
Abstract
Background
Hypertension is a crucial risk factor for developing cardiovascular disease and reducing life expectancy. We aimed to detect DNA methylation (DNAm) variants potentially related to systolic blood pressure (SBP) and diastolic blood pressure (DBP) by conducting epigenome-wide association studies in 60 and 59 Chinese monozygotic twin pairs, respectively.
Methods
Genome-wide DNA methylation profiling in whole blood of twins was performed using Reduced Representation Bisulfite Sequencing, yielding 551,447 raw CpGs. Association between DNAm of single CpG and blood pressure was tested by applying generalized estimation equation. Differentially methylated regions (DMRs) were identified by comb-P approach. Inference about Causation through Examination of Familial Confounding was utilized to perform the causal inference. Ontology enrichment analysis was performed using Genomic Regions Enrichment of Annotations Tool. Candidate CpGs were quantified using Sequenom MassARRAY platform in a community population. Weighted gene co-expression network analysis (WGCNA) was conducted using gene expression data.
Results
The median age of twins was 52 years (95% range 40, 66). For SBP, 31 top CpGs (p < 1 × 10–4) and 8 DMRs were identified, with several DMRs within NFATC1, CADM2, IRX1, COL5A1, and LRAT. For DBP, 43 top CpGs (p < 1 × 10–4) and 12 DMRs were identified, with several DMRs within WNT3A, CNOT10, and DAB2IP. Important pathways, such as Notch signaling pathway, p53 pathway by glucose deprivation, and Wnt signaling pathway, were significantly enriched for SBP and DBP. Causal inference analysis suggested that DNAm at top CpGs within NDE1, MYH11, SRRM1P2, and SMPD4 influenced SBP, while SBP influenced DNAm at CpGs within TNK2. DNAm at top CpGs within WNT3A influenced DBP, while DBP influenced DNAm at CpGs within GNA14. Three CpGs mapped to WNT3A and one CpG mapped to COL5A1 were validated in a community population, with a hypermethylated and hypomethylated direction in hypertension cases, respectively. Gene expression analysis by WGCNA further identified some common genes and enrichment terms.
Conclusion
We detect many DNAm variants that may be associated with blood pressure in whole blood, particularly the loci within WNT3A and COL5A1. Our findings provide new clues to the epigenetic modification underlying hypertension pathogenesis.
Similar content being viewed by others

Introduction
Hypertension is a chronic disease condition, and approximately 90% of the cases are considered as essential hypertension without a definitive cause. The prevalence of hypertension is 28.9%, and the rates of treatment and control of hypertension are only 35.3% and 13.4% in China. Hypertension has been a considerable risk factor for developing cardiovascular disease and reducing life expectancy and has become a huge burden on the nationwide health system [1, 2].
As a complex phenotype, hypertension is controlled by both genetic and environmental factors through the interface of epigenetics. At present, the magnitude of genetic sources of variance in hypertension has been extensively explored. Wu et al. found that blood pressure had a moderate heritability with 0.61 for systolic blood pressure (SBP) and 0.58 for diastolic blood pressure (DBP) in Chinese population [3]. Additionally, genome-wide association studies (GWASs) have reported some genetic variants that were responsible for susceptibility to blood pressure variation, such as the genetic loci within ADRB1, ATP2B1, SOX6, CHIC2, IGFBP3, and KCNK3 [4,5,6,7,8]. However, the previous reported genetic variants only partially contributed to the pathogenesis of hypertension.
In recent years, increasingly strong evidence has supported the significant role of epigenetic mechanisms with altered gene expression in the increased susceptibility to diseases. Currently, a large number of epigenome-wide association studies (EWASs) have been conducted to explore the underlying association between genomic DNA methylation (DNAm) variants and complex traits, such as heart failure [9] and permanent atrial fibrillation [10]. Meanwhile, accumulating evidence has also demonstrated a functional role of DNAm variants in the regulation of blood pressure or the development of hypertension [11, 12]. However, to date, very few studies have investigated the blood pressure or hypertension-related DNAm loci by applying an EWAS approach [13,14,15,16], and few results are replicated. In addition, the causal nature of the association, i.e., if DNAm exerts a causal effect on blood pressure or vice versa, is unknown. Hence, it is essential to further perform EWAS as well as causal inference analysis to investigate the association and causal relationship between DNAm and blood pressure.
Furthermore, the previous EWASs were most performed using samples from unrelated individuals, where the confounding effects from different genetic backgrounds were not well controlled for. Nowadays, the trait or disease-discordant twin design has been a popular and powerful tool for EWAS while controlling for individual genetic make-up [17, 18]. In this study based on a sample of blood pressure-discordant Chinese monozygotic twins, we conducted an EWAS to explore the association between the DNAm at CpGs and blood pressure as well as their causality and validated the candidate CpGs in a community population. Additionally, we further integrated the differentially methylated results with gene expression data.
Materials and methods
The primary materials and methods of this study were in accord with our previously published studies [19,20,21,22].
Participants
The sample collection was carried out through the Qingdao Twin Registry [23], and details of study recruitment have been previously described [24]. Participants who were pregnant and breastfeeding, who suffered from cardiovascular disease, stroke and/or tumor, and who were regularly taking any medications within one month before participation were excluded. Meanwhile, participants who were unconscious, unable, or unwilling to cooperate were also dropped. Considering that we used trait-discordant monozygotic twin design, the twins with intra-pair blood pressure difference ≥ 2 mmHg for SBP or intra-pair blood pressure difference ≥ 1 mmHg for DBP were separately chosen. A total of 60 SBP-discordant monozygotic twin pairs and 59 DBP-discordant twin pairs were included in the methylation analysis. The median of absolute values of intra-pair blood pressure difference was 18 mmHg (95% range 2, 55) for SBP and 10 mmHg (95% range 2, 28) for DBP, respectively. Additionally, a subsample of 12 monozygotic twin pairs were included in the gene expression analysis. All co-twin pairs completed a questionnaire and undertook a health examination after a 10–12-h overnight fast.
This study was approved by the Regional Ethics Committee of the Qingdao CDC Institutional Review Boards. The ethical principles of the Helsinki Declaration were also followed. Prior written informed consent was achieved.
Zygosity determination
We first identified potential monozygotic and dizygotic twins through sex and ABO blood types. Twins with opposite sex and/or different blood types were classified as dizygotic twins. Then, the zygosity of twins with same sex and blood types was further determined by DNA testing using 16 short tandem repeat markers [23, 25, 26].
Measurement of blood pressure
Blood pressure was measured in a sitting position following standard procedure using a mercurial table stand model sphygmomanometer. SBP was measured as Korotkoff phase I (appearance of sound) and DBP as Korotkoff phase V (disappearance of sound). Each subject took three repeated measurements, with at least one-minute interval. The mean value of these three measurements was calculated and used in subsequent analysis. All measurements greater than three standard deviations above or below the means were assigned as missing values.
Reduced representation bisulfite sequencing (RRBS) data preparation
The total DNA extracted from whole blood was used in RRBS experiment. Briefly, genomic DNA was first digested to generate short fragments. Then the CpG-rich DNA fragments was bisulfite-converted. Finally, the cDNA library was obtained and sequenced. The raw methylation data covered 551,447 CpGs across the genome of each individual. We mapped the raw sequencing data to the human GRCh37 by Bismark [27] and then imported data to BiSeq to smooth the methylation level [28]. We controlled the coverage to 90% quantile and dropped CpGs with average methylation β-values less than 0.01 or more than 10 missing observations. After quality control, a total of 248,262 CpGs for SBP and 248,955 CpGs for DBP remained for subsequent analyses. The methylation β-value was transformed to M-value by applying log2 transformation.
Since total DNA was extracted from whole blood, different methylation profiles of distinct cell-types may lead to false discoveries [29]. In our analysis, we applied ReFACTor method, a reference-free method to account for cell-type heterogeneity, and we used the top five components to correct for the cell-type composition effect on DNAm [30].
Gene expression data preparation
Briefly, the total mRNA was first extracted from whole blood. Subsequently, the RNA-Seq library was constructed and sequenced to obtain the sequenced data, which was then mapped to the human genome by TopHat2 [31]. The gene expression level was evaluated by FPKM value through Cufflinks software [32].
Epigenome-wide association analysis
The association between the DNAm M-value at each CpG and SBP or DBP was tested by using generalized estimating equation (GEE) approach through the geeglm function in R-package geepack, adjusting for age, sex, and cell-type composition. Moreover, in order to address the paired structure of the twin data, we included a vector which identified the clusters of twins within a pair into the GEE model. To correct for multiple testing, we calculated false discovery rate (FDR) [33] and defined FDR < 0.05 as genome-wide significance. For CpGs with FDR ≥ 0.05, we defined p < 1 × 10–6 as suggestive significance and 1 × 10–6 ≤ p < 1 × 10–5 as weaker-than-suggestive significance [34]. The CpGs with p < 1 × 10–4 were reported as top CpGs of this EWAS [35]. The identified genomic CpGs (p < 0.05) were annotated to the nearest genes by using R-package biomaRt [36, 37].
Causal inference analysis
For the top CpGs (p < 1 × 10–4), the causal relationship with blood pressure was investigated by the Inference about Causation through Examination of Familial Confounding (ICE FALCON) method which was a regression based method for causal inference in association studies using twins or family data [38, 39]. In this method, ‘familial’ meant both genetic and shared environmental factors in twins, which was essential to make explicit causal inference. The GEE model was applied for parameter estimation with correction for twin pairing. Estimations of βself, βco-twin as well as β’self and β’co-twin were calculated, where βself was the estimation of overall correlation including the causal proportion and family confounding proportion, βco-twin estimated only the family confounding proportion of the correlation, and β’self and β’co-twin was the estimation of full model. If |βco-twin – β’co-twin| was similar to |βself – β’self|, then the association was due to family confounding; and if |βco-twin – β’co-twin| was much larger than |βself—β’self| (ratio > 1.5), then it indicated a causal effect.
Region-based analysis
We applied the comb-p approach to detect the blood pressure-associated differentially methylated regions (DMRs) [40]. The significant enriched DMRs were determined by Stouffer–Liptak–Kechris (slk) corrected p < 0.05.
Ontology enrichments analysis
We submitted the identified CpGs (p < 0.05) to the Genomic Regions Enrichment of Annotations Tool (GREAT) online to analyze the ontology enrichments [41]. Annotation was based on the human GRCh37, and the default “basal plus extension” association rule was used. The false discovery rate (FDR) < 0.05 was considered as statistically significant in ontology enrichments analysis.
EWAS power estimation
We have recently published a computer simulation study on the power of EWAS using twin design [17]. According to this study, if one trait/disease had a heritability (h2) of 0.6 and there was a low correlation between environmental factors and DNAm (R2M,E = 0.1), the sample size required for statistical power to exceed 80% in trait or disease-discordant twin design ranged from 22 (when the correlation within twin pair due to either shared genetic background or common environment, denoted as ρε = 0.8) to 63 (when ρε = 0.1) pairs, which was an immense improvement over the ordinary case–control design. Hence, we speculated that our study based on nearly 60 twin pairs would get a statistical power of about 80%.
We also estimated the correlation between environmental factors (i.e., blood pressure) and DNAm based on the top CpGs identified in this EWAS. We tested the correlation between intra-pair blood pressure difference and intra-pair DNAm difference of each top CpG in EWAS by using partial correlation analysis model, adjusting for age and sex. The median of absolute values of partial correlation coefficients was 0.34 (range 0.03, 0.47) for SBP and 0.27 (range 0.04, 0.46) for DBP (Additional file 1: Table S1), indicating that the R2M, E of our study was likely to be greater than 0.1 and close to 0.3. The heritability of SBP and DBP was about 0.60 in the same twin population as our study [3]. According to the computer simulation study [17], for SBP and DBP with h2 = 0.6 and R2M, E = 0.3, the sample size required for statistical power to exceed 80% in our twin design would range from 17 (when ρε = 0.8) to 25 (when ρε = 0.1) pairs, which were much less than 60 pairs. Hence, our study based on nearly 60 twin pairs would get an enough statistical power.
Quantitative methylation analysis of COL5A1 and WNT3A
We randomly recruited 118 hypertension cases and 149 health controls from the community to validate the CpGs mapped to COL5A1 and WNT3A in EWAS. The cases were defined as those with SBP ≥ 140 mmHg and DBP ≥ 90 mmHg. The subjects with a history of diabetes, obesity, cancer, stroke, and cardiovascular disease were excluded. The participants were interviewed when blood samples were taken and stored under − 80 °C for DNA methylation analysis. We designed the primers for COL5A1 and WNT3A gene to cover the region with the most CpGs (p < 0.05) in EWAS. The mass spectra of cleavage products were collected using the MALDI-TOF mass spectrometry based on the MassARRAY System (Bio Miao Biological Technology, Beijing, China), and the spectra’s methylation ratio was generated by MassARRAY EpiTYPER software (Agena Bioscience, San Diego, California). The DNAm of CpGs between the two independent groups was compared by Wilcoxon rank-sum test. Binary logistic regression model was applied to evaluate the association of each CpG with hypertension while adjusting for BMI, triglyceride (TG), and fasting blood glucose (FBG). The p < 0.05 was set as statistically significant.
Weighted gene co-expression network analysis (WGCNA)
We conducted the weighted gene co-expression network analysis (WGCNA) [42, 43] to identify the specific modules and genes potentially associated with blood pressure. Briefly, a weighted adjacency matrix using gene expression profile data was established, and then, a topological overlap matrix was constructed and used as input for hierarchical clustering analysis. Gene modules were detected by the dynamic tree cutting algorithm, and module eigengenes were correlated with SBP or DBP to detect the module of interest. Enrichment analysis was conducted for the genes clustered in the specific module by DAVID tool [44, 45]. The significant enriched terms were identified with p < 0.05 from a modified Fisher’s exact test.
Results
Epigenome-wide association analysis
A total of 60 twin pairs with a median value of 134.00 mmHg (95% range 102.05, 184.90) for SBP and 59 twin pairs with a median value of 80.00 mmHg (95% range 62.00, 105.03) for DBP were included in the methylation analysis. The median age of twins was 52 years (95% range 40, 66). The other clinical indicators, i.e., BMI, serum uric acid, FBG, high-density lipoprotein cholesterol (HDLC), low-density lipoprotein cholesterol (LDLC), and TG, showed statistically intra-pair correlated, indicating the co-twin design beneficial (Additional file 2: Table S2).
The Manhattan plot of EWAS on SBP is shown in Additional file 3: Fig. S1 (a), and we identified 31 SBP-related top CpGs with p < 1 × 10–4 (Table 1). After correcting for multiple testing, no CpG reached genome-wide significance as defined by FDR < 0.05. The four strongest associations (β = − 0.01, p = 5.76 × 10–6–9.58 × 10–6) were detected for the CpGs (chr3: 84,330,415–84,330,441 bp) located at SRRM1P2, showing weaker-than-suggestive significance (1 × 10–6 ≤ p < 1 × 10–5). All these top CpGs were located at/near 15 genes, including SRRM1P2, COL5A1, MIR1268A, NFATC1, NDE1, MYH11, SMPD4, TXNL1P1, MIR3147, PIP5K1C, TNK2, CACHD1, SLC47A1, etc.
The association between DNAm of 43 top CpGs and DBP reached p < 1 × 10–4 level (Additional file 3: Fig. S1 (b) and Table 2). There were four CpGs (chr1: 228,195,260–228,195,292 bp) within WNT3A and one CpG (chr1: 2,391,479 bp) within PLCH2 detected as showing genome-wide significance (FDR < 0.05). Seven CpGs within SIM1, PLCH2, ATXN7L3B, and LOC646588 showed weaker-than-suggestive significance with 1 × 10–6 ≤ p < 1 × 10–5. All the top CpGs were located at/near 16 genes, and there was more than one CpG located at/near genes ATXN7L3B, DAB2IP, WNT3A, GNA14, EYS, KCNT1, LOC646588, MGEA5, PGR, PLCH2, SAE1, and SIM1.
We found 21 common CpGs (p < 1 × 10–3) between SBP and DBP, and these CpGs were annotated at genes CACNA1B, LARP4B, CSNK1G2, LOC646588, HES4, PPIAP45, GPX1, METRNL, ROBO3, and LINC00943.
We also compared previously reported significant blood pressure or hypertension-associated differentially methylated genes in EWASs [13,14,15] with our results. We defined our genes where CpGs with p < 0.05 were located as supportive to the reported results. The genes CDC42BPB, ALDH3B2, DAB2IP, SLC7A5, VPS25, SLC43A1, SKOR2, ATXN1, ZMIZ1, and CPT1A for SBP and MAN2A2, CFLAR, CPT1A, DAB2IP, SLC7A5, PHGDH, SKOR2, and ZMIZ1 for DBP could be replicated (Additional file 4: Table S3).
Causal inference analysis
The results of causal inference on the top CpGs (p < 1 × 10–4) with SBP and DBP are provided in Table 3. Interestingly, a causal effect of DNAm to SBP was clearly supported for 9 CpGs located at/near NDE1 and MYH11, TXNL1P1, SMPD4, SRRM1P2, TNK2, and CACHD1, respectively. Out of these 9 CpGs, a causal effect of SBP to DNAm of 4 CpGs located at/near TXNL1P1 and SMPD4 was also observed.
As for DBP, the causal effect of DNAm to DBP was obviously found for 6 CpGs, with 4 at WNT3A and 2 at LOC646588. A causal effect of DBP influencing DNAm was also observed for another 8 CpGs, with 4 CpGs at GNA14, 2 CpGs at EYS, 1 CpG at SAE1, and 1 CpG at TMEM114, respectively.
Region-based analysis
A total of 8 DMRs were identified for SBP (Table 4). As illustrated in Fig. 1, the methylation levels of 4 DMRs (A, C, D, and G) at/near NFATC1, LRAT, TUBA3C, and SLC6A10P were positively and 3 DMRs (B, E, and F) at/near CADM2, LOC100507377, and DMRTA2 negatively correlated with SBP, whereas the trend of association between one DMR (H) at IRX1 and SBP was uncertain.

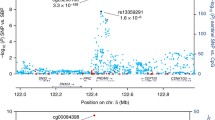
Differential methylation patterns from the identified differentially methylated regions for systolic blood pressure. The dots represent the CpGs. The x-axis shows the position of CpGs on chromosome and the y-axis shows regression coefficients. BP, base pair; DMR, differentially methylated region
Out of the 12 DMRs identified for DBP (Fig. 2; Table 4), the methylation level of 6 DMRs (A, C, E, F, K, and L) showed positive associations and two DMRs (G and H) showed negative associations with DBP. But it was difficult to determine the trend of association between 4 DMRs (B, D, I, and J) and DBP. These DMRs were annotated within 12 genes, such as WNT3A, CNOT10, and DAB2IP.

Differential methylation patterns from the identified differentially methylated regions for diastolic blood pressure. The dots represent the CpGs. The x-axis shows the position of CpGs on chromosome and the y-axis shows regression coefficients. BP, base pair; DMR, differentially methylated region
Ontology enrichments analysis
Lots of important ontology enrichments potentially associated with SBP were found, such as nicotinic acetylcholine receptor signaling pathway, p53 pathway by glucose deprivation, notch signaling pathway, Hedgehog signaling pathway, and PI3 kinase pathway (Table 5). For DBP, the ontology enrichments mainly highlighted inflammation mediated by chemokine and cytokine signaling pathway, notch signaling pathway, angiogenesis, Wnt signaling pathway, TGF-beta signaling pathway, etc. (Table 6).
Many common ontology enrichments for SBP and DBP were observed, such as nicotinic acetylcholine receptor signaling pathway, p53 pathway by glucose deprivation, Notch signaling pathway, Hedgehog signaling pathway, and PI3 kinase pathway (Additional file 5: Table S4).
We found that 2 pathways (PKA-mediated phosphorylation of CREB, regulation of insulin secretion) for SBP and 2 pathways (NCAM1 interactions, dorso-ventral axis formation) for DBP were also enriched in our previous GWAS of blood pressure in twins [8].
Quantitative methylation analysis of COL5A1 and WNT3A
Eight CpGs (p < 0.05) mapped to COL5A1 in EWAS of SBP were quantified using the Sequenom MassARRAY platform. As shown in Additional file 6: Table S5, just one CpG (Chr9: 137,673,907) was validated to be hypomethylated (β = -0.439, p = 0.048) in hypertension cases, and this CpG was also regarded as top signal as in Table 1.
Among the 5 CpGs (p < 0.05) mapped to WNT3A in EWAS of DBP, 3 were quantified using the Sequenom MassARRAY platform. As shown in Additional file 7: Table S6, all of the 3 CpGs were validated in the same direction as in EWAS and also regarded as top signal as in Table 2. Overall, the validation analysis showed clear consistency of hypermethylation in 3 CpGs within WNT3A associated with DBP in a community population.
Weighted gene co-expression network analysis (WGCNA) and gene expression analysis
We included 12 twin pairs (including 7 male pairs) with a median age of 53 years (95% range 43–65), a median SBP of 126 mmHg (95% range 94–195), and a median DBP of 81 mmHg (95% range 64–100) in the analyses.
Additional file 8: Fig. S2 illustrates the genes clustered in mediumpurple3 module (including 4,380 genes) were both negatively correlated with SBP (r = − 0.45, p = 0.03) and DBP (r = − 0.45, p = 0.03). Among the genes where the top CpGs (p < 1 × 10–4) and DMRs were annotated in methylation analysis, 3 genes (MYH11, NFATC1, and PIP5K1C) for SBP and 7 genes (WNT3A, EYS, GNA14, SAE1, CNOT10, APC2, and CFAP74) for DBP were also clustered in mediumpurple3 module in WGCNA.
The genes in methylation analysis and genes clustered in mediumpurple3 module were involved in some common enrichment terms, such as voltage-gated calcium channel activity, NADH dehydrogenase (ubiquinone) activity, PPAR signaling pathway, and acetylcholine receptor activity (Additional file 9: Table S7).
Discussion
It has been demonstrated that epigenetics plays a crucial part in the development hypertension; hence, looking for the specific DNAm variants potentially associated with blood pressure is still a research hotspot [46]. In this study, we detected multiple CpGs, genes, DMRs, and pathways that could not only elucidate the mechanisms of blood pressure variation but also have important implications for the intervention and treatment of hypertension.
In our EWAS on SBP, many genes where the top CpGs and DMRs were located, such as SRRM1P2, COL5A1, NFATC1, NDE1, MYH11, SMPD4, LRAT, CADM2, IRX1, and TNK2, may play important roles in regulating blood pressure. The SNP rs6794880 (chr3:84,402,361) in SRRM1P2 was reported to be related to obesity [47], and we suspected that this locus might influence the development of obesity through regulating the DNAm at one CpG (chr3:84,330,462) in SRRM1P2 we identified. Moreover, the association between obesity and hypertension has clearly been confirmed [48]. It was indicated that the SNPs rs4841895 in COL5A1 [49], rs4799055 in NFATC1 (from dbGaP database), rs1449386 in CADM2 [50], and rs954767 in IRX1 [51] might play a role in blood pressure regulation, and we suspected that these loci might influence the development of hypertension through regulating the DNAm in these genes. NDE1 gene was involved in the signaling pathway by Rho GTPases, which could play a critical role in the pathogenesis of hypertension [52]. The protein encoded by MYH11 is a smooth muscle myosin in vascular smooth muscle cell (SMC) whose principal functions were contraction and regulation of blood pressure and blood flow distribution. The DNAm variation of MYH11 might influence the function of SMC and hence took part in the pathogenesis of hypertension [53]. The protein encoded by SMPD4 was a sphingomyelinase involved in sphingolipid metabolism pathway, and mounting evidence pointed toward that a derangement of this pathway might trigger the precursor clinical conditions of hypertension and hypertension itself [54]. It was found that LRAT may be a critical biomarker of vitamin A deficiency in target organs and may regulate blood pressure through affecting renin angiotensin system biomarkers [55]. TNK2 gene was involved in the oxidative damage response pathway, and it was demonstrated that inflammation and oxidative stress significantly contributed to the vascular dysfunction and renal damage associated with hypertension [56]. However, the mechanism of other genes, such as TXNL1P1, PIP5K1C, MIR3147, and SLC47A1, underlining hypertension requires further investigation.
As for DBP, several interesting genes were also found, including DAB2IP, WNT3A, GNA14, KCNT1, PGR, PLCH2, SIM1, and CNOT10. It was previously reported that the SNPs rs35061590 and rs13290547 in DAB2IP might be associated with heart rate [57] and hence might influence the pathogenesis of hypertension. WNT3A gene was a member of the WNT gene family, and Wnt signaling pathway played an emerging role in regulating blood pressure [58]. The protein encoded by GNA14 was involved in the regulation of insulin secretion pathway, and the relationship of insulin, insulin sensitivity, and hypertension had been clearly confirmed [59]. It was reported that the genetic knockout mouse strain lacking KNa channels (KCNT1 and KCNT2) showed a modest hypertensive phenotype [60]. The SNP rs61892344 in PGR was previously reported to be associated with DBP [51]. The protein encoded by PLCH2 was involved in the inositol phosphate metabolism pathway, and the inositol phosphate production in blood vessels differed in normotensive and spontaneously hypertensive rats [61]. An association of SIM1 variants with early-onset obesity in children was demonstrated [62], but the association of SIM1 with hypertension was currently unknown. The CNOT10 gene was probably associated with left ventricular remodeling in hypertension by bioinformatics-based analysis [63]. Up until now, the association of other genes, such as ATXN7L3B, LOC646588, EYS, MGEA5, and SAE1, with hypertension had not been extensively researched, but they may also serve as candidates to be further verified.
There is a particular challenge regarding the causal effects in observational epidemiological studies using high-dimensional omics data [64]. Our study provides evidence for the causation underlying the blood pressure–DNA methylation association using ICE FALCON method. We found the causal effect that SBP was in response to the DNAm at CpGs located at several genes. NDE1 and MYH11 were involved in the Rho GTPase effectors pathway whose important role in the pathogenesis of vasospasm, hypertension, pulmonary hypertension, and heart failure had been demonstrated [65]. TNK2 was involved in the oxidative damage response pathway that could cause vascular dysfunction and renal damage associated with hypertension [56]. As for DBP, clear causal effect from DNAm to DBP was found for CpGs within WNT3A and LOC646588. WNT3A was involved in Wnt signaling pathway whose role in regulating blood pressure had previously been reported [58]. However, the mechanism of DNAm variation response to blood pressure changes was currently unclear, and further research was needed.
As additional validation, we quantified candidate CpGs mapped to WNT3A and COL5A1 using Sequenom MassARRAY platform in a community population, and three CpGs mapped to WNT3A and one CpG mapped to COL5A1 were successfully validated. As additional replication, we also compared previously reported results in EWASs with ours. A list of differentially methylated genes could be replicated, especially the well-known hypertension-associated gene DAB2IP [57]. We also compared the results from methylation and gene expression analyses and found a list of common genes. For SBP, these genes were involved in various biological pathways, such as nicotinic acetylcholine receptor signaling pathway (MYH11), Wnt signaling pathway (NFATC1), and RhoA signaling pathway (PIP5K1C). For DBP, these common genes took part in Wnt signaling pathway (WNT3A, APC2, and GNA14), ubiquitin proteasome pathway (SAE1), and RNA degradation pathway (CNOT10), etc. Moreover, we also found many common enrichment terms, such as voltage-gated calcium channel activity [66], NADH dehydrogenase (ubiquinone) activity [67], PPAR signaling pathway [68], and acetylcholine receptor activity [69], for which the relationships with hypertension were clear. All of these indicated that the DNAm variants we identified Additional file 8 played a significant role in the development of hypertension.
Several strengths can be noticed in our study. First, the trait or disease-discordant twin design we adopted has been revealed as a powerful tool for detecting the epigenetic variation underling complex diseases [18]. Second, we also performed causal inference to investigate the causation underlying the cross-sectional epigenetic associations and found that blood pressure changes had a causal effect on the DNAm variants at some CpGs, and vice versa. Third, given the various genetic constitutions, environmental exposures, and a multitude of lifestyles in different ethnic populations worldwide, our findings will specifically help elucidate the underlying pathogenesis of hypertension in the Chinese population.
Nevertheless, the sample size of the present study was relatively limited due to the challenges of recruiting and identifying qualified twins. However, the trait or disease-discordant twin design we adopted had greater statistical power over the traditional cross-sectional or case–control design. For blood pressure with a moderate heritability, this design would allow for large sample size reductions comparing to the traditional designs. According to our previous study [17], this study based on nearly 60 twin pairs would get a statistical power of about 80%.
Conclusions
In summary, we found evidence that in peripheral blood from middle and old-aged Chinese twins, the DNAm at several loci within WNT3A and COL5A1 is associated with blood pressure. Additionally, we also found evidence that blood pressure has a causal effect on peripheral blood DNAm, and vice versa. Our findings provide new clues to the epigenetic modification underlying hypertension pathogenesis.
Availability of data and materials
The data used or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ICE FALCON:
-
Causation through examination of familial confounding
- DMR:
-
Differentially methylated region
- DBP:
-
Diastolic blood pressure
- EWAS:
-
Epigenome-wide association study
- FDR:
-
False discovery rate
- GREAT:
-
Genomic Regions Enrichment of Annotations Tool
- GEE:
-
Generalized estimating equation
- RRBS:
-
Reduced-representation bisulfite sequencing
- SBP:
-
Systolic blood pressure
- WGCNA:
-
Weighted gene co-expression network analysis
References
Bundy JD, He J. Hypertension and related cardiovascular disease burden in China. Ann Glob Health. 2016;82(2):227–33.
Wang Y, Peng X, Nie X, Chen L, Weldon R, Zhang W, et al. Burden of hypertension in China over the past decades: systematic analysis of prevalence, treatment and control of hypertension. Eur J Prev Cardiol. 2016;23(8):792–800.
Wu Y, Zhang D, Pang Z, Jiang W, Wang S, Li S, et al. Multivariate modeling of body mass index, pulse pressure, systolic and diastolic blood pressure in Chinese twins. Twin Res Hum Genet. 2015;18(1):73–8.
Ganesh SK, Tragante V, Guo W, Guo Y, Lanktree MB, Smith EN, et al. Loci influencing blood pressure identified using a cardiovascular gene-centric array. Hum Mol Genet. 2013;22(8):1663–78.
Franceschini N, Fox E, Zhang Z, Edwards TL, Nalls MA, Sung YJ, et al. Genome-wide association analysis of blood-pressure traits in African-ancestry individuals reveals common associated genes in African and non-African populations. Am J Hum Genet. 2013;93(3):545–54.
Wain LV, Verwoert GC, O’Reilly PF, Shi G, Johnson T, Johnson AD, et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet. 2011;43(10):1005–11.
Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet. 2015;47(11):1282–93.
Chen J, Wang W, Li Z, Xu C, Tian X, Zhang D. Heritability and genome-wide association study of blood pressure in Chinese adult twins. Mol Genet Genomic Med. 2021;9(11):e1828-e.
Li B, Feng ZH, Sun H, Zhao ZH, Yang SB, Yang P. The blood genome-wide DNA methylation analysis reveals novel epigenetic changes in human heart failure. Eur Rev Med Pharmacol Sci. 2017;21(8):1828–36.
Zhao G, Zhou J, Gao J, Liu Y, Gu S, Zhang X, et al. Genome-wide DNA methylation analysis in permanent atrial fibrillation. Mol Med Rep. 2017;16(4):5505–14.
Demura M, Saijoh K. The role of DNA methylation in hypertension. Adv Exp Med Biol. 2017;956:583–98.
Liang M. Epigenetic mechanisms and hypertension. Hypertension. 2018;72(6):1244–54.
Boström AE, Mwinyi J, Voisin S, Wu W, Schultes B, Zhang K, et al. Longitudinal genome-wide methylation study of Roux-en-Y gastric bypass patients reveals novel CpG sites associated with essential hypertension. BMC Med Genomics. 2016;9:20.
Huang Y, Ollikainen M, Muniandy M, Zhang T, van Dongen J, Hao G, et al. Identification, heritability, and relation with gene expression of novel DNA methylation loci for blood pressure. Hypertension. 2020;76(1):195–205.
Richard MA, Huan T, Ligthart S, Gondalia R, Jhun MA, Brody JA, et al. DNA methylation analysis identifies loci for blood pressure regulation. Am J Hum Genet. 2017;101(6):888–902.
Wang X, Falkner B, Zhu H, Shi H, Su S, Xu X, et al. A genome-wide methylation study on essential hypertension in young African American males. PLoS ONE. 2013;8(1): e53938.
Li W, Christiansen L, Hjelmborg J, Baumbach J, Tan Q. On the power of epigenome-wide association studies using a disease-discordant twin design. Bioinformatics. 2018;34(23):4073–8.
Tan Q, Christiansen L, Von Bornemann HJ, Christensen K. Twin methodology in epigenetic studies. J Exp Biol. 2015;218(1):134–9.
Li W, Zhang D, Wang W, Wu Y, Mohammadnejad A, Lund J, et al. DNA methylome profiling in identical twin pairs discordant for body mass index. Int J Obes (Lond). 2019;43(12):2491–9.
Wang W, Li W, Jiang W, Lin H, Wu Y, Wen Y, et al. Genome-wide DNA methylation analysis of cognitive function in middle and old-aged Chinese monozygotic twins. J Psychiatr Res. 2021;136:571–80.
Wang W, Jiang W, Hou L, Duan H, Wu Y, Xu C, et al. Weighted gene co-expression network analysis of expression data of monozygotic twins identifies specific modules and hub genes related to BMI. BMC Genomics. 2017;18(1):872.
Wang W, Li W, Wu Y, Tian X, Duan H, Li S, et al. Genome-wide DNA methylation and gene expression analyses in monozygotic twins identify potential biomarkers of depression. Transl Psychiatry. 2021;11(1):416.
Duan H, Ning F, Zhang D, Wang S, Zhang D, Tan Q, et al. The Qingdao twin registry: a status update. Twin Res Hum Genet. 2013;16(1):79–85.
Xu C, Zhang D, Tian X, Wu Y, Pang Z, Li S, et al. Genetic and environmental basis in phenotype correlation between physical function and cognition in aging Chinese twins. Twin Res Hum Genet. 2017;20(1):60–5.
Becker A, Busjahn A, Faulhaber HD, Bähring S, Robertson J, Schuster H, et al. Twin zygosity. Automated determination with microsatellites. J Reprod Med. 1997;42(5):260–6.
Jackson RW, Snieder H, Davis H, Treiber FA. Determination of twin zygosity: a comparison of DNA with various questionnaire indices. Twin Res. 2001;4(1):12–8.
Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–2.
Hebestreit K, Dugas M, Klein HU. Detection of significantly differentially methylated regions in targeted bisulfite sequencing data. Bioinformatics. 2013;29(13):1647–53.
Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15(2):R31.
Rahmani E, Zaitlen N, Baran Y, Eng C, Hu D, Galanter J, et al. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods. 2016;13(5):443–5.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300.
Li S, Wang W, Zhang D, Li W, Lund J, Kruse T, et al. Differential regulation of the DNA methylome in adults born during the Great Chinese Famine in 1959–1961. Genomics. 2021;113(6):3907–18.
Mohammadnejad A, Soerensen M, Baumbach J, Mengel-From J, Li W, Lund J, et al. Novel DNA methylation marker discovery by assumption-free genome-wide association analysis of cognitive function in twins. Aging Cell. 2021;20(2): e13293.
Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, et al. BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21(16):3439–40.
Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4(8):1184–91.
Li S, Wong EM, Bui M, Nguyen TL, Joo JE, Stone J, et al. Inference about causation between body mass index and DNA methylation in blood from a twin family study. Int J Obes (Lond). 2019;43(2):243–52.
Li W, Baumbach J, Larsen MJ, Mohammadnejad A, Lund J, Christensen K, et al. Differential long noncoding RNA profiling of BMI in twins. Epigenomics. 2020;12(17):1531–41.
Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28(22):2986–8.
McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501.
Langfelder P, Horvath S. Fast R functions for robust correlations and hierarchical clustering. J Stat Softw. 2012;46(11): i11.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13.
Raftopoulos L, Katsi V, Makris T, Tousoulis D, Stefanadis C, Kallikazaros I. Epigenetics, the missing link in hypertension. Life Sci. 2015;129:22–6.
Justice AE, Winkler TW, Feitosa MF, Graff M, Fisher VA, Young K, et al. Genome-wide meta-analysis of 241,258 adults accounting for smoking behaviour identifies novel loci for obesity traits. Nat Commun. 2017;8:14977.
DeMarco VG, Aroor AR, Sowers JR. The pathophysiology of hypertension in patients with obesity. Nat Rev Endocrinol. 2014;10(6):364–76.
Simino J, Shi G, Bis JC, Chasman DI, Ehret GB, Gu X, et al. Gene-age interactions in blood pressure regulation: a large-scale investigation with the CHARGE, Global BPgen, and ICBP Consortia. Am J Hum Genet. 2014;95(1):24–38.
Giri A, Hellwege JN, Keaton JM, Park J, Qiu C, Warren HR, et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat Genet. 2019;51(1):51–62.
Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50(10):1412–25.
Loirand G, Pacaud P. The role of Rho protein signaling in hypertension. Nat Rev Cardiol. 2010;7(11):637–47.
Gomez D, Swiatlowska P, Owens GK. Epigenetic control of smooth muscle cell identity and lineage memory. Arterioscler Thromb Vasc Biol. 2015;35(12):2508–16.
Kulkarni H, Mamtani M, Blangero J, Curran JE. Lipidomics in the study of hypertension in metabolic syndrome. Curr Hypertens Rep. 2017;19(1):7.
Liang X, Chen M, Wang D, Wen J, Chen J. Vitamin A deficiency indicating as low expression of LRAT may be a novel biomarker of primary hypertension. Clin Exp Hypertens. 2021;43(2):151–63.
Small HY, Migliarino S, Czesnikiewicz-Guzik M, Guzik TJ. Hypertension: focus on autoimmunity and oxidative stress. Free Radic Biol Med. 2018;125:104–15.
Jeff JM, Ritchie MD, Denny JC, Kho AN, Ramirez AH, Crosslin D, et al. Generalization of variants identified by genome-wide association studies for electrocardiographic traits in African Americans. Ann Hum Genet. 2013;77(4):321–32.
Abou Ziki MD, Mani A. Wnt signaling, a novel pathway regulating blood pressure? State of the art review Atherosclerosis. 2017;262:171–8.
Ferrari P, Weidmann P. Insulin, insulin sensitivity and hypertension. J Hypertens. 1990;8(6):491–500.
Li P, Halabi CM, Stewart R, Butler A, Brown B, Xia X, et al. Sodium-activated potassium channels moderate excitability in vascular smooth muscle. J Physiol. 2019;597(20):5093–108.
Vila E, Macrae IM, Reid JL. Differences in inositol phosphate production in blood vessels of normotensive and spontaneously hypertensive rats. Br J Pharmacol. 1991;104(2):296–300.
Zegers D, Beckers S, Hendrickx R, Van Camp JK, de Craemer V, Verrijken A, et al. Mutation screen of the SIM1 gene in pediatric patients with early-onset obesity. Int J Obes (Lond). 2014;38(7):1000–4.
Pang B, Hu C, Wu G, Zhang Y, Lin G. Identification of target genes in hypertension and left ventricular remodeling. Medicine (Baltimore). 2020;99(28): e21195.
Richmond RC, Hemani G, Tilling K, Davey Smith G, Relton CL. Challenges and novel approaches for investigating molecular mediation. Hum Mol Genet. 2016;25(R2):R149–56.
Shimokawa H, Sunamura S, Satoh K. RhoA/Rho-kinase in the cardiovascular system. Circ Res. 2016;118(2):352–66.
Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. 2015;67(4):821–70.
Chen X, He XY, Zhu C, Zhang Y, Li Z, Liu Y, et al. Interaction between mitochondrial NADH dehydrogenase subunit-2 5178 C > A and clinical risk factors on the susceptibility of essential hypertension in Chinese population. BMC Med Genet. 2019;20(1):121.
Toral M, Romero M, Pérez-Vizcaíno F, Duarte J, Jiménez R. Antihypertensive effects of peroxisome proliferator-activated receptor-β/δ activation. Am J Physiol Heart Circ Physiol. 2017;312(2):H189–200.
Liu C, Su D. Nicotinic acetylcholine receptor α7 subunit: a novel therapeutic target for cardiovascular diseases. Front Med. 2012;6(1):35–40.
Acknowledgements
Not applicable.
Funding
This work is funded by the National Natural Science Foundation of China (31741063), Natural Science Foundation of Shandong Province (ZR2020QH304), and China Postdoctoral Science Foundation (2020M682129).
Author information
Authors and Affiliations
Contributions
Material preparation, data collection, and analysis were performed by WJW, JY, WLL, and DFZ. Investigation, resource, and data curation were performed by YLW, HPD, CSX, and XCT. The first draft of the manuscript was written by WJW and JY. The draft was revised by SXL, QHT, and DFZ. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Regional Ethics Committee of the Qingdao CDC Institutional Review Boards. Prior written informed consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. The results of partial correlation analysis model between intra-pair blood pressure difference and intra-pair DNA methylation difference of each top CpG in epigenome-wide association analysis
Additional file 2: Table S2
. Basic characteristics of the participants
Additional file 3: Fig. S1
. Circular Manhattan plot for epigenome-wide association studies of systolic blood pressure (a) and diastolic blood pressure (b). The numbers of chromosome and the -log10 of p-values for statistical significance are shown. The dots represent the observed CpGs.
Additional file 4: Table S3
. Comparison between our results and other previously reported blood pressure or hypertension-associated differentially methylated genes
Additional file 5: Table S4
. Common ontology enrichments by GREAT tool between systolic blood pressure and diastolic blood pressure
Additional file 6: Table S5
. The results of validation analysis for the CpGs mapped to COL5A1 on systolic blood pressure
Additional file 7: Table S6
. The results of validation analysis for the CpGs mapped to WNT3A on diastolic blood pressure
Additional file 8: Fig. S2
. Relationships of consensus module eigengenes and external trait of blood pressure. Numbers in the table report the correlations with the p-values printed in parentheses. The table is color coded by correlation according to the color legend.
Additional file 9: Table S7
. Common enrichment terms for blood pressure between methylation analysis and weighted gene co-expression network analysis
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Wang, W., Yao, J., Li, W. et al. Epigenome-wide association study in Chinese monozygotic twins identifies DNA methylation loci associated with blood pressure. Clin Epigenet 15, 38 (2023). https://doi.org/10.1186/s13148-023-01457-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-023-01457-1