mutant gliomas [68].
scATAC-seq proves to be a valuable tool for investigating alterations in cellular response to therapeutic interventions throughout the course of tumor treatment. To improve the clinical outcome of CAR-T cell therapy, a study by scATAC-seq of sorted T-cell subsets from seven patient, it was found that IRF7-regulated chronic IFN signaling was associated with poor persistence of CAR-T cells in T-cell subsets and that TCF7 regulators were relevant not only in maintaining naive and early memory T-cell states, but also in maintaining a good phenotype in effector cell lineages play a role [69]. Another study, by scATAC-seq of 12 breast cancer patients, found that the transcription factor GRHL2 cooperates with FOXA1 to initiate endocrine resistance and that epigenetic heterogeneity may contribute to endocrine resistance in breast cancer patients [70].
Table 2 presents the unique advantages and limitations of single-cell chromatin accessibility in terms of characteristics, throughput, and applicability.
DNA methylation
DNA methylation is a modification in which –CH3 methylation modification occurs on cytosine bases [71], specifically 5mC (5-methylcytosine) in this article. This methylation process is catalyzed by the DNA methyltransferase (DNMT) family, which adds methyl to the cytosine of the 5′ terminal CpG dinucleotide of human genes. A CpG island is a region in the 5′ → 3′ direction of a DNA sequence that is rich in CG dinucleotides. CpG islands can be found in the promoters of more than two-thirds of all genes and in most cases these CpG islands are in an unmethylated state. CpG islands act as definitive markers of DNA methylation and act as key switches in epigenetic regulation [72], restricting gene expression in the presence of methylation. Studies have shown that methylation of CpG islands plays an important role in transcriptional regulation and is usually altered during malignant transformation. Approximately 5–10% of normal unmethylated CpG island promoters are aberrantly methylated in various cancer genomes. Methylation of promoter CpG islands mediates the phenomenon of gene silencing observed during tumorigenesis [73, 74]. In addition, dysregulated DNA methylation is considered a hallmark of cancer. Genomic demethylation and gene-specific hypermethylation are prevalent in oncogenes and tumor suppressor genes [75,76,77] (Fig. 5).

Insights into the dynamic regulation of DNA methylation and methods for single-cell DNA methylation sequencing
With the advancement of next-generation sequencing (NGS) technologies, high-throughput methylation sequencing methods have made significant progress, enhancing the accessibility and efficiency of sequencing. Among these methods, bisulfite sequencing is considered the “gold standard” for DNA methylation analysis due to its high accuracy and single-base resolution [78, 79]. This technology includes whole-genome bisulfite sequencing (WGBS), which evaluates the extent of methylation within CpG islands by converting unmethylated cytosine © to uracil (U) while leaving 5-methylcytosine (5mC) unchanged [80]. WGBS provides in-depth understanding of the DNA methylation patterns across the entire genome and has revolutionized our understanding of DNA methylation. To reduce costs and increase sample throughput, researchers have developed methods that target specific regions for methylation sequencing, such as reduced representation bisulfite sequencing (RRBS) [81]. RRBS utilizes restriction enzymes to enzymatically cleave the genome, reducing its complexity during sequencing and enriching the analysis for important regulatory regions like promoters and CpG islands where detailed methylation analysis can be performed.
However, previous methods relied on bulk sequencing, which averaged the methylation information of cell populations, unable to resolve the heterogeneity present within individual cells. With the development of single-cell sequencing technologies, single-cell methylation sequencing has become feasible. The first method based on single-cell bisulfite sequencing was single-cell methylation genome sequencing (scRRBS), which employs enzymatic cleavage to generate CpG-rich DNA fragments for subsequent library construction and sequencing [82]. However, the harsh conditions used in bisulfite conversion can lead to DNA degradation, resulting in DNA loss and reduced sequencing quality, ultimately affecting the data yield. To address this issue, researchers have developed PBAT (post-bisulfite adaptor tagging) to mitigate the loss caused by degradation. Other single-cell methylation sequencing methods have also been developed, as shown in Table 3.
Methylation events have a major impact on the regulation of cell fate, and single-cell DNA methylation sequencing has provided key new insights into the important issue of tumor heterogeneity. For instance, a study utilized single-cell bisulfite sequencing (scBS-seq) technology to characterize partial methylation domains (PMDs) within individual cells of colorectal cancer. The results revealed that over half of the genome was covered by PMDs, and Gain-PMDs, a specific subtype, exhibited a higher coverage of protein-coding genes. Furthermore, the study unveiled substantial epigenetic heterogeneity among different cells within the same tumor and demonstrated that DNA methylation in cells is influenced by the tumor microenvironment [97].
Another study emphasizes the significance of genetic and epigenetic heterogeneity within tumors and its impact on the evolutionary trajectory of cancer. The researchers utilized single-cell bisulfite sequencing analysis (MscRRBS) to investigate this heterogeneity in chronic lymphocytic leukemia (CLL). Their findings revealed that CLL cells exhibit high rates of epigenetic mutations, while showing minimal variation in mutation rates among individual cells. Through comprehensive single-cell analyses, the study elucidated the lineage diversity of CLL cells and their evolutionary patterns following treatment. Notably, the researchers observed specific lineage biases during therapy. By integrating genetic, epigenetic, and transcriptional information at the single-cell level, this study reconstructed the genealogical history of CLL, thereby providing valuable insights into the understanding of tumor development [98].
There are also articles that investigated the DNA methylation profiles of circulating tumor cells (CTCs) using the scBS-seq technique and revealed the subclonal structure, evolutionary history and classification of tissue-specific DNA methylation profiles in CTCs. The results indicate the heterogeneity of DNA methylation in CTCs and reveal the epigenetic regulatory mechanisms in cancer metastasis [99].
Prior epidemiological studies have established a significant association between the consumption of food contaminated with aflatoxin B1 and the incidence of hepatocellular carcinoma. Scientists utilized single-cell RRBS technology to investigate the hepatotoxic mechanism induced by aflatoxin B1 (AFB1) in S phase-arrested L02 cells. The study found that AFB1 caused apoptosis and S phase arrest in L02 cells, reduced mitochondrial membrane potential, increased reactive oxygen species generation, and led to an increase in DNA methylation levels. Through single-cell RRBS analysis, it was revealed that DNA methylation, regulated by the gonadotropin-releasing hormone receptor pathway, Wnt signaling pathway, and TGF-beta signaling pathway, was involved in the hepatotoxic mechanism induced by AFB1 in S phase-arrested L02 cells [1].
Hannah Demond et al. utilized single-cell bisulfite sequencing (scBS-seq) to investigate DNA methylation abnormalities caused by maternal effect mutations in the subcortical maternal complex (SCMC) of human oocytes. These mutations are associated with early embryonic failure, gestational abnormalities, and recurrent pregnancy loss. The researchers observed a genome-wide deficiency of DNA methylation in the oocytes of patients with SCMC mutations compared to normal oocytes. Both the germline differentially methylated regions (gDMRs) of imprinted genes and other sequence features that are normally methylated in oocytes were affected, indicating a lack of selectivity toward imprinted genes. The degree of methylation loss varied across different genomic features. Furthermore, analysis of a preimplantation embryo and molar tissue from the same patient revealed persistent methylation defects at imprinted genes after fertilization, while non-imprinted regions of the genome showed near-normal methylation levels after implantation. These findings emphasize the critical role of the SCMC in de novo methylation in the female germline and provide valuable insights into imprinting defects and potential therapeutic strategies [100].
Nevertheless, single-cell methylation sequencing does have limitations. Firstly, due to the limited amount of DNA within a single cell, single-cell sequencing often faces challenges of low coverage and sparse results. Secondly, compared to traditional bulk cell sequencing, single-cell sequencing may exhibit higher technological noise. For example, the DNA amplification process can introduce biases, resulting in oversampling or undersampling of certain methylation sites and impacting the final analysis results. Additionally, during the single-cell collection process, there may be loss of cellular information, such as spatial relationships between cells, and some cells may not be successfully sequenced due to isolation process-related damage. In conclusion, single-cell methylation sequencing is crucial in uncovering the methylation heterogeneity within individual cells. However, challenges and opportunities for improvement remain in terms of throughput, sequencing depth, and accuracy. As technology continues to advance and improve, we can expect wider applications of single-cell methylation sequencing in life science research.
Table 3 presents the unique advantages and limitations of single-cell DNA methylation in terms of characteristics, throughput, and applicability.
Histone modification
Within a cell, genomic DNA is not just a string of linear sequences but exhibits a highly complex three-dimensional (3D) spatial structure. This structure is largely dependent on histones, octamers composed of two units each of four core components (H2A, H2B, H3, H4) [101]. These histones possess protruding “tail” structures, which can be regulated via a series of chemical modifications.
Histone modifications represent a key epigenetic mechanism, encompassing, but not limited to, acetylation, methylation, phosphorylation, glycosylation, ubiquitination, and nitrosylation. These modifications are usually catalyzed by specific enzymes, such as histone methyltransferase (HMT) and histone acetyltransferase (HAT). These enzymes add specific chemical groups to the amino acid residues of the histones or remove them, thus altering the state of the chromatin [102].
HMT and HAT can be perceived as “writers,” responsible for adding chemical marks. In turn, histone demethylase (HDM) and histone deacetylase (HDAC) can be seen as “erasers,” tasked with erasing these marks [103]. This series of modifications affects the compactness of the chromatin structure, thereby changing its interaction with DNA, and ultimately leading to the activation or silencing of genes.
The advent of next-generation sequencing (NGS) technologies has been a significant leap forward in understanding gene regulation and epigenetic mechanisms. Early techniques such as chromatin immunoprecipitation (ChIP) microarrays paved the way for more advanced methods, most notably Chromatin Immunoprecipitation Sequencing (ChIP-Seq). Compared to its predecessors, ChIP-Seq offers unparalleled resolution down to the single base-pair level, reduced methodological artifacts, and comprehensive genomic coverage [104]. ChIP-Seq is the gold standard for genome-wide analyses of DNA–protein interactions, histone modifications, and nucleosome positioning. While it offers a wealth of data, traditional or ‘bulk’ ChIP-Seq falls short in assessing the variability in chromatin states across individual cells. This limitation was addressed in 2015, when a landmark paper by David Weitz and Bradley E Bernstein in Nature Biotechnology introduced the concept of single-cell ChIP-Seq108. This innovation permitted the analysis of histone modifications at the single-cell level, thereby unveiling a new dimension of chromatin state heterogeneity. Subsequent to this pioneering work, various methods have emerged to probe chromatin states at single-cell resolution. Techniques such as scCUT&Tag [105], CoBATCH [106], and scChIC-seq [107] have further refined our understanding of chromatin dynamics. Each of these methods summarized in Table 4 and Fig. 6.

Insights into the dynamic regulation of Histone Modification and methods for single-cell Histone Modification sequencing
The significance of single-cell protein modifications lies in revealing the identity and differentiation state of cells, unraveling cellular heterogeneity, studying disease mechanisms, and guiding treatment response and drug discovery. Analyzing protein modification patterns in individual cells helps to understand cellular function and regulation, identify cell-to-cell differences, and provide insights into disease mechanisms for potential therapeutic targets.
A study utilized high-throughput single-cell ChIP-seq technology to investigate chromatin heterogeneity and drug resistance in breast cancer. The researchers employed a microdroplet microfluidic platform to sequence and analyze the chromatin states of thousands of individual cells at single-cell resolution. The study revealed that in untreated drug-resistant tumors, there exists a subset of cells that share chromatin markers with drug-resistant cells, and these cells have lost the chromatin marker H3K27me3 associated with genes promoting drug resistance. This single-cell ChIP-seq technology offers a novel approach to studying the role of chromatin heterogeneity in cancer and other diseases, and aids in uncovering the regulatory mechanisms involved in cellular differentiation and development [116].
There are also study through scCUT&Tag method, combined with scalable nanopore and droplet-based single-cell platforms, was employed to analyze specific chromatin regions in individual cells. The focus was on analyzing the polycomb group (PcG) silencing regions marked by the histone modification H3K27me3 as an orthogonal approach to identify cell states based on chromatin accessibility. Results showed that scCUT&Tag analysis of H3K27me3 could distinguish different cell types in human blood and generate cell type-specific PcG landscapes in heterogeneous tissues. Furthermore, the study utilized scCUT&Tag to analyze H3K27me3 in brain tumor patients before and after treatment, identifying cell types in the tumor microenvironment and revealing heterogeneity in PcG activity between primary samples and post-treatment [117].
Another study utilized automated CUT&Tag chromatin profiling to investigate the impact of KMT2A oncofusion proteins in leukemias. By mapping fusion-specific targets across the genome, the researchers identified common and tumor-subtype-specific sites of aberrant chromatin regulation. They found that certain binding sites for KMT2A oncofusion proteins exhibited cell-to-cell heterogeneity and were associated with lineage plasticity. Additionally, they discovered that abnormal enrichment of H3K4me3 in gene bodies could be targeted by Menin inhibitors. The integration of automated and single-cell CUT&Tag techniques enabled the identification of epigenomic heterogeneity within patient samples and the prediction of therapeutic sensitivity [114].
Over the years, we have gradually come to recognize the complexity and importance of histone modifications and chromatin states in gene regulation. The emergence of single-cell technologies is a significant breakthrough, allowing us to explore previously uncharted layers of epigenetic regulation. New technologies are expected to emerge that integrate the advantages of existing methods, providing higher resolution and throughput, and possibly reducing costs as well. Furthermore, the continued development of computational analysis and machine learning will help in parsing increasingly complex datasets. In short, the field of single-cell histone modification is still evolving and holds the promise of providing us with a deeper understanding of gene regulation. It may also inspire our insights into cellular processes and disease mechanisms.
Table 4 presents the unique advantages and limitations of Histone Modification sequencing in terms of characteristics, throughput, and applicability.
Nucleosome localization
Nucleosome positioning refers to the precise localization of nucleosomes, which are structural units composed of an octamer of histone proteins and the wrapped DNA, on the genome [109]. In eukaryotic chromosomes, the binding of DNA to histones is not static, and the accurate determination of nucleosome positions on the genome, known as nucleosome positioning, is crucial for maintaining genome structure and function.
The regulation of nucleosome positioning is closely associated with the spatial organization of chromatin, DNA replication, transcription, and gene expression regulation [118]. Nucleosome positioning can influence the chromatin state and accessibility, thereby impacting the transcriptional activity of genes. The positions of nucleosomes on the genome can affect the binding of transcription factors and the accessibility of promoters, determining the transcriptional levels and patterns of genes.
The study of nucleosome positioning can be conducted using various experimental techniques and computational methods. Among them, micrococcal nuclease sequencing (MNase-seq) is a widely used approach that involves the digestion of chromatin with micrococcal nuclease to degrade nucleosome structures, releasing the core DNA of nucleosomes, which can then be sequenced using high-throughput sequencing technology [56]. By analyzing the sequencing data, the positions and positioning patterns of nucleosomes can be determined.
Recent studies have demonstrated that even within a homogeneous population of cells, different cells exhibit significant heterogeneity in chromatin states [119, 120], which may be related to heterogeneity in chromatin accessibility. scMNase-seq is a high-throughput sequencing technique used for studying nucleosome positioning at the single-cell level. It can uncover the heterogeneity of nucleosomes between cells and investigate cell-specific chromatin states and gene regulatory mechanisms [56].
In one research, through single-cell MNase-seq analysis, researchers identified distinct accessible chromatin regions in all lymphoid progenitor cells (ALP), early ILC progenitors (EILP), and ILC progenitors (ILCP). Within EILP, different subpopulations were identified, indicating their potential to differentiate into either dendritic cell lineage or ILC lineage based on epigenetic profiles. The researchers found that the transcription factors TCF-1 and GATA3 co-bound with lineage defining sites (LDS-Is) associated with ILC, while PU.1 binding was enriched in LDS (LDS-As) associated with alternative dendritic cell fate. TCF-1 and GATA3 were found to be essential for the epigenetic priming of LDS at the EILP stage. These findings suggest that the fate of multipotent progenitors during the differentiation into ILC and hematopoietic stem cells is pre-defined by their epigenetic state. The presence of distinct subpopulations within multipotent progenitors and their regulation by key transcription factors highlight the heterogeneity of cells that contribute to lineage specification. The application of single-cell MNase-seq technology enables a deeper understanding of the epigenetic changes during cellular differentiation, shedding light on the molecular mechanisms and role of transcription factors in determining cell fate [121].
Genomics
With the advancement of technology, researchers are able to study cells in a more detailed manner through methods such as single-cell genomics sequencing, rather than relying on bulk tissue samples alone. This provides us with a unique perspective that allows us to gain a deeper understanding of the underlying mechanisms of complex diseases, particularly cancer. Cancer can be understood as a genomic disorder in which the accumulation of somatic mutations and epigenetic modifications plays a crucial role in its development. Somatic mutations include various types of genomic alterations introduced into the genome, such as single nucleotide variants (SNVs), structural variations, gene splicing variants, and copy number variations [122]. These genomic alterations contribute to the occurrence of cancer and other diseases. Considering the frequencies of these changes in the human genome, it becomes particularly important to comprehensively understand the gene expression patterns of individual cells.
Traditional genomics sequencing involves sequencing DNA or RNA from populations or tissues. However, this method typically results in the average sequencing of the genome from the population and loses the genetic information of individual cells, as well as cellular heterogeneity, the ability to detect rare variations, distinguish between cell types, and lack of cellular functional information [123].
To overcome these limitations, the field of single-cell genomics (SCG) has emerged as an important technology within the genomics field, aiming to perform high-throughput sequencing of individual cells' genomes to reveal their genetic characteristics and structures, providing a new approach to understanding tumor development and disease progression [124]. Through single-cell genomics sequencing, researchers can obtain genetic information about individual cells, including gene mutations, copy number variations, chromosomal structures, and rearrangements. Additionally, it can reveal important features such as genomic-level cellular heterogeneity, cell type differentiation, and development.
Currently, the most common application of SCG is the analysis of copy number variations (CNVs). For example, in a study on hepatocellular carcinoma (HCC), single-cell DNA sequencing (scDNA-seq) revealed that the accumulation of CNVs follows a biphasic copy number evolution model, confirming for the first time at the single-cell level that multiploid hepatocellular carcinoma originates from diploid cells [125]. Another study examining colorectal cancer patients found that fibroblasts with somatic copy number alterations (SCNAs) in tumor tissues were significantly more abundant than in adjacent normal tissues, suggesting potential functional consequences or effects of these chromosomal-level SCNAs in tumor development. Fibroblasts with SCNA may interact with cancer cells, contributing synergistically to tumor development [126].
Mutation detection is another significant application of single-cell genomics (SCG), which holds great potential but can be costly. To enhance cost-effectiveness, a strategic approach involves initially conducting bulk sequencing, followed by targeted sequencing with increased depth focusing on specific loci of interest for mutation analysis. This sequential approach allows for a broader survey of genomic information through bulk sequencing, followed by a more in-depth analysis of specific regions of interest, optimizing both resource utilization and mutation detection efficiency.
This article employs single-cell mutational profiling to investigate myeloid malignancies, particularly acute myeloid leukemia (AML). The study reveals that AML is primarily driven by a small number of dominant clones, often carrying co-occurring mutations in epigenetic regulatory genes. Conversely, mutations in signaling genes tend to occur in distinct subclones, contributing to increased clonal diversity. Through single-cell analysis, interactions between mutations are unveiled, along with the relationship between immunophenotype and somatic genotype/clonal architecture. This research provides valuable insights into the pathogenesis of myeloid malignancies and the evolution of disease progression.
With the continuous advancement of sequencing technologies, single-cell whole-genome sequencing (scWGS) has become possible. scWGS is a technique for analyzing the DNA of individual cells, enabling the sequencing of their complete genomes at the single-cell resolution [127]. It involves amplifying the DNA of single cells through methods such as multiple displacement amplification (MDA) or polymerase chain reaction (PCR), and the amplified DNA is then subjected to high-throughput sequencing technologies such as Illumina sequencing. The sequencing data obtained through scWGS can provide valuable information about the genomic features of individual cells within heterogeneous cell populations. It can help identify genetic variations within single cells, including single nucleotide variants (SNVs), copy number variations (CNVs), and structural variations (SVs).
For example, a study utilized scWGS to analyze recurrent liver metastatic lesions in patients with metastatic colorectal cancer. The study found that treatment resulted in a severe reduction in the number of tumor cells in the liver metastatic lesions, but previously differentiated cell lineages remained alive and potentially survived through migration to different sites within the liver. These cell lineages underwent slow evolution under adjuvant drug treatment for 2 years and then rapidly diversified within a short period of time. The study also identified several non-silent mutations specific to these cell lineages and speculated that chemotherapy contributed significantly to the overall genomic mutational burden. Overall, the study revealed that subclones of metastatic colorectal cancer could undergo local migration and escape surgical resection, continue to evolve under chemotherapy, and exhibit explosive re-expansion [128].
Another study used scWGS to analyze cells from a Chinese female patient with cervical cancer before and after radiotherapy. They discovered a deleterious mutation in the NFKB12 gene that increased in frequency after radiotherapy. Functional analysis revealed that NFKB430 acts as a tumor suppressor, and the mutation in NFKB1 weakened its tumor suppressive function. NFKB1 enhanced radiation sensitivity, and its mutation reduced this effect. The study suggests that NFKB1 could be a potential molecular target for future cervical cancer radiotherapy [129].
However, scWGS has certain limitations when it comes to detecting copy number variations (CNVs), such as lower accuracy and insufficient amplification fidelity. To overcome these limitations, a series of new techniques, including MALBAC, eMDA, LIANTI, SISSOR, and META-CS, have emerged to improve the accuracy of CNV detection and reduce false-positive rates. Table 5 provides an overview of single-cell genomics sequencing methods.
Table 5 presents the unique advantages and limitations of single-cell genomic sequencing in terms of characteristics, throughput, and applicability.
Multi-omics
Recent advances in single-cell genomics technology have allowed us to analyze the epigenome, genome, and transcriptome of individual cells with single-cell resolution, greatly enhancing our ability to study tumor heterogeneity. Tumors can be viewed as ecosystems with complex interactions between different phenotypes [140]. However, analyzing cells at the individual level may only provide a partial picture of the intricate regulatory networks, limiting our understanding of the mechanisms driving cancer initiation and progression. Currently, advanced analysis techniques and experimental methods are being developed to effectively capture information from multiple epigenomic layers within individual cells. By integrating epigenomics with other omics technologies, we can gain a more comprehensive understanding of these intricate interactions, thus unraveling the nature of tumor heterogeneity. In cancer research, the epigenome, transcriptome, and genome exhibit significant heterogeneity. The integration of single-cell epigenomics with multi-omics analysis holds promise in better elucidating the mechanisms driving cancer initiation and development, as well as the regulatory heterogeneity [141] (Table 6).
In recent years, multi-omics technologies have emerged as crucial tools for deciphering the complexity of biological systems, particularly at the single-cell level. Independently, single-cell transcriptomics and single-cell epigenomics provide rich information about gene expression and epigenetic regulation. However, integrating these two single-cell-level holistic analytical methods has the potential to reveal deeper insights. This integration is especially critical in the context of tumor biology. Individually, these technologies can describe the transcriptional states, expression profiles, and epigenetic features of individual cells within tumor tissues. However, by jointly analyzing these two types of data, we can not only identify epigenetic patterns within specific cell populations but also unravel how these patterns directly influence or are influenced by transcriptional activity. Specifically, such multi-omics analysis enables us to explore in detail how epigenetic regulation affects the maintenance and transition of cell identity. Furthermore, it helps us understand how these epigenetic mechanisms manipulate cellular fate, driving cells into specific lineage trajectories and generating varying levels of heterogeneity within tumor tissues [142].
For example, scCAT-seq, which stands for single-cell chromatin accessibility and transcriptomics sequencing, is an advanced multi-omics technique that enables simultaneous analysis of chromatin accessibility and gene expression within individual cells [143]. It combines the strengths of single-cell chromatin accessibility analysis such as ATAC-seq or DNase-seq, with single-cell RNA sequencing (scRNA-seq), providing a comprehensive view of the epigenetic and transcriptional landscape within single cells. By integrating chromatin accessibility and transcriptomic data obtained from scCAT-seq, researchers can study the relationship between chromatin structure and gene expression at single-cell resolution. This integrative analysis allows for the identification of regulatory elements involved in specific transcriptional programs and characterization of cellular heterogeneity based on epigenetic and transcriptional states. The scCAT-seq technique successfully acquired precise chromatin accessibility (CA) and gene expression (GE) information in lung, cervical, and colorectal cancers [143]. Other tools for joint transcriptome chromatin accessibility analysis include sci-CAR-seq [144], SHARE-seq [145], SNARE-seq [146], ASTAR-seq [147], and Joint scATAC-Seq/scRNA-seq [148].
Smart-RRBS is an advanced single-cell multi-omics technique that provides single-cell-level information on genomic DNA methylation and transcriptome. This technology is of great significance in exploring cellular heterogeneity, developmental processes, and epigenetic regulation in disease mechanisms [149, 150]. By integrating genomic DNA methylation and gene expression data, Smart-RRBS enables in-depth analysis of the functionality and regulatory patterns of gene regulatory networks within cells.
One notable study utilized this technique to examine chronic lymphocytic leukemia (CLL) and glioblastoma cells, uncovering the crucial role of intratumoral epigenetic heterogeneity in cancer progression. For instance, Gaiti et al. [150] employed Smart-RRBS to map CLL lineage history and predict its evolution after therapy by leveraging epigenetic information. Another study by Chaligne et al. focused on glioma cells, revealing the heritability of epigenetic patterns within this population. They discovered distinct variations in cell plasticity states between IDH-mutant and IDH-wild-type glioblastoma, demonstrating the significance of epigenetic traits in differentiating glioblastoma subtypes [1]. Methods for simultaneous analysis of DNA methylome and transcriptome from the same cell at single-cell resolution also include scMT-seq [151], scmCT-seq [152], scM&T-seq [153], and scGEM [154]. Single-cell transcriptomes can also be combined with histone modifications and protein-DNA interactions, such as Paired-Tag [155], scDAM&T-seq [156].
The regulation of gene expression involves a dynamic interplay in vivo among various epigenomic layers, including DNA methylation, chromatin accessibility, and others, rather than functioning independently of one another. cNMT-seq [157], scNOMe-seq [158], scChaRM-seq [159], and snmCAT-seq [160] can combine single-cell transcriptomics and multiple epigenomic layers to analyze the relationship between multiple epigenetic features and gene expression at single-cell resolution and play a crucial role in further understanding the epigenetic-dependent associations on transcription and cellular states. For instance, Bian et al. used scTrio-seq to study colorectal cancer tumors and metastases from 10 patients. They found that DNA methylation levels were consistent within genetic lineages but varied significantly among clones. The technique provided insights into tumor evolution and the relationship between DNA methylation and genetic lineages. Overall, scTrio-seq allowed for a comprehensive analysis of mutations, transcriptome, and methylome in individual cells, shedding light on tumor heterogeneity and metastasis [161].
There are many other tools that can combine single-cell proteomics with single-cell epigenomics such as Pi-ATAC [162] and ASAP-seq [163]. Pi-ATAC can identify both epigenomic and proteomic heterogeneity in a single cell. A study by quantifying the protein levels of Pi-ATAC on TFs NF-kB and HIF1α in mouse mammary tumors and measuring the DNA occupancy of both found that the primary role of HIF1α protein in the tumor microenvironment of tumor hypoxia is through shaping the regulatory groups in parenchymal tumor cells and infiltrating immune cell subpopulations [162].
Some tools can explore the epigenome of several layers of a single cell simultaneously to achieve a more comprehensive epigenomic analysis, such as scNOME-seq (chromatin accessibility + DNA methylation status) [164], scMethyl-HiC [164] and sn-m3C-seq (chromatin 3D structure + methylome) [165].
Other multi-omics analysis solutions include scCOOL-seq [166] and iscCOOL-seq [167] that allow simultaneous analysis of chromatin state/nucleosome localization, DNA methylation, copy number variation and chromosome ploidy in individual cells, enabling the combination of different epigenomic sequencing layers in a single cell at the same time. Although integrated multi-omics technologies such as iscCOOL-seq in single-cell analysis have tremendous potential in exploring the interplay between multiple layers of the epigenome and the genome within individual cells, their application in tumor research is still not widely adopted. This relatively new technology faces several challenges and limitations. Firstly, the maturity of these techniques is relatively low, requiring more laboratories to become familiar with and master the experimental and data analysis workflows. Secondly, the experimental procedures of these techniques are complex and require optimization of experimental conditions and coordination of multiple steps. Additionally, the complexity of data interpretation and analysis poses a challenge, necessitating the development of more reliable multi-omics data analysis methods and resources. Lastly, sample requirements and feasibility are also limiting factors for the application of these technologies, particularly in obtaining a sufficient quantity and quality of single-cell samples from tumors. Despite these challenges, we can anticipate further research efforts to overcome the technical challenges, improve data interpretation and analysis, and expand feasibility, thereby promoting the widespread application of technologies like iscCOOL-seq in tumor research to uncover mechanisms of tumor development and facilitate precision therapeutics.
Table 6 presents the unique advantages and limitations of multi-omics sequencing in terms of characteristics, throughput, and applicability.
Concluding remarks and future perspectives
Nowadays, cancer medicine has entered the era of precision medicine, and single-cell sequencing technologies based on next-generation sequencing are rapidly advancing. Traditional bulk sequencing methods often provide analysis based on the average values of multiple cells, which cannot provide a high-resolution view of the cellular composition in the tumor ecosystem. Compared to bulk sequencing methods that provide averaged data, single-cell sequencing has significant advantages. It allows analysis of the transcriptome, genome, and epigenome of individual tumor cells, revealing the heterogeneity within the tumor.
The interplay of various "omics" within a cell follows a complex regulatory mechanism, starting from the genome and epigenome, extending to the transcriptome, proteome, and back again [22]. This intricate process highlights the crucial role of epigenetic regulation in the gene regulatory network. Cancer can be viewed retrospectively as an epigenetic disease, wherein the identity and function of different cell types within various tumor cells are determined by the epigenetic landscape of those cells. Consequently, non-genetic factors significantly contribute to tumor progression [141]. The study of epigenetics has greatly enhanced our understanding of tumor heterogeneity. In recent years, numerous cutting-edge techniques have been developed to analyze epigenetic modifications at the single-cell resolution level. These techniques encompass the examination of chromatin accessibility, DNA/RNA methylation, histone modifications, and nucleosome localization. They provide invaluable research methods for investigating the intricate relationship between tumor epigenetic modifications and tumor heterogeneity.
To comprehensively understand the complex epigenetic regulatory mechanisms driving tumor cell development, progression, and drug resistance within the tumor microenvironment, it is often insufficient to rely solely on single-cell epigenomic analysis. Rather, the integration of multiple omics data at a single-cell resolution is necessary. This entails considering genomic, epigenomic, transcriptomic information.
By employing single-cell multi-omics techniques and analyses, it becomes possible to simultaneously capture multiple layers of epigenetic information together with other omics data. For instance, a commonly used approach involves combining single-cell epigenomics and transcriptomics, allowing researchers to investigate the relationship between the epigenome and gene expression at the genomic level in individual cells [168]. Furthermore, multi-omics studies that incorporate DNA methylation patterns, open chromatin regions, nucleosome positioning, DNA–protein interactions, genomic alterations (mutations and copy number variations), as well as mRNA or protein abundance at the single-cell level, have been employed in tumor heterogeneity research. These comprehensive analyses enable a more in-depth understanding of the epigenome and highlight the significance of epigenetic heterogeneity within tumors in cancer progression [168].
However, the application of single-cell multi-omics analysis to investigate tumor heterogeneity is still in its early stages and faces technical and computational challenges. These challenges include limitations in obtaining valid information, which can result in missing values, systematic noise, and issues with sample coverage. These factors can impact the final analysis of the data, thereby affecting our understanding of tumor heterogeneity.
For example, traditional bisulfite-based methods used to measure single-cell DNA methylation can introduce DNA damage and compromise the accuracy of DNA methylome sequencing. To overcome this limitation, the application of third-generation/real-time single molecule sequencing (TGS) can be employed [169]. TGS has the advantage of directly detecting epigenetic modifications, including DNA 5 mC and 6 mA, without the need for bisulfite conversion or PCR amplification. Additionally, recent advancements in nanopore technology, such as SMAC-seq, Fiber-seq, and nanoNOMe, have enabled long-range detection of single-molecule chromatin states by combining nanopore sequencing with m6A methyltransferase or M. CviPI GpC methyltransferase enzyme accessibility [170,171,172]. These innovations reduce DNA damage and improve the accuracy of DNA methylome sequencing, providing researchers with a more precise tool for studying epigenetic modifications.
The integration of spatial omics with single-cell omics such as (spatial proteomics, spatial transcriptomics, spatial epigenomics) has improved our understanding of the evolution of cancer in temporal and spatial dimensions. However, the study of tumors by spatial omics is still in its infancy, and the next step is to extend spatial mono-omic to spatial multi-omics by converting spatial information into DNA barcodes, or imaging-based methods to preserve spatial information of each cell [23]. The spatial distribution of epigenome and transcriptome is a key determinant of the cellular characteristics, and the extension of spatial transcriptomic approaches to epigenomic analysis is a critical step forward in our efforts to unravel the epigenetic drivers underlying tumor heterogeneity.
A 3D tumor-like organ model with certain spatial structure constructed by culturing tumor stem cells in vitro has the ability to stably preserve the epigenomic, proteomic, transcriptomic, morphological and pharmacological features of the parent tumor [173]. Although a tumor-like organ is not a genuine human tumor, it has received widespread attention for its ability to simulate real organs structurally and functionally, which closely reflects the pathophysiological characteristics of tumorigenesis and metastasis in humans. With the advent of the era of spatial multi-omics technology, biomarkers based on peripheral blood and bulk tissue biopsies will not be able to meet the demand of transferring laboratory results to medical and clinical research. Tumor-like organs can mimic the real environment of tumors in human body, with the advantages of closer physiological cell composition and behavior, more stable genome, more suitable for biotransfection and high-throughput screening, providing a fast and effective way for tissue spatial biomarker discovery and drug screening. It provides a rapid and effective platform for tissue spatial biomarker discovery and drug screening and offers a validation model for us to explore the key epigenetic mechanisms behind the prognosis and therapeutic resistance of tumor patients.
Although single-cell sequencing has improved our understanding of the relationship between epigenetics and tumor heterogeneity, we have to face the following obstacles. Firstly, due to the large amount of information in the epigenome (tens of fold larger than the transcriptome) which is distributed throughout the genome, the coverage of the epigenome of a single cell by current methods is still relatively thin, and research errors arise when distinguishing technical noise from intercellular differences, and novel biochemistry or strategies may be needed to overcome this limitation in the future. In addition, multi-omics analysis of a single cell at a time, although providing a comprehensive molecular profile, generates very complex data that needs to be analyzed both to match information between omics and to reduce errors and biases generated in data matching, thus requiring simplified and targeted analysis of the data of interest. Finally, large-scale single-cell analysis of complex tumor samples can be expensive due to the low throughput and high single-cell cost of single-cell sequencing technologies. These characteristics determine that single-cell technology still has a long way to go before it can be widely used in clinical cancer research, but it is certain that the future of single-cell epigenetics in clinical applications is bright, and perhaps in the near future, further understanding of single-cell epigenetics will undoubtedly improve our knowledge of tumor cell heterogeneity and the prognosis and drug resistance mechanisms of tumor patients, providing patients with more individualized clinical applications.
Availability of data and materials
Not applicable; all information in this review can be found in the reference list.
Abbreviations
- TME:
-
Tumor microenvironment
- CTC:
-
Circulating tumor cell
- CSC:
-
Cancer stem cell
- SNV:
-
Single-nucleotide variation
- CNV:
-
Copy number variation
- Sv:
-
Structural variation
- HCC:
-
Hepatocellular carcinoma
- M1:
-
Classically activated macrophages
- M2:
-
Alternatively activated macrophages
References
Waddington CH. The epigenotype. Int J Epidemiol. 2012;41(1):10–3. https://doi.org/10.1093/ije/dyr184.
Harvey ZH, et al. Protein-based inheritance: epigenetics beyond the chromosome. Mol Cell. 2018;69(2):195–202. https://doi.org/10.1016/j.molcel.2017.10.030.
Buenrostro JD, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8. https://doi.org/10.1038/nmeth.2688.
McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613–28. https://doi.org/10.1016/j.cell.2017.01.018.
McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27(1):15–26. https://doi.org/10.1016/j.ccell.2014.12.001.
Turajlic S, et al. Resolving genetic heterogeneity in cancer. Nat Rev Genet. 2019;20(7):404–16. https://doi.org/10.1038/s41576-019-0114-6.
Shaffer SM, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546(7658):431–5. https://doi.org/10.1038/nature22794.
Chen JF, Yan Q. The roles of epigenetics in cancer progression and metastasis. Biochem J. 2021;478(17):3373–93. https://doi.org/10.1042/BCJ20210084.
Chen Y, et al. ALKBH5 suppresses malignancy of hepatocellular carcinoma via m6A-guided epigenetic inhibition of LYPD1. Mol Cancer. 2020;19(1):123. https://doi.org/10.1186/s12943-020-01239-w.
Lu C, et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer. 2019;18(1):130. https://doi.org/10.1186/s12943-019-1047-6.
Qu S, et al. A positive-feedback loop between HBx and ALKBH5 promotes hepatocellular carcinogenesis. BMC Cancer. 2021;21(1):686. https://doi.org/10.1186/s12885-021-08449-5.
Ma J-Z, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6 -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65(2):529–43. https://doi.org/10.1002/hep.28885.
Zhang C, et al. YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene. 2020;39(23):4507–18. https://doi.org/10.1038/s41388-020-1303-7.
Chen M, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–70. https://doi.org/10.1002/hep.29683.
Lin Z, et al. RNA m6 A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. 2020;39(12):e103181. https://doi.org/10.15252/embj.2019103181.
Hou J, et al. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Mol Cancer. 2019;18(1):163. https://doi.org/10.1186/s12943-019-1082-3.
Lei Y, et al. Applications of single-cell sequencing in cancer research: progress and perspectives. J Hematol Oncol. 2021;14(1):91. https://doi.org/10.1186/s13045-021-01105-2.
Anna S, et al. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet. 2021;22(1):3–18. https://doi.org/10.1038/s41576-020-0265-5.
Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82. https://doi.org/10.1038/nmeth.1315.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. https://doi.org/10.1016/j.cell.2012.06.013.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Nat Rev Genet. 2021;22(4):235–50. https://doi.org/10.1038/s41576-020-00300-0.
Vandereyken K, et al. Methods and applications for single-cell and spatial multi-omics. Nat Rev Genet. 2023. https://doi.org/10.1038/s41576-023-00580-2.
Zhu C, et al. Single-cell multimodal omics: the power of many. Nat Methods. 2020;17(1):11–4. https://doi.org/10.1038/s41592-019-0691-5.
Jovic D, et al. Single-cell RNA sequencing technologies and applications: a brief overview. Clin Transl Med. 2022;12(3): e694. https://doi.org/10.1002/ctm2.694.
Wu H, et al. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol. 2019;30(1):23–32. https://doi.org/10.1681/ASN.2018090912.
Peng J, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019;29(9):725–38. https://doi.org/10.1038/s41422-019-0195-y.
Coons AH, et al. Immunological properties of an antibody containing a fluorescent group. Proc Soc Exp Biol Med. 1941;47:200–2.
Li P-H, et al. Recent developments in application of single-cell RNA sequencing in the tumour immune microenvironment and cancer therapy. Military Med Res. 2022;9(1):52. https://doi.org/10.1186/s40779-022-00414-y.
Ding S, et al. Single-cell RNA sequencing in breast cancer: understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun (Lond Engl). 2020;40(8):329–44. https://doi.org/10.1002/cac2.12078.
Zhang Z, et al. Integrated analysis of single-cell and bulk RNA sequencing data reveals a pan-cancer stemness signature predicting immunotherapy response. Genome Med. 2022;14(1):45. https://doi.org/10.1186/s13073-022-01050-w.
Zhang Y, et al. Single-cell RNA sequencing in cancer research. J Exp Clin Cancer Res. 2021. https://doi.org/10.1186/s13046-021-01874-1.
Maynard A, et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell. 2020;182(5):1232-1251.e22. https://doi.org/10.1016/j.cell.2020.07.017.
Li X, et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics. 2022;12(2):620–38. https://doi.org/10.7150/thno.60540.
Hashimshony T, et al. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2(3):666–73. https://doi.org/10.1016/j.celrep.2012.08.003.
Hashimshony T, et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016;28(17):77.
Jaitin DA, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343(6172):776–9. https://doi.org/10.1126/science.1247651.
Keren-Shaul H, et al. MARS-seq2.0: an experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat Protoc. 2019;14(6):1841–62.
Sasagawa Y, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013;14(4):R31.
Sasagawa Y, et al. Quartz-Seq2: a high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads. Genome Biol. 2018;19(1):29. https://doi.org/10.1186/s13059-018-1407-3.
Bagnoli JW, et al. Sensitive and powerful single-cell RNA sequencing using mcSCRB-seq. Nat Commun. 2018;9(1):2937. https://doi.org/10.1038/s41467-018-05347-6.
Ramsköld D, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30(8):777–82. https://doi.org/10.1038/nbt.2282.
Picelli S, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10(11):1096–8. https://doi.org/10.1038/nmeth.2639.
Hagemann-Jensen M, et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat Biotechnol. 2020;38(6):708–14. https://doi.org/10.1038/s41587-020-0497-0.
Goldstein LD, et al. Massively parallel nanowell-based single-cell gene expression profiling. BMC Genom. 2017;18(1):519. https://doi.org/10.1186/s12864-017-3893-1.
Macosko EZ, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–14. https://doi.org/10.1016/j.cell.2015.05.002.
Klein AM, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161(5):1187–201. https://doi.org/10.1016/j.cell.2015.04.044.
Stoeckius M, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–8. https://doi.org/10.1038/nmeth.4380.
Datlinger P, et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods. 2017;14(3):297–301. https://doi.org/10.1038/nmeth.4177.
Hill AJ, et al. On the design of CRISPR-based single-cell molecular screens. Nat Methods. 2018;15(4):271–4. https://doi.org/10.1038/nmeth.4604.
Spanjaard B, et al. Simultaneous lineage tracing and cell-type identification using CRISPR-Cas9-induced genetic scars. Nat Biotechnol. 2018;36(5):469–73. https://doi.org/10.1038/nbt.4124.
Recillas-Targa F. Cancer epigenetics: an overview. Arch Med Res. 2022;53(8):732–40. https://doi.org/10.1016/j.arcmed.2022.11.003.
Klemm SL, et al. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet. 2019;20(4):207–20. https://doi.org/10.1038/s41576-018-0089-8.
Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010;2010(2):pdb.prot5384. https://doi.org/10.1101/pdb.prot5384.
Buenrostro JD, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–90. https://doi.org/10.1038/nature14590.
Becker WR, et al. Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat Genet. 2022;54(7):985–95. https://doi.org/10.1038/s41588-022-01088-x.
Lai B, et al. Principles of nucleosome organization revealed by single-cell micrococcal nuclease sequencing. Nature. 2018;562(7726):281–5. https://doi.org/10.1038/s41586-018-0567-3.
Cusanovich DA, et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348(6237):910–4. https://doi.org/10.1126/science.aab1601.
Wang K, et al. Simple oligonucleotide-based multiplexing of single-cell chromatin accessibility. Mol Cell. 2021;81(20):4319-4332.e10. https://doi.org/10.1016/j.molcel.2021.09.026.
Thornton CA, et al. Spatially mapped single-cell chromatin accessibility. Nat Commun. 2021;12(1):1274. https://doi.org/10.1038/s41467-021-21515-7.
Tedesco M, et al. Chromatin velocity reveals epigenetic dynamics by single-cell profiling of heterochromatin and euchromatin. Nat Biotechnol. 2022;40(2):235–44. https://doi.org/10.1038/s41587-021-01031-1.
De Rop FV, et al. Hydrop enables droplet-based single-cell ATAC-seq and single-cell RNA-seq using dissolvable hydrogel beads. Elife. 2022;11:e73971. https://doi.org/10.7554/eLife.73971.
Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352(6282):169–75. https://doi.org/10.1126/science.aaf2784.
Hunter KW, et al. Genetic insights into the morass of metastatic heterogeneity. Nat Rev Cancer. 2018;18(4):211–23. https://doi.org/10.1038/nrc.2017.126.
Xu K, et al. Integrative analyses of scRNA-seq and scATAC-seq reveal CXCL14 as a key regulator of lymph node metastasis in breast cancer. Hum Mol Genet. 2021;30(5):370–80. https://doi.org/10.1093/hmg/ddab042.
Wang H, et al. Single-cell analyses reveal mechanisms of cancer stem cell maintenance and epithelial-mesenchymal transition in recurrent bladder cancer. Clin Cancer Res. 2021;27(22):6265–78. https://doi.org/10.1158/1078-0432.CCR-20-4796.
Long Z, et al. Single-cell multiomics analysis reveals regulatory programs in clear cell renal cell carcinoma. Cell Discov. 2022;8(1):68. https://doi.org/10.1038/s41421-022-00415-0.
Satpathy AT, et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol. 2019;37(8):925–36. https://doi.org/10.1038/s41587-019-0206-z.
Babikir H, et al. ATRX regulates glial identity and the tumor microenvironment in IDH-mutant glioma. Genome Biol. 2021;22(1):311. https://doi.org/10.1186/s13059-021-02535-4.
Chen GM, et al. Integrative bulk and single-cell profiling of premanufacture T-cell populations reveals factors mediating long-term persistence of CAR T-cell therapy. Cancer Discov. 2021;11(9):2186–99. https://doi.org/10.1158/2159-8290.CD-20-1677.
Kumegawa K, et al. GRHL2 motif is associated with intratumor heterogeneity of cis-regulatory elements in luminal breast cancer. NPJ Breast Cancer. 2022;8(1):70. https://doi.org/10.1038/s41523-022-00438-6.
Yang B, et al. RNA methylation and cancer treatment. Pharmacol Res. 2021;174:105937. https://doi.org/10.1016/j.phrs.2021.105937.
Yuan Y. Spatial heterogeneity in the tumor microenvironment. Cold Spring Harb Perspect Med. 2016;6(8): a026583. https://doi.org/10.1101/cshperspect.a026583.
Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610. https://doi.org/10.1038/nrg1655.
Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer. 2011;11(10):726–34. https://doi.org/10.1038/nrc3130.
Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. 2019;20(2):109–27. https://doi.org/10.1038/s41576-018-0074-2.
Sharma S, et al. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. https://doi.org/10.1093/carcin/bgp220.
Haldrup C, et al. DNA methylation signatures for prediction of biochemical recurrence after radical prostatectomy of clinically localized prostate cancer. J Clin Oncol. 2013;31(26):3250–8. https://doi.org/10.1200/JCO.2012.47.1847.
Stevens M, et al. Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods. Genome Res. 2013;23(9):1541–53. https://doi.org/10.1101/gr.152231.112.
Stuart T, et al. Approaches for the analysis and interpretation of whole genome bisulfite sequencing data. Methods Mol Biol. 2018;1767:299–310. https://doi.org/10.1007/978-1-4939-7774-1_17.
Gong T, et al. Analysis and performance assessment of the whole genome bisulfite sequencing data workflow: currently available tools and a practical guide to advance DNA methylation studies. Small Methods. 2022;6(3): e2101251. https://doi.org/10.1002/smtd.202101251.
Gu H, et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6(4):468–81. https://doi.org/10.1038/nprot.2010.190.
Guo H, et al. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat Protoc. 2015;10(5):645–59. https://doi.org/10.1038/nprot.2015.039.
Smallwood SA, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11(8):817–20. https://doi.org/10.1038/nmeth.3035.
Li X, et al. BRIF-Seq: bisulfite-converted randomly integrated fragments sequencing at the single-cell level. Mol Plant. 2019;12(3):438–46. https://doi.org/10.1016/j.molp.2019.01.004.
Arlik M, Sheffield NC, Nuzzo A, et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015;10(8):1386–97. https://doi.org/10.1016/j.celrep.2015.02.001.
Wei CJY, Zhang K. RETrace: simultaneous retrospective lineage tracing and methylation profiling of single cells. Genome Res. 2020;30(4):602–10. https://doi.org/10.1101/gr.255851.119.
Mooijman D, et al. Single-cell 5hmC sequencing reveals chromosome-wide cell-to-cell variability and enables lineage reconstruction. Nat Biotechnol. 2016;34(8):852–6. https://doi.org/10.1038/nbt.3598.
Zhu C, et al. Single-cell 5fC sequencing. Methods Mol Biol. 2019;1979:251–67. https://doi.org/10.1007/978-1-4939-9240-9_16.
Wu X, et al. Simultaneous mapping of active DNA demethylation and sister chromatid exchange in single cells. Genes Dev. 2017;31(5):511–23. https://doi.org/10.1101/gad.294843.116.
Hunt KV, et al. scTEM-seq: single-cell analysis of transposable element methylation to link global epigenetic heterogeneity with transcriptional programs. Sci Rep. 2022;12(1):5776. https://doi.org/10.1038/s41598-022-09765-x.
Bianchi A, et al. scTAM-seq enables targeted high-confidence analysis of DNA methylation in single cells. Genome Biol. 2022;23(1):229. https://doi.org/10.1186/s13059-022-02796-7.
Pixberg CF, et al. Analysis of DNA methylation in single circulating tumor cells. Oncogene. 2017;36(23):3223–31. https://doi.org/10.1038/onc.2016.480.
Han L, et al. Bisulfite-independent analysis of CpG island methylation enables genome-scale stratification of single cells. Nucleic Acids Res. 2017;45(10): e77. https://doi.org/10.1093/nar/gkx026.
Luo C, et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science. 2017;357(6351):600–4. https://doi.org/10.1126/science.aan3351.
Luo C, et al. Robust single-cell DNA methylome profiling with snmC-seq2. Nat Commun. 2018;9(1):3824. https://doi.org/10.1038/s41467-018-06355-2.
Niemöller C, et al. Bisulfite-free epigenomics and genomics of single cells through methylation-sensitive restriction. Commun Biol. 2021;4(1):153. https://doi.org/10.1038/s42003-021-01661-w.
Huang Y, et al. Comprehensive analysis of partial methylation domains in colorectal cancer based on single-cell methylation profiles. Brief Bioinform. 2021;22(6):bbab267. https://doi.org/10.1093/bib/bbab267.
Gaiti F, Chaligne R, Gu H, et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature. 2019;569:576–80. https://doi.org/10.1038/s41586-019-1198-z.
Chen H, et al. Single-cell DNA methylome analysis of circulating tumor cells. Chin J Cancer Res. 2021;33(3):391–404. https://doi.org/10.21147/j.issn.1000-9604.2021.03.10.
Demond H, et al. A KHDC3L mutation resulting in recurrent hydatidiform mole causes genome-wide DNA methylation loss in oocytes and persistent imprinting defects post-fertilisation. Genome Med. 2019. https://doi.org/10.1186/s13073-019-0694-y.
Millán-Zambrano G, et al. Histone post-translational modifications—cause and consequence of genome function. Nat Rev Genet. 2022;23(9):563–80. https://doi.org/10.1038/s41576-022-00468-7.
Demetriadou C, et al. Histone N-alpha terminal modifications: genome regulation at the tip of the tail. Epigenetics Chromatin. 2020;13(1):29. https://doi.org/10.1186/s13072-020-00352-w.
Zhuang J, et al. Perspectives on the role of histone modification in breast cancer progression and the advanced technological tools to study epigenetic determinants of metastasis. Front Genet. 2020;11: 603552. https://doi.org/10.3389/fgene.2020.603552.
Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10(10):669–80. https://doi.org/10.1038/nrg2641.
Kaya-Okur HS, et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019;10(1):1930. https://doi.org/10.1038/s41467-019-09982-5.
Wang Q, et al. CoBATCH for high-throughput single-cell epigenomic profiling. Mol Cell. 2019;76(1):206-216.e7. https://doi.org/10.1016/j.molcel.2019.07.015.
Ku WL, et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat Methods. 2019;16(4):323–5. https://doi.org/10.1038/s41592-019-0361-7.
Rotem A, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015;33(11):1165–72. https://doi.org/10.1038/nbt.3383.
Ai S, et al. Profiling chromatin states using single-cell itChIP-seq. Nat Cell Biol. 2019;21(9):1164–72. https://doi.org/10.1038/s41556-019-0383-5.
Carter B, et al. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat Commun. 2019;10(1):3747. https://doi.org/10.1038/s41467-019-11559-1.
Harada A, et al. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat Cell Biol. 2019;21(2):287–96. https://doi.org/10.1038/s41556-018-0248-3.
Hainer SJ, et al. Profiling of pluripotency factors in single cells and early embryos. Cell. 2019;177(5):1319-1329.e11. https://doi.org/10.1016/j.cell.2019.03.014.
Artlett DA, et al. High-throughput single-cell epigenomic profiling by targeted insertion of promoters (TIP-seq). J Cell Biol. 2021;220(12): e202103078. https://doi.org/10.1083/jcb.202103078.
Janssens DH, et al. Automated CUT&Tag profiling of chromatin heterogeneity in mixed-lineage leukemia. Nat Genet. 2021;53(11):1586–96. https://doi.org/10.1038/s41588-021-00941-9.
Ku WL, et al. Profiling single-cell histone modifications using indexing chromatin immunocleavage sequencing. Genome Res. 2021;31(10):1831–42. https://doi.org/10.1101/gr.260893.120.
Grosselin K, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet. 2019;51(6):1060–6. https://doi.org/10.1038/s41588-019-0424-9.
Wu SJ, Furlan SN, Mihalas AB, et al. Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat Biotechnol. 2021;39:819–24. https://doi.org/10.1038/s41587-021-00865-z.
Lai WKM, Pugh BF. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol. 2017;18(9):548–62. https://doi.org/10.1038/nrm.2017.47.
Cooper J, et al. Genome-wide mapping of DNase I hypersensitive sites in rare cell populations using single-cell DNase sequencing. Nat Protoc. 2017;12(11):2342–54. https://doi.org/10.1038/nprot.2017.099.
Jin W, et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 2015;528(7580):142–6. https://doi.org/10.1038/nature15740.
Ren G, et al. Transcription factors TCF-1 and GATA3 are key factors for the epigenetic priming of early innate lymphoid progenitors toward distinct cell fates. Immunity. 2022;55(8):1402-1413.e4. https://doi.org/10.1016/j.immuni.2022.06.019.
Vijg J, Suh Y. Genome instability and aging. Annu Rev Physiol. 2013;75:645–68. https://doi.org/10.1146/annurev-physiol-030212-183715.
Navin NE. Delineating cancer evolution with single-cell sequencing. Sci Transl Med. 2015;7(296):296fs29. https://doi.org/10.1126/scitranslmed.aac8319.
Paolillo C, et al. Single-cell genomics. Clin Chem. 2019;65(8):972–85. https://doi.org/10.1373/clinchem.2017.283895.
Guo L, et al. Single-cell DNA sequencing reveals punctuated and gradual clonal evolution in hepatocellular carcinoma. Gastroenterology. 2022;162(1):238–52. https://doi.org/10.1053/j.gastro.2021.08.052.
Zhou Y, et al. Single-cell multiomics sequencing reveals prevalent genomic alterations in tumor stromal cells of human colorectal cancer. Cancer Cell. 2020;38(6):818-828.e5. https://doi.org/10.1016/j.ccell.2020.09.015.
Franco I, et al. Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type. Genome Biol. 2019;20(1):285. https://doi.org/10.1186/s13059-019-1892-z.
Alves JM, et al. Clonality and timing of relapsing colorectal cancer metastasis revealed through whole-genome single-cell sequencing. Cancer Lett. 2022;543: 215767. https://doi.org/10.1016/j.canlet.2022.215767.
Yang D, et al. Single cell whole genome sequencing reveals that NFKB1 mutation affects radiotherapy sensitivity in cervical cancer. Oncotarget. 2017;9(7):7332–40. https://doi.org/10.18632/oncotarget.23587.
Telenius H, et al. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13(3):718–25. https://doi.org/10.1016/0888-7543(92)90147-k.
Dean FB, et al. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11(6):1095–9. https://doi.org/10.1101/gr.180501.
Chen C, et al. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science. 2017;356(6334):189–94. https://doi.org/10.1126/science.aak9787.
Xing D, et al. Accurate SNV detection in single cells by transposon-based whole-genome amplification of complementary strands. Proc Natl Acad Sci U S A. 2021;118(8): e2013106118. https://doi.org/10.1073/pnas.2013106118.
Zong C, et al. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338(6114):1622–6. https://doi.org/10.1126/science.1229164.
Fu Y, et al. Uniform and accurate single-cell sequencing based on emulsion whole-genome amplification. Proc Natl Acad Sci U S A. 2015;112(38):11923–8. https://doi.org/10.1073/pnas.1513988112.
Chu WK, et al. Ultraaccurate genome sequencing and haplotyping of single human cells. Proc Natl Acad Sci U S A. 2017;114(47):12512–7. https://doi.org/10.1073/pnas.1707609114.
Huang L, et al. Single-cell whole-genome amplification and sequencing: methodology and applications. Annu Rev Genom Hum Genet. 2015;16:79–102. https://doi.org/10.1146/annurev-genom-090413-025352.
Wang R, et al. Single-cell genomic and transcriptomic landscapes of primary and metastatic colorectal cancer tumors. Genome Med. 2022. https://doi.org/10.1186/s13073-022-01093-z.
Evrony GD, et al. Applications of single-cell DNA sequencing. Annu Rev Genom Hum Genet. 2021;22:171–97. https://doi.org/10.1146/annurev-genom-111320-090436.
Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017;16(1):41. https://doi.org/10.1186/s12943-017-0600-4.
Nam AS, et al. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet. 2021;22(1):3–18. https://doi.org/10.1038/s41576-020-0265-5.
Moris N, et al. Transition states and cell fate decisions in epigenetic landscapes. Nat Rev Genet. 2016;17(11):693–703. https://doi.org/10.1038/nrg.2016.98.
Liu L, et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat Commun. 2019;10(1):470. https://doi.org/10.1038/s41467-018-08205-7.
Cao J, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361(6409):1380–5. https://doi.org/10.1126/science.aau0730.
Ma S, et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell. 2020;183(4):1103-1116.e20. https://doi.org/10.1016/j.cell.2020.09.056.
Plongthongkum N, et al. Scalable dual-omics profiling with single-nucleus chromatin accessibility and mRNA expression sequencing 2 (SNARE-seq2). Nat Protoc. 2021;16(11):4992–5029. https://doi.org/10.1038/s41596-021-00507-3.
Xing QR, et al. Parallel bimodal single-cell sequencing of transcriptome and chromatin accessibility. Genome Res. 2020;30(7):1027–39. https://doi.org/10.1101/gr.257840.119.
Reyes M, et al. Simultaneous profiling of gene expression and chromatin accessibility in single cells. Adv Biosyst. 2019;3(11):1900065. https://doi.org/10.1002/adbi.201900065.
Gu H, et al. Smart-RRBS for single-cell methylome and transcriptome analysis. Nat Protoc. 2021;16(8):4004–30. https://doi.org/10.1038/s41596-021-00571-9.
Gaiti F, et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature. 2019;569(7757):576–80. https://doi.org/10.1038/s41586-019-1198-z.
Hu Y, et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 2016;17:88. https://doi.org/10.1186/s13059-016-0950-z.
Luo C, et al. Multi-omic profiling of transcriptome and DNA methylome in single nuclei with molecular partitioning. bioRxiv. 2018. https://doi.org/10.1101/434845.
Angermueller C, et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods. 2016;13(3):229–32. https://doi.org/10.1038/nmeth.3728.
Cheow LF, et al. Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat Methods. 2016;13(10):833–6. https://doi.org/10.1038/nmeth.3961.
Rooijers K, et al. Simultaneous quantification of protein-DNA contacts and transcriptomes in single cells. Nat Biotechnol. 2019;37(7):766–72. https://doi.org/10.1038/s41587-019-0150-y.
Zhu C, et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat Methods. 2021;18(3):283–92. https://doi.org/10.1038/s41592-021-01060-3.
Clark SJ, et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun. 2018;9(1):781. https://doi.org/10.1038/s41467-018-03149-4.
Wang Y, et al. Single-cell multiomics sequencing reveals the functional regulatory landscape of early embryos. Nat Commun. 2021;12(1):1247. https://doi.org/10.1038/s41467-021-21409-8.
Yan R, et al. Decoding dynamic epigenetic landscapes in human oocytes using single-cell multi-omics sequencing. Cell Stem Cell. 2021;28(9):1641-1656.e7. https://doi.org/10.1016/j.stem.2021.04.012.
Luo C, et al. Single nucleus multi-omics identifies human cortical cell regulatory genome diversity. Cell Genom. 2022;2(3): 100107. https://doi.org/10.1016/j.xgen.2022.100107.
Bian S, et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science (New York, NY). 2018;362(6418):1060–3. https://doi.org/10.1126/science.aao3791.
Chen X, et al. Joint single-cell DNA accessibility and protein epitope profiling reveals environmental regulation of epigenomic heterogeneity. Nat Commun. 2018;9(1):4590. https://doi.org/10.1038/s41467-018-07115-y.
Mimitou EP, et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat Biotechnol. 2021;39(10):1246–58. https://doi.org/10.1038/s41587-021-00927-2.
Pott S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. Elife. 2017;6: e23203. https://doi.org/10.7554/eLife.23203.
Li G, et al. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat Methods. 2019;16(10):991–3. https://doi.org/10.1038/s41592-019-0502-z.
Guo F, et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017;27(8):967–88. https://doi.org/10.1038/cr.2017.82.
Gu C, et al. Integrative single-cell analysis of transcriptome, DNA methylome and chromatin accessibility in mouse oocytes. Cell Res. 2019;29(2):110–23. https://doi.org/10.1038/s41422-018-0125-4.
Lee J, et al. Single-cell multiomics: technologies and data analysis methods. Exp Mol Med. 2020;52(9):1428–42. https://doi.org/10.1038/s12276-020-0420-2.
Liu Y, et al. DNA methylation-calling tools for Oxford Nanopore sequencing: a survey and human epigenome-wide evaluation. Genome Biol. 2021;22(1):295. https://doi.org/10.1186/s13059-021-02510-z.
Shipony Z, et al. Long-range single-molecule mapping of chromatin accessibility in eukaryotes. Nat Methods. 2020;17(3):319–27. https://doi.org/10.1038/s41592-019-0730-2.
Stergachis AB, et al. Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science. 2020;368(6498):1449–54. https://doi.org/10.1126/science.aaz1646.
Lee I, et al. Simultaneous profiling of chromatin accessibility and methylation on human cell lines with nanopore sequencing. Nat Methods. 2020;17(12):1191–9. https://doi.org/10.1038/s41592-020-01000-7.
LeSavage BL, et al. Next-generation cancer organoids. Nat Mater. 2022;21(2):143–59. https://doi.org/10.1038/s41563-021-01057-5.
Acknowledgements
This work was supported by Jiangsu Medical Innovation Team of Jiangsu Provincial Health Planning Commission, Project No. CXTDA2017042; The Fifth “311 project” of Taizhou City, Project No. RCPY202116; Nanjing Medical University Taizhou Clinical School Project, Project No. TZKY20220101.
Author information
Authors and Affiliations
Contributions
YHH, FS, and XY wrote the manuscript. TTH, ZWL, JLW, JXH, JFS, and QG designed and revised the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hu, Y., Shen, F., Yang, X. et al. Single-cell sequencing technology applied to epigenetics for the study of tumor heterogeneity. Clin Epigenet 15, 161 (2023). https://doi.org/10.1186/s13148-023-01574-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-023-01574-x