
Abstract
Genomic imprinting is an epigenetic phenomenon of monoallelic gene expression pattern depending on parental origin. In humans, congenital imprinting disruptions resulting from genetic or epigenetic mechanisms can cause a group of diseases known as genetic imprinting disorders (IDs). Genetic IDs involve several distinct syndromes sharing homologies in terms of genetic etiologies and phenotypic features. However, the molecular pathogenesis of genetic IDs is complex and remains largely uncharacterized, resulting in a lack of effective therapeutic approaches for patients. In this review, we begin with an overview of the genomic and epigenomic molecular basis of human genetic IDs. Notably, we address ethical aspects as a priority of employing emerging techniques for therapeutic applications in human IDs. With a particular focus, we delineate the current field of emerging therapeutics for genetic IDs. We briefly summarize novel symptomatic drugs and highlight the key milestones of new techniques and therapeutic programs as they stand today which can offer highly promising disease-modifying interventions for genetic IDs accompanied by various challenges.
Similar content being viewed by others
Background
In mammals, the term genomic imprinting is an epigenetic phenomenon that, in some autosomal genes, gene expression depends on the parent-of-origin so that only one gene copy from the two parental alleles is preferentially active, either maternally or paternally [1]. In humans, there are approximately 150 imprinted genes residing on chromosomes 6, 7, 11, 14, 15, and 20 [2]. The realization has emerged that imprinted genes can function as pivotal regulators in prenatal and postnatal growth and development control, brain function, body composition, and energy homeostasis [3]. Disturbances in gene dosage, epigenetic regulation, and genomic sequences of imprinted genes may result in their function loss and can cause pathological conditions in humans—imprinting disorders (IDs).
Genetic IDs are a subset of congenital diseases caused by common molecular disturbances in genomic imprinting. Since the first human ID—Prader–Willi syndrome (PWS)—was identified in 1989 [4], there have been several other genetic IDs recognized according to genotype–phenotype studies: Angelman syndrome (AS), Beckwith–Wiedemann syndrome (BWS), Silver–Russell syndrome (SRS), pseudohypoparathyroidism types 1a (PHP1a) and 1b (PHP1b), transient neonatal diabetes mellitus (TNDM), Temple syndrome (TS14), Kagami–Ogata syndrome (KOS14), and Schaaf–Yang syndrome (SYS).
Currently, there is no dedicated and radical therapy for patients with genetic IDs, and all available first-line therapies per se are mainly supportive of the management and mitigation of partial existing symptoms. These symptomatic treatments usually cannot offer completely satisfactory symptom resolution for ID patients and have limited benefits to improve their quality of life. In the past decades, there has been a broad consensus on developing novel symptomatic drugs as candidate pharmacological approaches for genetic IDs. The pharmacotherapeutic armamentariums in progress, aiming at different pathophysiological pathways of every ID, hold great potential to optimize or renew present combinatorial therapies for treating certain symptom domains for ID patients.
Moreover, a great interest has been sparked regarding a subset of new and improved techniques and therapeutic programs that, as disease-modifying interventions, could render higher future potentials for treating genetic IDs. These therapies are developed based on the strategies of genetic precision medicine, which mainly target correcting or counteracting the defects caused by the function loss of associated imprinted genes: (1) gene replacement; (2) molecular reinstatement of the normal expression of candidate imprinted genes; (3) silencing the related inhibitory transcripts of imprinted genes, such as clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated endonuclease (Cas) (CRISPR–Cas)-mediated gene editing; and (4) epigenetic circuit reprogramming. These therapies could represent relatively superior and more promising opportunities for managing genetic IDs. However, the majority of these novel therapeutic methods are still at the initial discovery level; therefore, further studies are needed to not only unveil detailed imprinting mechanisms from genetics and epigenetics backdrops but also perform more preclinical and clinical trials on these new interventions. In this review, we summarize recent findings on novel symptomatic drugs for IDs and highlight the advances in the fields of innovative promising therapeutic techniques and treatment programs in progress.
The genomic and epigenomic basis of imprinting
Imprinted genes can be marked by different epigenetic machinery including DNA methylation, histone modifications, and chromatin structure. These modifications are set up during germline development and can be maintained as the memory of germline-derived parental-specific origin after fertilization, eluding genome-wide reprogramming [5]. Imprinted genes display the allelic parental expression pattern ubiquitously and permanently in nearly all cell types; however, some imprinted genes can exhibit imprinted expression patterns restricted to specific cell/tissue types [6, 7] or specific developmental windows [8].
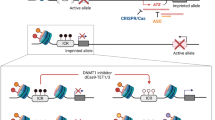
Throughout the mammalian genome, the majority of imprinted genes are clustered together in imprinted domains, spanning 20–3,700 kb of DNA, and generally include several protein-coding genes and non-coding RNAs (ncRNAs) of different types (long non-coding RNAs (lncRNAs), microRNAs (miRNAs), and small nucleolar RNAs (snoRNAs)) [9, 10] (Fig. 1). The clustered imprinted genes in one imprinted domain are under the shared and coordinated regulation of an independent imprinting control region (ICR)—a germline differentially methylated region (DMR) [11]. Notably, four ICRs are paternally germline methylated, all within intergenic regions, and over twenty ICRs are of maternal germline origin, all comprising promoters of imprinted genes [12, 13]. The DNA methylation state of a cis-acting regulatory ICR determines the activity state of the imprinted genes within the entire domain—an ICR can actively direct monoallelic gene expression when unmethylated and become inactive when acquiring parent-of-origin-specific methylation.

Schematic illuminations of representative imprinted gene clusters in humans. a The imprinted gene clusters within Prader–Willi syndrome (PWS)–Angelman syndrome (AS) (PWS–AS) region are shown. b The imprinted genes of potassium voltage-gated channel subfamily Q member 1 (KCNQ1) and H19 (which encodes an imprinted maternally expressed non-coding transcript)–insulin-like growth factor 2 (IGF2) (H19–IGF2) clusters, associated with Beckwith–Wiedemann syndrome (BWS) and Silver–Russell syndrome (SRS), are shown. c The GNAS (which encodes the G protein α-subunit Gsα) cluster of imprinted genes associated with pseudohypoparathyroidism is shown. Different transcripts originate from alternative 5’ exons display parental-specific expression pattern in certain tissues. d The delta-like 1 homologue (DLK1)–iodothyronine deiodinase 3 (DIO3) (DLK1–DIO3) cluster of imprinted genes associated with Temple syndrome (TS14) and Kagami–Ogata syndrome (KOS14) is shown. Chr, chromosome; ICR, imprinting control region; DMR, differentially methylated region, CTCF, CCCTC-binding factor; snoRNAs, small nucleolar RNAs; and miRNAs, microRNAs
The mechanisms by which unmethylated ICRs regulate imprinted gene expression are largely unknown. To date, two major mechanisms have been established: lncRNAs with silencing capacity and CCCTC-binding factor (CTCF)-dependent insulators. The lncRNAs, including KCNQ1 opposite strand 1 (KCNQ1OT1), UBE3A antisense transcript (UBE3A-ATS), gene-trap locus 2 (GTL2), and Airn, being imprinted themselves, can originate from the promoters within or near the ICRs. They have been shown to play an important role in silencing flanking imprinted genes in cis by different mechanisms: (1) The lncRNA product itself can directly suppress imprinted genes, such as UBE3A-ATS [14]; (2) the antisense transcriptional overlap of the lncRNA, rather than the lncRNA product, can confer transcriptional interference on the adjacent imprinted genes by disturbing RNA polymerase II recruitment, for example, the repressor function of Airn transcription in suppressing the transcription of the insulin-like growth factor type 2 receptor (Igf2r) gene[15]; and (3) lncRNAs can associate with local chromosome regions and recruit repressive histone modification machinery to the imprinted cluster, for example, GTL2 and KCNQ1OT1 which can form a discrete suppressive complex with Polycomb proteins [16, 17] to mediate the silencing of imprinted genes.
Moreover, CTCF, the zinc finger insulator protein, can participate in modulating the monoallelic expression of imprinted genes within the locus of H19 (which encodes an imprinted maternally expressed non-coding transcript)–insulin-like growth factor 2 (IGF2) (H19–IGF2) whose ICR harbors multiple CTCF binding sites [18] (Fig. 1b). CTCF can specifically bind to the unmethylated maternally inherited chromosome and form a higher-order chromatin structure, inducing a methylation-sensitive promoter–enhancer interaction: On the maternal chromosome, CTCF acts to prevent downstream enhancers from accessing IGF2 promoters and results in maternal IGF2 silencing, driving activity from H19 instead; in contrast, on the paternally inherited ICR, which is methylated, IGF2 promoters can be activated without CTCF binding.
Types of molecular defects of genomic imprinting disturbances
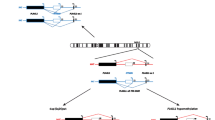
Molecular defects underlying genetic IDs, causing unbalanced expression and functional disruption of imprinted genes, comprise four types of mutations and epimutations: (1) genomic mutations within imprinted genes, (2) chromosomal imbalances (deletions, duplications, and translocations), (3) uniparental disomy (UPD), and (4) epigenetic dysregulation of imprinted loci–imprinting defects (Fig. 2). The frequency of these molecular disturbances varies remarkably between different IDs. Moreover, it is noteworthy that somatic mosaicism can occur in individuals with IDs, in which cells with imprinting disruptions and cells with normal imprints are both contained in tissues. Mosaic distribution can account for somatic asymmetry and may obscure genotype–phenotype correlations.

Exemplary schematic of four molecular subtypes in genetic imprinting disorders. The pathological conditions affecting the expression pattern of one maternal-inherited imprinted gene involve chromosomal deletion, paternal uniparental disomy (UPD) (where two paternal-inherited chromosomes are present losing the maternally expressed gene copy), imprinting defect (which is resulted from epigenetic dysregulation with identical DNA sequences), and gene mutation. For clarity, the paternal and maternal homologous chromosome ideograms (in blue and pink, respectively), one paternally expressed imprinted gene (yellow hexagram), and one maternally expressed imprinted gene (red square) are shown, respectively
Imprinted gene mutation
IDs caused by pathogenic gene sequence variants of imprinted genes have been reported in BWS (inhibiting CDKN1C mutations [19]), SRS (activating CDKN1C mutation [20]), AS (UBE3A mutations [21]), PHP (inactivating GNAS (which encodes the G protein α-subunit Gsα) mutations [22]), and SYS (MAGEL2 mutation [23]). These intragenic mutations can occur de novo in specific germline or may be inherited from the father/mother who carries the disease-causing allele on the silent paternal chromosome. Indeed, the latter case shows an autosomal dominant inheritance mode with parent-of-origin-dependent penetrance of either maternal or paternal transmission and can account for familial reoccurring phenomena of IDs.
Chromosomal imbalance
Chromosomal rearrangements producing copy number variants can cause the expression and complete function loss of imprinted genes by chromosomal deletions, translocations, or duplications. In some IDs, chromosomal deletion accounts for the majority of cases, for example, AS and PWS—with a maternal and paternal 15q11–q13 deletion, respectively. They can occur de novo or result from inherited chromosomal rearrangements.
Uniparental disomy
UPD, mostly resulting from meiotic and mitotic nondisjunction errors, is a detectable genomic situation in which the inheritances of two both homologous chromosomes or chromosomal segments are derived from one parent. It can cause a double dose of imprinted genes and a complete function deficiency and has been reported in nearly all IDs associated with chromosomes 6, 7, 11, 14, 15, and 20 [24]. Specifically, for the fact that chromosomes 14 and 15 are predisposed to Robertsonian translocation (RobT) due to nondisjunction events, the RobT formation containing chromosome 14 and/or 15 is at increased risk of UPD, leading to related ID issues [25].
Imprinting defects
An imprinting defect can cause incorrect activation of an allele-specific gene that should be silent or incorrect silencing of an allele-specific gene that should be active. Isolated or multi-locus epimutations can occur in gametes or often after fertilization, resulting in somatic mosaicism in ID carriers. The majority of epimutations can be involved in incorrect imprints of DMRs without evident alterations in genomic DNA sequence (primary imprinting defect). Moreover, epigenetic alterations can also indirectly result from genetic causes that may be traced back to mutations in cis- or trans-acting factors regulating the erasure, establishment, and maintenance of germline epigenetic imprints (secondary imprinting defect) [26]. In particular, a rare subset of imprinting disruptions occurring at multiple genome loci leads to multi-locus imprinting disturbances (MLIDs) in ID patients [27]. Notably, the involved loci can be linked with one known ID, for example, the BWS and SRS cases caused by the highest epimutation frequency in the ICR1 on chromosome 11p15.5 [28]. However, some epimutations may not have obvious correlations with phenotypes, possibly due to the specifically involved spectrum or their somatic mosaic nature.
Environmental contributions to imprinting disturbances
In addition to genetic abnormalities, environmental factors can also cause imprinting disruptions. Numerous studies in humans have implicated an association between assisted reproductive technology (ART) and an increased risk of IDs, particularly BWS and AS [29,30,31]. ART manipulations, classically including in vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI), take place during the time window of epigenome reprogramming and can induce epigenetic variation in isolated hypomethylation on the KCNQ1OT1 and small nuclear ribonucleoprotein polypeptide N (SNRPN) genes [31, 32]. Other environmental predispositions, such as maternal nutritional status, paternal metabolic disorders, and prenatal exposure to endocrine disruptors, are reported to disturb imprinting marks affecting imprinted methylation patterns and may change the long-term epigenetic regulation of imprinted genes [33,34,35].
Human genetic imprinting disorders
Genetic IDs usually affect multiple systems and produce spectrum disorders, often causing growth restriction during the fetal period and after birth, abnormal neuronal function and development, metabolic disorders, and in some cases, increased cancer susceptibility [36] (Table 1). The clinical phenotypes of these congenital conditions mainly depend on the affected parental allele, whereas different IDs can have overlapping phenotypes, for example, PWS, SRS, and TS14. Particularly noteworthy is that some of the IDs that result from the same dysregulated ICR are “mirror disorders,” for example, BWS and SRS, characterized by opposite gene expression patterns and clinical phenotypes.
OMIM, Online Mendelian Inheritance in Man; SNRPN, small nuclear ribonucleoprotein polypeptide N; SNORD116, small nucleolar RNA, C/D box 116 cluster; mat, maternal; UPD, uniparental disomy; UBE3A, ubiquitin protein ligase E3A; pat, paternal; IGF2, insulin-like growth factor 2; CDKN1C, cyclin-dependent kinase inhibitor 1C; H19 encodes an imprinted maternally expressed non-coding transcript; IG-DMR, intergenic differentially methylated region; KCNQ1OT1, KCNQ1 opposite strand 1; TSS-DMR, transcription start site differentially methylated region; IUGR, intrauterine growth restriction; MEST, mesoderm-specific transcript; GRB10, growth factor receptor-bound protein 10; PTH, parathyroid hormone; GNAS encodes the G protein α-subunit Gsα; PLAGL1, pleomorphic adenoma gene‑like 1; HYMAI, hydatidiform mole-associated and imprinted; DMRs, differentially methylated regions; DLK1, delta-like 1 homologue; RTL1, retrotransposon-like 1; GTL2, gene-trap locus 2; and MAGEL2, MAGE family member L2.
The therapeutic strategies for ID patients are substantially different between the conditions in which unbalanced imprinted gene expression modes appear before lineage commitment (paternally/maternally inherited or occurring de novo before fertilization) and the somatic mosaicism conditions occurring after fertilization (resulting from epimutations and/or UPDs). Effective and complex coordination of health care targeting multisystem manifestations constitutes a common consensus for the former condition. Regarding ID mosaicism conditions, which can be seen in BWS and SRS, limb or body asymmetry can often present in these cases as an isolated finding, and treatment choices for these patients should determine whether the asymmetry condition represents decreased growth (hemihypoplasia) or overgrowth (hemihyperplasia) [39]. Surgical corrections are usually planned for subjects with lateralized overgrowth; tumor management is required for BWS children with any kind of tumor, mainly Wilms tumor, hepatoblastoma, and adrenal carcinoma. Indeed, these therapeutic strategies are all symptomatic and cannot prevent the high recurrence risk of related pathological conditions.
Emerging pharmacotherapies for genetic imprinting disorders
Over the decades, the lack of known molecular targets related to genetic IDs has hindered the development of specific pharmacotherapies for patients. However, a recent surge of treatments with novel symptomatic drugs that traditionally have curative effects in other disorders has begun to suggest their therapeutic potential for patients with genetic IDs because the molecular modes of action of these new therapeutic agents can have correlations with those in genetic IDs (Tables 2, 3, 4). These emerging drug candidates, despite not being disease-modifying, are developing at different research phases and hold promise to counter molecular defects in genetic IDs and afford clear therapy benefits for clinically treating certain phenotypes in patients.
Indeed, for PWS patients, several pharmacological treatments in the pipeline can exert significant effects on specific symptom domains, for example, life-threatening hyperphagia and morbid obesity, aberrant body composition, and cognitive and behavioral abnormalities. In addition, for AS patients, the present exploration of pharmaceuticals is a great attempt to manage seizures with the goal of reducing the side effects of current antiepileptic drugs application. New drugs have also been developed to improve the therapeutic outcomes of ameliorating cognitive impairment, sleep disturbance, and motor deficits in AS patients. Regarding other genetic IDs, novel symptomatic drugs could also facilitate the rescue of functional phenotypes to some degree in a few affected domains. These novel symptomatic drugs indeed have the potential to improve patients’ quality of life and be further used as major therapy methods or, more likely, combined therapeutic interventions for treating patients.
Moreover, a subset of precision therapy approaches employing highly appealing therapeutic techniques has come into view and renewed our understanding of treating genetic IDs (Fig. 3). Indeed, they could represent relatively valid and superior approaches beyond the reach of traditional therapy options for the management of genetic IDs.

Novel therapeutic strategies for treating human genetic imprinting disorders. AAV, adeno-associated virus; ASOs, antisense oligonucleotides; ATFs, artificial transcription factors; CRISPR–Cas, clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein (Cas)
Ethical considerations and concerns regarding novel ID therapeutic strategies
Like any new therapeutics for human diseases, ethical aspects warrant great concern before ensuring responsible use of the emerging techniques to treat and cure IDs in human patients. Several essential but controversial ethical issues need careful consideration.
First, the broad consensus is that studies on these novel therapeutic strategies for IDs should primarily provide scientific evidence on potential health benefits with compelling medical needs. This necessitates considering whether a partial or complete phenotypic correction is of considerable curative significance for ID patients. Second, rigorous evaluations must be carried out on the issues of safety, tolerability, feasibility, and efficacy [80]. In this regard, some forms of therapeutic technologies leading to higher risks of genetic modifications of the human genome, such as the CRISPR–Cas9-mediated genome-editing approach, cannot meet basic safety and efficacy standards and have proven to be ethically unacceptable thus far, as reviewed and discussed by an international committee cosponsored by the US National Academy of Sciences and the National Academy of Medicine [81]. Moreover, phenotypic toxicity and undesired “off-target” effects are recurrent questions producing potential risks and adverse effects in many novel therapeutic choices for IDs, such as virus-mediated gene replacement therapy, pharmacological small-molecule approaches, and epi-drugs, and the prevention of these risks and adverse effects must be emphasized in preclinical studies. Third, choosing the appropriate treatment window can be an important factor in achieving satisfactory effects, especially for ID patients with disturbed growth and development. Basically, restorative interventions for IDs should be administered early because missing a critical treatment period may lead to less-than-expected therapeutic effects. Lastly, further establishment of exact standards for future clinical applications under credible and concomitant ongoing surveillance and administration is urgently necessary. Indeed, there is a long way to go for setting corresponding bioethical guidelines and regulatory policies to protect ID patients, which should be prepared for advanced therapeutics before translating into clinical practice.
Gene replacement therapy
Gene therapy, defined as introducing functional genetic materials into the target cells of patients as a therapeutic pathway, is considered a highly promising treatment for many genetic diseases and also some previously untreatable diseases [82, 83]. These findings provide an excellent proof-of-concept of gene replacement therapy for treating genetic IDs. In some ID cases resulting from the mutation and deficiency of imprinted genes, the therapeutic strategy of sole reinstatement of the expression of the candidate gene might ameliorate or cure disease-related phenotypes. The two basic delivery strategies for therapeutic genes are adeno-associated virus (AAV)-based in vivo gene delivery and ex vivo stem cell transduction pathway.
Adeno-associated virus-mediated gene replacement strategy
As a gene delivery vector, AAV can transfer genes through recognizing and infecting cells efficiently and has been increasingly successful with the properties of long-term efficacy, low-level pathogenicity, and a strong safety profile, making it suitable for therapeutic application. Considerable clinical potential has been established for the treatment of Leber congenital amaurosis (LCA) [84], hemophilia B [85], cancers [86, 87], etc.
Regarding AAV-based gene therapy for genetic IDs, AAV vectors can be engineered to carry the normal gene and replace the pathogenic mutated one in patients’ cells. Taking AS cases as an example, the majority of which lack maternal UBE3A, an early attempt utilized an AAV vector carrying an exogenous UBE3A copy injected into the hippocampus of adult AS mice to restore local UBE3A expression [88]. The therapeutic consequences involve enhanced hippocampus-dependent associative learning and memory ability, but motor deficits show slight improvement due to the limited distribution beyond the hippocampus [88]. In another optimized study, a dual-isoform of human UBE3A (hUBE3A) enabling the translation of both short and long hUBE3A isoforms was packaged into a recombinant AAV9-derived PHP.B vector and was under intracerebroventricular administration into the developing mouse brain [89]. Upon widespread UBE3A re-expression in the brain, more significant therapeutic outcomes were supported, especially improvements in motor learning and behavioral performance [89].
Notably, a reliable AAV-mediated gene replacement strategy should provide two fundamental therapeutic aspects: efficacious relief of symptoms and tolerance to transgene overexpression in vivo. Thus, before being translated into clinical benefits for patients with genetic IDs, more attempts focusing on engineering AAV vectors and devising therapeutic transgenes with better bioavailability potential should be further explored.
Stem cell transduction
Stem cell gene therapy is a promising therapeutic strategy for genetic disorders. It exploits hematopoietic stem and progenitor cells (HSPCs) engineered and modified ex vivo to transfer a healthy gene copy by integrating a viral vector–like lentiviral vector (LV) or to function as cell vehicles to deliver therapeutic molecules into the targeted cells or tissues [90]. Particularly noteworthy is that autologous transplanted HSPCs can differentiate into physiological cell lines such as immune cells, making it possible for them to cross the blood–brain barrier (BBB) to reach the central nervous system (CNS), thus offering wider application in the treatment of CNS diseases. Moreover, this gene therapy pathway involving autologous transplantation can reduce the risks of immune reactions; at the same time, due to the genome integration ability of LVs, it also provides a theoretically permanent treatment for patients. Many treatments of gene-corrected HSPCs have shown favorable safety profiles and excellent therapy efficacy in clinical use for various diseases, including primary immune deficiencies [91], erythrocyte disorders [92], and inherited metabolic diseases [93].
Recently, one study utilized this gene therapy approach for treating AS by transplanting genetically modified HSPCs transduced with a functional UBE3A LV into an immunodeficient AS mouse model [94]. Following transplantation, UBE3A-expressing microglia could be derived from CD34+ HSPCs and further provided UBE3A protein at therapeutic levels to the affected neurons in the brain. A significant enhancement of cognitive behaviors and motor functions was observed in both neonatal and adult mice. This autologous stem cell-based gene therapy suggests a broadened promising treatment to achieve life-long UBE3A delivery in the CNS for AS patients, independent of a critical treatment window.
Therapeutic methods of reactivating imprinted genes
Because inhibitory lncRNAs within imprinted domains are responsible for silencing flanking imprinted genes in cis, the reinstatement of candidate imprinted genes through targeting these lncRNAs conceptually harbors certain feasibility and utility. This raises the possibility that suppressing the expression of lncRNAs with silencing capacity can be realized and become a potential therapy for ID patients. Some exploratory attempts have been made to validate this treatment strategy via various methods, such as high-content small-molecule screening, antisense oligonucleotides (ASOs), and artificial transcription factors (ATFs).
Pharmacological small-molecule approaches
Small molecules, with the advantages of favorable chemical stability and bioavailability, can effectively regulate different signaling pathways [95]. Through large-scale, high-content screening, certain modulatory compounds can be discovered and selected from a chemical library of small molecules. The past decades have witnessed significant advances in small-molecule drugs as molecular tools for deciphering mechanisms of human diseases and normal biology, and as new therapeutics for treating cancers [96, 97] and monogenic diseases [98] and addressing drug resistance (as in infectious diseases [99] and cancers [100]). Hence, the therapeutic potential of pharmacological small molecules for treating IDs is notable.
For AS patients, due to tissue-specific genomic imprinting, the maternal expression of the UBE3A gene is absent in neurons, while the equivalent paternal copy is intact but repressed by the transcription of the lncRNA UBE3A-ATS in the antisense direction [14]. Through an unbiased, high-content screen, topoisomerase inhibitors, such as topotecan, a chemotherapeutic anticancer drug, have been selected and identified to inhibit Ube3a-ATS transcription and increase paternal expression of catalytically active Ube3a in the cortical neurons of mice [101]. In addition, transient topotecan treatment also showed enduring effects of restoring the paternal Ube3a allele in vivo. However, topoisomerase inhibitors exhibit toxicity and lack specificity in that they can concomitantly affect the transcription of many other long genes across the genome in addition to Ube3a-ATS [102], raising the concern of “off-target” effects and suggesting the limitations of their further advancement in AS treatment.
Antisense oligonucleotide (ASO)
ASOs are chemically modified short single-stranded nucleic acids that can specifically suppress the expression of certain genes or modulate splice switching [103]. ASOs can bind to targeted RNA through Watson–Crick base-pairing and cause RNA degradation by recruiting RNase H endonuclease. Emerging as a novel class of drugs, antisense drugs have been widely investigated for their curative effects in many human diseases, several of which have already been implemented in the clinic to treat patients with hereditary transthyretin-mediated (hATTR) amyloidosis, Duchenne muscular dystrophy (DMD), and spinal muscular atrophy (SMA) [104]. Moreover, ASO drugs can be delivered to the target system via many methods and especially exhibit therapeutic advantages in neurological diseases following central administration [105, 106].
ASOs can also be considered suitable candidates for AS treatment strategies. It has been demonstrated that ASO treatment is generally well tolerated and can effectively achieve specific inhibition of Ube3a-ats to sustain the expression of paternal Ube3a, both at the molecular level in neurons and in brains in vivo [107, 108]. Importantly, the optimal ASO treatment for reactivating Ube3a expression in murine AS models requires a critical developmental window of the earlier neurodevelopmental phase around birth [109, 110]. Through intracerebroventricular injection in neonatal AS mice, brain-wide Ube3a distribution can be achieved and AS-related neurocognitive deficits, and epilepsy can also be partially but persistently ameliorated [108]. Therefore, the ASO approach could represent a promising disease-modifying treatment to rescue most disease-associated phenotypes for patients with AS.
To date, three ASO compounds for AS treatment have entered phase 1 clinical trials (GTX-102, NCT04259281; RO7248824, NCT04428281; and ION582, NCT05127226). However, we should also note that the direct translation of ASO treatment for ID patients is still limited. One crucial problem remains for preclinical studies: The precise alignment of brain development between mice and humans is largely unknown; therefore, the phenotypic assessment among mice, especially behavioral tests, cannot be applied to humans with adequate clinical value [110]. Additionally, when proceeding into clinical tests, another concern is about the most efficient mode and route of the ASO delivery system for humans, combined with its dose, duration, and distribution throughout targeted tissues and systems in vivo. Moreover, as ASOs have short half-lives, they need to be administered on a regular schedule to achieve long-term effects on gene regulation in patients. Indeed, before translating into a clinically feasible treatment for IDs, more detailed information is necessary regarding the issues of ASO management regimens and evaluations of efficacy, safety, and tolerability.
Artificial transcription factors (ATFs)
An ATF is a genome interrogation-mediated tool to modulate gene expression in trans. It can be designed as a binary system composed of a DNA-binding domain (DBD), such as zinc fingers, transcription activator-like effectors (TALEs) or CRISPR–Cas systems, and an effector region that can decide the up- or downregulatory transcription of endogenous target genes [111]. When the DBD is linked to an activating domain, the ATF becomes a transcriptional activator to increase targeted gene expression, whereas the linkage of a repressive domain makes a transcriptional repressor to downregulate targeted gene expression. Moreover, the effector domain can also exhibit enzymatic activities involved in epigenetic editing and chromatin remodeling [112].
In one study, an injectable ATF was established, functioning in suppressing Ube3a-ATS production to restore Ube3a expression, which supports a therapeutic evidence line for AS [113]. Structurally, this factor was engineered to involve a zinc finger domain to bind to the promoter region of small nuclear ribonucleoprotein polypeptide N (SNRPN). Through subcutaneous or intraperitoneal injection in adult AS mice, the ATF could cross the BBB and distribute diffusely throughout the hippocampus and cerebellum [103]. Although utilizing ATF biologics can show short-term rescue effects in the brain from molecular aspects, the phenotypic investigations on behavior and cognition remain unassessed and unknown in treated mice.
CRISPR–Cas9-mediated genome-editing approach
The genome-editing system of RNA-guided CRISPR–Cas, functionally introducing DNA double-strand breaks (DSBs) and stimulating non-homologous end joining (NHEJ) and homologous-directed repair (HDR), can correct gene mutations and modulate gene expression in eukaryotic cells [114]. Representing genetic precision medicine, CRISPR–Cas9 technology holds great promise to offer permanent reversal of pathologic DNA mutations for genetic disorders. In particular, the CNS has been recognized as a potential target for therapeutic genome-editing methods following focal delivery of CRISPR–Cas9 into different brain regions. Accumulating lines of therapeutic evidence of gene editing in the CNS have been reported in mouse models of various diseases, such as amyotrophic lateral sclerosis [115], Huntington’s disease [116], and familial Alzheimer’s disease [117].
As recently demonstrated, an established CRISPR–Cas9 system targeting the UBE3A-ATS transcript can block its expression and reactivate paternally inherited UBE3A expression in primary human neurons and in an AS mouse model [118, 119]. In addition, this efficacious gene-editing tool can be delivered via AAV into the mouse brain during early postnatal stages, and due to the functional restoration of paternal UBE3A throughout the nervous system, long-lasting therapeutic benefits in anatomical and behavioral deficits of AS can be achieved [118].
Although CRISPR–Cas9-mediated gene-editing technology has been implicated to have optimistic therapeutic benefits in genetic IDs, no gene-specific therapy exists for ID patients. Considering the genome and phenotype differences between humans and mice, numerous obstacles limiting the therapeutic outcome of this approach still lie ahead, the main aspects of which involve (1) design and screening of appropriate single-guide RNAs (sgRNAs) for human mutant alleles; (2) efficient and safe transport and delivery pathways of the editing system; (3) genome-editing efficiency; (4) unintended off-target activity; (5) therapeutic threshold of editing; (6) toxic reactions; and (7) other unknown potential risks. Beyond any doubt, surmounting these pitfalls is an essential prerequisite for future translational studies.
Epigenetics-based intervention
Within the past decades, reprogramming the epigenetic landscape to modulate the disease-causing defects present in the genome has revolutionized the research field of developing innovative treatments for different diseases. The so-called epi-drugs can function as activators or inhibitors of epigenetic regulatory proteins to manipulate aberrant DNA methylation and histone modifications. Several epi-drugs, such as DNA methylation inhibitors and histone deacetylase inhibitors, have been approved for clinical applications to treat solid tumors and hematologic cancers and have afforded clear therapeutic benefits in patients [120,121,122]. Moreover, numerous investigations of potential epigenetic therapy in other pathologies, involving infectious diseases [123], cardiovascular disease [124], metabolic disorders [125], and neurodevelopmental and neuropsychiatric diseases [126, 127], either have received approval or are being tested in clinical trials.
Given that individuals with genetic IDs retain at least one intact but epigenetically silent gene copy, reversing their imprinting status to compensate for the deficiency from the affected parental copy is a potential opportunity for curing and treating IDs. Hence, reprogramming the related epigenetic circuits to induce the reinstatement of inactivated candidate genes can become a research hotspot.
Epigenetic therapy for PWS
Within the PWS region on chromosome 15q11–q13, there are 15 known disease-related genes, among which the small nucleolar C/D box RNA 116 (SNORD116) cluster may be the paramount causative player in PWS [128]. Unveiling related epigenetic mechanisms and reactivating functionally dormant genes on maternal alleles can offer potential therapeutic targets. At present, several groups have proposed new methods of epigenetics-based intervention for the treatment of PWS.
An epigenetic complex comprised of zinc finger protein 274 (ZNF274) and SET domain bifurcated 1 (SETDB1)—a histone H3 lysine 9 (H3K9) methyltransferase, has been demonstrated to be a separate imprinting mark to induce the expression silence of maternal PWS genes [129]. Studies knocking down SETDB1 [129] or knocking out ZNF274 [130] in neurons from PWS induced pluripotent stem cells (iPSCs) found that H3K9 tri-methylation (H3K9me3), a featured histone mark of closed chromatin, at the SNORD116 locus decreases and maternal SNORD116 expression can be partially restored. In contrast to SETDB1-deficiency, ZNF274 inactivation can acquire specificity without affecting DNA methylation at the PWS-ICR [129, 130].
Additionally, through a high-content screen, two selective small-molecule inhibitors of euchromatic histone-lysine N-methyltransferase-2 (EHMT2/G9a), UNC0642 and UNC0638, have been discovered to reactivate maternal PWS imprinted genes both in a PWS mouse model and in PWS-derived iPSCs [131]. The two G9a inhibitors can selectively reduce H3K9 di-methylation (H3K9me2) at the PWS-ICR locus without affecting DNA methylation [131]. Moreover, therapeutic effects have been indicated in PWS newborn mice managed by G9a inhibitors, and survival and growth have been significantly improved [131]. This result provides a functionally optimistic basis for developing small-molecule-based epigenetic therapy for PWS patients, and further investigation of this approach is warranted.
Moreover, versatile CRISPR–Cas9 technology has emerged as a powerful leading platform for engineering the epigenome with the advent of catalytically dead Cas9 (dCas9) (without DNA cleavage function). Via fusing to various epigenetic effectors, such as DNA demethylases/methyltransferases and histone-modifying enzymes [132], the CRISPR–dCas9 system can target the specific promoter/exon 1 sequences to modulate gene expression sterically. This site-specific epigenome-editing tool could broaden the targeted therapeutic space in human diseases because several proof-of-principle studies in vivo have provided its therapeutic effects in the management of chronic pain [133] and neurological disorders [134, 135]. Indeed, as an alternative to pharmacological small-molecule approaches, the utilization of CRISPR–dCas9 in epigenetic reprogramming also provides novel therapeutic directions for genetic IDs based on the strategies of reactivating the imprinted genes from the other parent-of-origin chromosome or suppressing the improper biallelically expressed genes. There was a study fusing dCas9 with an epigenetic effector–DNA plus dioxygenase 1 (TET1) (dCas9–TET1) to effectively edit the targeted epigenetic marks of the imprinted PWS region, allowing the activation of the SNRPN gene in vivo [136].
Future prospects in epigenetic treatments for other genetic imprinting disorders
From the view of treating genetic IDs, advances in understanding disease-related imprinting mechanisms and the deregulation of epigenetic processes will provide an opportunity for using an epigenetic approach to functionally induce the restoration of silenced genes. Therefore, by targeting these syndromes at their “roots” to reinstate the normal transcriptional activity of imprinted genes, epigenetics-based intervention toward new drug discovery could arguably be a game-changer for the therapeutic strategy of IDs.
Ideally, for IDs patients harboring only one intact but inactive candidate gene, using epigenetic modulating agents against the activity of epigenetic modifiers or remodelers may offer a valid opportunity to restore the silenced copy, for instance, IGF2 re-expression for SRS patients, GNAS re-expression for PHP patients, and UBE3A re-expression for AS patients. In detail, for AS cases with maternal UBE3A deletion, sgRNA-guided dCas9 may tether with epigenetic modifiers or regulators, such as DNA methyltransferase writers, to target the CpG islands at the UBE3A-ATS promoter. This could theoretically result in increased UBE3A expression through mimicking the maternal silencing methylation pattern. As another case in point, for SRS patients caused by a loss of methylation (LOM) on paternal H19–IGF2 DMR in chromosome 11p15, epi-editors of DNA methyltransferase may function to reverse the imprinted status of the IGF2 gene. Besides, in some pathological situations in which interstitial duplications cause imprinted gene overexpression, complete methylation-induced silencing of candidate genes may not be favorable; rather, a decrease in expression to ordinary levels should be considered the main goal of epigenetic therapy.
Challenges in epi-drugs development
The development of epi-based applications for genetic IDs may face several formidable challenges.
The first challenge stems from the scientific aspect—the complexity of epigenetic regulatory processes and the heterogeneity of genome and epigenome landscapes, which remain elusive for most genetic IDs. Actually, these open questions are fundamental and pivotal for determining the right and most beneficial genomic and epigenomic programs needing regeneration in ID patients.
Second, as a rescue option, efficacy evaluation is also a critical issue: whether epigenetic manipulation could largely or completely restore imprinted gene expression to sufficient levels to render global effects on phenotypic correction or whether merely partial reinstatement is enough to mitigate phenotypes. Additionally, we should note that many investigations on the therapeutic strategies in disease mouse models clarify only a part of the relieved phenotypes, which could be because the researchers conducted only a part of the treatment evaluations or, more importantly, because the murine models have their own shortcomings for phenotypic assays. In fact, a high fraction of phenotypes in mice and human ID patients do not align well, for example, a subset of motor-functional deficits, neurodevelopmental and neurocognitive deficits involving intellectual impairments, behavioral disability, and sleep disorders.
Third, for potentially feasible management before clinical tests, the issues of the safety and tolerability of epigenetic drugs should be considered. In principle, the epigenetic reprogramming pathway modulating common epigenetic enzymes might generate a combination of opposite alterations depending on the consequences of the gain or loss of methylation in different genes. The low-specificity-caused general changes in the epigenetic pattern on non-targeted genes can lead to pleiotropic effects and chaotic disorders, which can be quite risky in humans. Therefore, epigenetic regulating agents and CRISPR-based epigenetic editing methods should be explored to collect detailed specificity information for humans and further narrow down potential off-target effects. While speculatively, risk–benefit assessments are necessary for the clinical use of novel epi-drugs. Another safety question is how to precisely control the restoration level of imprinted genes in the target tissue of patients because excessive gene dosages can introduce another risk factor for disruptions in the body; for example, UBE3A overexpression in the CNS can actually cause autism spectrum disorders (ASDs) [137, 138]. Hence, when the experimental epi-drugs move into the clinical study stage, they should be primarily utilized under careful supervision, and the sufficient therapeutic benefits of ameliorating or curing disease phenotypes with minimal adverse events should be monitored and evaluated precisely.
Fourth, the critical treatment window should be addressed when prescribed for ID patients with disturbed growth and development. Basically, epigenetics-based intervention would be an early-onset therapy because missing the critical treatment period may lead to less-than-expected therapeutic effects. To some extent, for CNS-related IDs, treatment during distinct neurodevelopmental windows can be a decisive factor in completely correcting pathological defects and acquiring sufficient rescue benefits. However, current studies are puzzling when using cell and animal models. On the one hand, although patient-derived iPSCs are reliable cell models reflecting the pathogenic conditions of early development, they cannot anticipate the situation in vivo. On the other hand, regarding disease-related mouse models, it is crucial to understand how the critical time window in humans compares to that in mice, which is not yet clear. Further investigation is needed to clarify the timing of therapeutic intervention in humans for each individual disease through clinical trials.
Conclusions and future perspectives
In the past two decades, an improved understanding of both the fundamental mechanisms of genetic imprinting and genotype–phenotype correlations has enabled and driven unparalleled progress in developing novel therapeutics for human genetic IDs. The potential of innovative treatments to enhance patient benefit concentrates on overriding the limitations of current therapeutic strategies that are merely supportive and symptom-addressed. As focused upon in our review, these new and improved therapeutic approaches can be summarized in two primary directions: (1) exploiting symptomatic pharmacologic drugs targeting related pathophysiological pathways to treat specific symptom domains in genetic IDs, a large proportion of which have been developed in clinical trials and (2) exploring disease-modifying interventions of new techniques and therapeutic programs, such as gene replacement, molecular reinstatement of imprinted genes (through restoring the normal transcriptional activity of candidate genes or silencing the related inhibitory transcripts of imprinted genes), and epigenetics-based interventions. The latter therapeutic direction theoretically provides a more valid and promising treatment option for patients with genetic IDs. Specifically, it is tempting to imagine that the epi-based treatment approach could revolutionize the therapeutic strategy for genetic IDs and that, in the near future, the first application of epi-drugs might emerge as a supportive combination therapy to manage symptoms and limit disease progression for patients.
However, these emerging therapeutics are still at the discovery level, coping with various formidable obstacles. Going forward, on the one hand, more insights into the detailed molecular mechanism of genetic imprinting are still needed, providing the mechanistic basis and determining the relevance of potential therapeutic targets; on the other hand, more preclinical and clinical tests on discovered novel interventions are crucial for progress in this area. Undoubtedly, these new avenues of therapeutics open the door for treating genetic IDs and hold great promise for future adoption as accepted management strategies for patients.
Availability of data and materials
All the information is included in this manuscript.
Abbreviations
- AAV:
-
Adeno-associated virus
- ART:
-
Assisted reproductive technology
- AS:
-
Angelman syndrome
- ASDs:
-
Autism spectrum disorders
- ASOs:
-
Antisense oligonucleotides
- ATFs:
-
Artificial transcription factors
- BBB:
-
Blood–brain barrier
- BK:
-
Calcium- and voltage-dependent big potassium
- BWS:
-
Beckwith–Wiedemann syndrome
- CBD:
-
Cannabidiol
- CB1R:
-
Cannabinoid type1 receptor
- CDKN1C:
-
Cyclin-dependent kinase inhibitor 1C
- CFE:
-
Caralluma fimbriata extract
- Chr:
-
Chromosome
- CIM6P:
-
Cation-independent mannose-6-phosphate
- CNS:
-
Central nervous system
- CRISPR–Cas:
-
Clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated endonuclease (Cas)
- CYP:
-
Cyproheptadine
- DBD:
-
DNA-binding domain
- dCas9:
-
Dead Cas9
- DCCR:
-
Diazoxide choline controlled release
- DIO3:
-
Iodothyronine deiodinase 3
- DLK1:
-
Delta-like 1 homologue
- DMD:
-
Duchenne muscular dystrophy
- DMRs:
-
Differentially methylated regions
- DPP4:
-
Dipeptidyl peptidase-4
- DSBs:
-
Double-strand breaks
- EEG:
-
Electroencephalography
- EHMT2/G9a:
-
Euchromatic histone-lysine N-methyltransferase-2
- GLP-1:
-
Glucagon-like peptide 1
- GNAS:
-
Which encodes the G protein α-subunit Gsα
- GOAT:
-
Ghrelin O-acyltransferase
- GRB10:
-
Growth factor receptor-bound protein 10
- GTL2:
-
Gene-trap locus 2
- hATTR:
-
Hereditary transthyretin-mediated
- HDR:
-
Homologous-directed repair
- HSPCs:
-
Hematopoietic stem and progenitor cells
- hUBE3A:
-
Human UBE3A
- HYMAI:
-
Hydatidiform mole-associated and imprinted
- H19:
-
Which encodes an imprinted maternally expressed non-coding transcript
- H3K9:
-
Histone H3 lysine 9
- H3K9me2:
-
H3K9 di-methylation
- H3K9me3:
-
H3K9 tri-methylation
- H19:
-
Which encodes an imprinted maternally expressed non-coding transcript
- ICR:
-
Imprinting control region
- ICSI:
-
Intracytoplasmic sperm injection
- IDs:
-
Imprinting disorders
- IGF2:
-
Insulin-like growth factor 2
- Igf2r:
-
Insulin-like growth factor type 2 receptor
- IG-DMR:
-
Intergenic differentially methylated region
- iPSCs:
-
Induced pluripotent stem cells
- IUGR:
-
Intrauterine growth restriction
- IVF:
-
In vitro fertilization
- KCNQ1:
-
Potassium voltage-gated channel subfamily Q member 1
- KCNQ1OT1:
-
KCNQ1 opposite strand 1
- KOS14:
-
Kagami–Ogata syndrome
- LCA:
-
Leber congenital amaurosis
- lncRNAs:
-
Long non-coding RNAs
- LOM:
-
Loss of methylation
- LV:
-
Lentiviral vector
- MAGEL2:
-
MAGE family member L2
- mat:
-
Maternal
- MC4R:
-
Melanocortin-4 receptor
- MEST:
-
Mesoderm-specific transcript
- MetAP2:
-
Methionine aminopeptidase 2
- miRNAs:
-
MicroRNAs
- MLIDs:
-
Multi-locus imprinting disturbances
- ncRNAs:
-
Non-coding RNAs
- NHEJ:
-
Non-homologous end joining
- NNZ-2591:
-
Cyclo-l-glycyl-l-2-allylproline
- NSI-189:
-
NSI-189 phosphate
- OMIM:
-
Online Mendelian Inheritance in Man
- pat:
-
Paternal
- PDE:
-
Phosphodiesterase
- PHP:
-
Pseudohypoparathyroidism
- PLAGL1:
-
Pleomorphic adenoma gene‑like 1
- PP2A:
-
Protein phosphatase 2A
- PTH:
-
Parathyroid hormone
- PWS:
-
Prader–Willi syndrome
- RobT:
-
Robertsonian translocation
- RTL1:
-
Retrotransposon-like 1
- SETDB1:
-
SET domain bifurcated 1
- sgRNAs:
-
Single-guide RNAs
- SMA:
-
Spinal muscular atrophy
- SNORD116:
-
Small nucleolar RNA, C/D box 116 cluster
- snoRNAs:
-
Small nucleolar RNAs
- SNRPN:
-
Small nuclear ribonucleoprotein polypeptide N
- SRS:
-
Silver–Russell syndrome
- SU:
-
Sulfonylurea
- SYS:
-
Schaaf–Yang syndrome
- TALEs:
-
Transcription activator-like effectors
- TET1:
-
DNA plus dioxygenase 1
- TNDM:
-
Transient neonatal diabetes mellitus
- TSS-DMR:
-
Transcription start site differentially methylated region
- TS14:
-
Temple syndrome
- UAG:
-
Unacylated ghrelin
- UBE3A:
-
Ubiquitin protein ligase E3A
- UBE3A-ATS:
-
UBE3A antisense transcript
- UPD:
-
Uniparental disomy
- ZNF274:
-
Zinc finger protein 274
References
Sagi I, De Pinho JC, Zuccaro MV, Atzmon C, Golan-Lev T, Yanuka O, Prosser R, Sadowy A, Perez G, Cabral T, Glaser B, Tsang SH, Goland R, Sauer MV, Lobo R, Benvenisty N, Egli D. Distinct imprinting signatures and biased differentiation of human androgenetic and parthenogenetic embryonic stem cells. Cell Stem Cell. 2019;25:419-32.e9.
Santoni FA, Stamoulis G, Garieri M, Falconnet E, Ribaux P, Borel C, Antonarakis SE. Detection of imprinted genes by single-cell allele-specific gene expression. Am J Hum Genet. 2017;100:444–53.
Tucci V, Isles AR, Kelsey G, Ferguson-Smith AC. Genomic imprinting and physiological processes in mammals. Cell. 2019;176:952–65.
Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. 1989;342:281–5.
Wang L, Zhang J, Duan J, Gao X, Zhu W, Lu X, Yang L, Zhang J, Li G, Ci W, Li W, Zhou Q, Aluru N, Tang F, He C, Huang X, Liu J. Programming and inheritance of parental DNA methylomes in mammals. Cell. 2014;157:979–91.
Hsiao JS, Germain ND, Wilderman A, Stoddard C, Wojenski LA, Villafano GJ, Core L, Cotney J, Chamberlain SJ. A bipartite boundary element restricts UBE3A imprinting to mature neurons. Proc Natl Acad Sci U S A. 2019;116:2181–6.
Laukoter S, Pauler FM, Beattie R, Amberg N, Hansen AH, Streicher C, Penz T, Bock C, Hippenmeyer S. Cell-type specificity of genomic imprinting in cerebral cortex. Neuron. 2020;107:1160-79.e9.
Terranova R, Yokobayashi S, Stadler MB, Otte AP, van Lohuizen M, Orkin SH, Peters AH. Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev Cell. 2008;15:668–79.
Kota SK, Llères D, Bouschet T, Hirasawa R, Marchand A, Begon-Pescia C, Sanli I, Arnaud P, Journot L, Girardot M, Feil R. ICR noncoding RNA expression controls imprinting and DNA replication at the Dlk1-Dio3 domain. Dev Cell. 2014;31:19–33.
Whipple AJ, Breton-Provencher V, Jacobs HN, Chitta UK, Sur M, Sharp PA. Imprinted maternally expressed microRNAs antagonize paternally driven gene programs in neurons. Mol Cell. 2020;78:85-95.e8.
Williamson CM, Turner MD, Ball ST, Nottingham WT, Glenister P, Fray M, Tymowska-Lalanne Z, Plagge A, Powles-Glover N, Kelsey G, Maconochie M, Peters J. Identification of an imprinting control region affecting the expression of all transcripts in the Gnas cluster. Nat Genet. 2006;38:350–5.
Kaufman Y, Heled M, Perk J, Razin A, Shemer R. Protein-binding elements establish in the oocyte the primary imprint of the Prader-Willi/Angelman syndromes domain. Proc Natl Acad Sci U S A. 2009;106:10242–7.
Hur SK, Freschi A, Ideraabdullah F, Thorvaldsen JL, Luense LJ, Weller AH, Berger SL, Cerrato F, Riccio A, Bartolomei MS. Humanized H19/Igf2 locus reveals diverged imprinting mechanism between mouse and human and reflects Silver–Russell syndrome phenotypes. Proc Natl Acad Sci U S A. 2016;113:10938–43.
Meng L, Person RE, Beaudet AL. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet. 2012;21:3001–12.
Latos PA, Pauler FM, Koerner MV, Şenergin HB, Hudson QJ, Stocsits RR, Allhoff W, Stricker SH, Klement RM, Warczok KE, Aumayr K, Pasierbek P, Barlow DP. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science. 2012;338:1469–72.
Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40:939–53.
Zhang H, Zeitz MJ, Wang H, Niu B, Ge S, Li W, Cui J, Wang G, Qian G, Higgins MJ, Fan X, Hoffman AR, Hu JF. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J Cell Biol. 2014;204:61–75.
Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–9.
Sparago A, Cerrato F, Pignata L, Cammarata-Scalisi F, Garavelli L, Piscopo C, Vancini A, Riccio A. Variable expressivity of the Beckwith–Wiedemann syndrome in four pedigrees segregating loss-of-function variants of CDKN1C. Genes. 2021;12:9.
Brioude F, Oliver-Petit I, Blaise A, Praz F, Rossignol S, Le Jule M, Thibaud N, Faussat AM, Tauber M, Le Bouc Y, Netchine I. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J Med Genet. 2013;50:823–30.
Bossuyt SNV, Punt AM, de Graaf IJ, van den Burg J, Williams MG, Heussler H, Elgersma Y, Distel B. Loss of nuclear UBE3A activity is the predominant cause of Angelman syndrome in individuals carrying UBE3A missense mutations. Hum Mol Genet. 2021;30:430–42.
Elli FM, deSanctis L, Ceoloni B, Barbieri AM, Bordogna P, Beck-Peccoz P, Spada A, Mantovani G. Pseudohypoparathyroidism type Ia and pseudo-pseudohypoparathyroidism: the growing spectrum of GNAS inactivating mutations. Hum Mutat. 2013;34:411–6.
Fountain MD, Aten E, Cho MT, Juusola J, Walkiewicz MA, Ray JW, Xia F, Yang Y, Graham BH, Bacino CA, Potocki L, van Haeringen A, Ruivenkamp CA, Mancias P, Northrup H, Kukolich MK, Weiss MM, van Ravenswaaij-Arts CM, Mathijssen IB, Levesque S, Meeks N, Rosenfeld JA, Lemke D, Hamosh A, Lewis SK, Race S, Stewart LL, Hay B, Lewis AM, Guerreiro RL, Bras JT, Martins MP, Derksen-Lubsen G, Peeters E, Stumpel C, Stegmann S, Bok LA, Santen GW, Schaaf CP. The phenotypic spectrum of Schaaf–Yang syndrome: 18 new affected individuals from 14 families. Genet Med. 2017;19:45–52.
Nakka P, Pattillo Smith S, O’Donnell-Luria AH, McManus KF, Mountain JL, Ramachandran S, Sathirapongsasuti JF. Characterization of prevalence and health consequences of uniparental disomy in four million individuals from the general population. Am J Hum Genet. 2019;105:921–32.
Wiland E, Olszewska M, Wozniak T, Kurpisz M. How much, if anything, do we know about sperm chromosomes of Robertsonian translocation carriers? Cell Mol Life Sci. 2020;77:4765–85.
Monteagudo-Sánchez A, Hernandez Mora JR, Simon C, Burton A, Tenorio J, Lapunzina P, Clark S, Esteller M, Kelsey G, López-Siguero JP, de Nanclares GP, Torres-Padilla ME, Monk D. The role of ZFP57 and additional KRAB-zinc finger proteins in the maintenance of human imprinted methylation and multi-locus imprinting disturbances. Nucleic Acids Res. 2020;48:11394–407.
Kagami M, Hara-Isono K, Matsubara K, Nakabayashi K, Narumi S, Fukami M, Ohkubo Y, Saitsu H, Takada S, Ogata T. ZNF445: a homozygous truncating variant in a patient with Temple syndrome and multilocus imprinting disturbance. Clin Epigenet. 2021;13:119.
Sparago A, Cerrato F, Riccio A. Is ZFP57 binding to H19/IGF2:IG-DMR affected in Silver–Russell syndrome? Clin Epigenet. 2018;10:23.
Fauque P, De Mouzon J, Devaux A, Epelboin S, Gervoise-Boyer MJ, Levy R, Valentin M, Viot G, Bergère A, De Vienne C, Jonveaux P, Pessione F. Reproductive technologies, female infertility, and the risk of imprinting-related disorders. Clin Epigenet. 2020;12:191.
Mussa A, Molinatto C, Cerrato F, Palumbo O, Carella M, Baldassarre G, Carli D, Peris C, Riccio A, Ferrero GB. Assisted reproductive techniques and risk of Beckwith–Wiedemann syndrome. Pediatrics. 2017;140:1.
Hattori H, Hiura H, Kitamura A, Miyauchi N, Kobayashi N, Takahashi S, Okae H, Kyono K, Kagami M, Ogata T, Arima T. Association of four imprinting disorders and ART. Clin Epigenet. 2019;11:21.
de Waal E, Yamazaki Y, Ingale P, Bartolomei M, Yanagimachi R, McCarrey JR. Primary epimutations introduced during intracytoplasmic sperm injection (ICSI) are corrected by germline-specific epigenetic reprogramming. Proc Natl Acad Sci U S A. 2012;109:4163–8.
Kochmanski JJ, Marchlewicz EH, Cavalcante RG, Perera BPU, Sartor MA, Dolinoy DC. Longitudinal effects of developmental bisphenol A exposure on epigenome-wide DNA hydroxymethylation at imprinted loci in mouse blood. Environ Health Perspect. 2018;126: 077006.
Cinquina V, Calvigioni D, Farlik M, Halbritter F, Fife-Gernedl V, Shirran SL, Fuszard MA, Botting CH, Poullet P, Piscitelli F, Máté Z, Szabó G, Yanagawa Y, Kasper S, Di Marzo V, Mackie K, McBain CJ, Bock C, Keimpema E, Harkany T. Life-long epigenetic programming of cortical architecture by maternal “Western” diet during pregnancy. Mol Psychiatry. 2020;25:22–36.
Soubry A, Schildkraut JM, Murtha A, Wang F, Huang Z, Bernal A, Kurtzberg J, Jirtle RL, Murphy SK, Hoyo C. Paternal obesity is associated with IGF2 hypomethylation in newborns: results from a Newborn Epigenetics Study (NEST) cohort. BMC Med. 2013;11:29.
Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet. 2019;20:235–48.
Tauber M, Hoybye C. Endocrine disorders in Prader–Willi syndrome: a model to understand and treat hypothalamic dysfunction. Lancet Diabetes Endocrinol. 2021;9:235–46.
Keute M, Miller MT, Krishnan ML, Sadhwani A, Chamberlain S, Thibert RL, Tan WH, Bird LM, Hipp JF. Angelman syndrome genotypes manifest varying degrees of clinical severity and developmental impairment. Mol Psychiatry. 2021;26:3625–33.
Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, Boonen SE, Cole T, Baker R, Bertoletti M, Cocchi G, Coze C, De Pellegrin M, Hussain K, Ibrahim A, Kilby MD, Krajewska-Walasek M, Kratz CP, Ladusans EJ, Lapunzina P, Le Bouc Y, Maas SM, Macdonald F, Õunap K, Peruzzi L, Rossignol S, Russo S, Shipster C, Skórka A, Tatton-Brown K, Tenorio J, Tortora C, Grønskov K, Netchine I, Hennekam RC, Prawitt D, Tümer Z, Eggermann T, Mackay DJG, Riccio A, Maher ER. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018;14:229–49.
Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM, Salem J, Bliek J, Canton AP, Chrzanowska KH, Davies JH, Dias RP, Dubern B, Elbracht M, Giabicani E, Grimberg A, Grønskov K, Hokken-Koelega AC, Jorge AA, Kagami M, Linglart A, Maghnie M, Mohnike K, Monk D, Moore GE, Murray PG, Ogata T, Petit IO, Russo S, Said E, Toumba M, Tümer Z, Binder G, Eggermann T, Harbison MD, Temple IK, Mackay DJ, Netchine I. Diagnosis and management of Silver–Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017;13:105–24.
Kagami M, Yanagisawa A, Ota M, Matsuoka K, Nakamura A, Matsubara K, Nakabayashi K, Takada S, Fukami M, Ogata T. Temple syndrome in a patient with variably methylated CpGs at the primary MEG3/DLK1:IG-DMR and severely hypomethylated CpGs at the secondary MEG3:TSS-DMR. Clin Epigenet. 2019;11:42.
Kagami M, Matsubara K, Nakabayashi K, Nakamura A, Sano S, Okamura K, Hata K, Fukami M, Ogata T. Genome-wide multilocus imprinting disturbance analysis in Temple syndrome and Kagami–Ogata syndrome. Genet Med. 2017;19:476–82.
Hollander E, Levine KG, Ferretti CJ, Freeman K, Doernberg E, Desilva N, Taylor BP. Intranasal oxytocin versus placebo for hyperphagia and repetitive behaviors in children with Prader–Willi syndrome: a randomized controlled pilot trial. J Psychiatr Res. 2021;137:643–51.
Tauber M, Boulanouar K, Diene G, Çabal-Berthoumieu S, Ehlinger V, Fichaux-Bourin P, Molinas C, Faye S, Valette M, Pourrinet J, Cessans C, Viaux-Sauvelon S, Bascoul C, Guedeney A, Delhanty P, Geenen V, Martens H, Muscatelli F, Cohen D, Consoli A, Payoux P, Arnaud C, Salles JP. The use of oxytocin to improve feeding and social skills in infants with Prader–Willi syndrome. Pediatrics. 2017;139:2.
Dykens EM, Miller J, Angulo M, Roof E, Reidy M, Hatoum HT, Willey R, Bolton G, Korner P. Intranasal carbetocin reduces hyperphagia in individuals with Prader–Willi syndrome. JCI Insight. 2018;3:12.
Kimonis V, Surampalli A, Wencel M, Gold JA, Cowen NM. A randomized pilot efficacy and safety trial of diazoxide choline controlled-release in patients with Prader–Willi syndrome. PLoS ONE. 2019;14: e0221615.
Allas S, Caixàs A, Poitou C, Coupaye M, Thuilleaux D, Lorenzini F, Diene G, Crinò A, Illouz F, Grugni G, Potvin D, Bocchini S, Delale T, Abribat T, Tauber M. AZP-531, an unacylated ghrelin analog, improves food-related behavior in patients with Prader–Willi syndrome: a randomized placebo-controlled trial. PLoS ONE. 2018;13: e0190849.
Salehi P, Hsu I, Azen CG, Mittelman SD, Geffner ME, Jeandron D. Effects of exenatide on weight and appetite in overweight adolescents and young adults with Prader–Willi syndrome. Pediatr Obes. 2017;12:221–8.
McCandless SE, Yanovski JA, Miller J, Fu C, Bird LM, Salehi P, Chan CL, Stafford D, Abuzzahab MJ, Viskochil D, Barlow SE, Angulo M, Myers SE, Whitman BY, Styne D, Roof E, Dykens EM, Scheimann AO, Malloy J, Zhuang D, Taylor K, Hughes TE, Kim DD, Butler MG. Effects of MetAP2 inhibition on hyperphagia and body weight in Prader–Willi syndrome: a randomized, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2017;19:1751–61.
Falls BA, Zhang Y. Insights into the allosteric mechanism of setmelanotide (RM-493) as a potent and first-in-class melanocortin-4 receptor (MC4R) agonist to treat rare genetic disorders of obesity through an in silico approach. ACS Chem Neurosci. 2019;10:1055–65.
Bischof JM, Van Der Ploeg LH, Colmers WF, Wevrick R. Magel2-null mice are hyper-responsive to setmelanotide, a melanocortin 4 receptor agonist. Br J Pharmacol. 2016;173:2614–21.
Teuffel P, Wang L, Prinz P, Goebel-Stengel M, Scharner S, Kobelt P, Hofmann T, Rose M, Klapp BF, Reeve JR Jr, Stengel A. Treatment with the ghrelin-O-acyltransferase (GOAT) inhibitor GO-CoA-Tat reduces food intake by reducing meal frequency in rats. J Physiol Pharmacol. 2015;66:493–503.
Knani I, Earley BJ, Udi S, Nemirovski A, Hadar R, Gammal A, Cinar R, Hirsch HJ, Pollak Y, Gross I, Eldar-Geva T, Reyes-Capo DP, Han JC, Haqq AM, Gross-Tsur V, Wevrick R, Tam J. Targeting the endocannabinoid/CB1 receptor system for treating obesity in Prader–Willi syndrome. Mol Metab. 2016;5:1187–99.
Scopinho AA, Guimarães FS, Corrêa FM, Resstel LB. Cannabidiol inhibits the hyperphagia induced by cannabinoid-1 or serotonin-1A receptor agonists. Pharmacol Biochem Behav. 2011;98:268–72.
Consoli A, Cabal Berthoumieu S, Raffin M, Thuilleaux D, Poitou C, Coupaye M, Pinto G, Lebbah S, Zahr N, Tauber M, Cohen D, Bonnot O. Effect of topiramate on eating behaviours in Prader–Willi syndrome: TOPRADER double-blind randomised placebo-controlled study. Transl Psychiatry. 2019;9:274.
East N, Maroney M. Topiramate in the treatment of Prader–Willi syndrome: a case report. Ment Health Clin. 2017;7:7–9.
Bird LM, Ochoa-Lubinoff C, Tan WH, Heimer G, Melmed RD, Rakhit A, Visootsak J, During MJ, Holcroft C, Burdine RD, Kolevzon A, Thibert RL. The STARS phase 2 study: a randomized controlled trial of gaboxadol in Angelman syndrome. Neurology. 2021;96:e1024–35.
Ciarlone SL, Grieco JC, D’Agostino DP, Weeber EJ. Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol Dis. 2016;96:38–46.
Carson RP, Herber DL, Pan Z, Phibbs F, Key AP, Gouelle A, Ergish P, Armour EA, Patel S, Duis J. Nutritional formulation for patients with Angelman syndrome: a randomized, double-blind, placebo-controlled study of exogenous ketones. J Nutr. 2021;151:3628–36.
Buonfiglio D, Hummer DL, Armstrong A, Christopher Ehlen J, DeBruyne JP. Angelman syndrome and melatonin: what can they teach us about sleep regulation. J Pineal Res. 2020;69: e12697.
Paprocka J, Kijonka M, Wojcieszek P, Pęcka M, Emich-Widera E, Sokół M. Melatonin and Angelman syndrome: implications and mathematical model of diurnal secretion. Int J Endocrinol. 2017;2017:5853167.
Tan WH, Bird LM, Sadhwani A, Barbieri-Welge RL, Skinner SA, Horowitz LT, Bacino CA, Noll LM, Fu C, Hundley RJ, Wink LK, Erickson CA, Barnes GN, Slavotinek A, Jeremy R, Rotenberg A, Kothare SV, Olson HE, Poduri A, Nespeca MP, Chu HC, Willen JM, Haas KF, Weeber EJ, Rufo PA. A randomized controlled trial of levodopa in patients with Angelman syndrome. Am J Med Genet A. 2018;176:1099–107.
Ruiz-Antoran B, Sancho-López A, Cazorla-Calleja R, López-Pájaro LF, Leiva Á, Iglesias-Escalera G, Marín-Serrano ME, Rincón-Ortega M, Lara-Herguedas J, Rossignoli-Palomeque T, Valiente-Rodríguez S, González-Marques J, Román-Riechmann E, Avendaño-Solá C. A randomized placebo controlled clinical trial to evaluate the efficacy and safety of minocycline in patients with Angelman syndrome (A-MANECE study). Orphanet J Rare Dis. 2018;13:144.
Liu Y, Johe K, Sun J, Hao X, Wang Y, Bi X, Baudry M. Enhancement of synaptic plasticity and reversal of impairments in motor and cognitive functions in a mouse model of Angelman syndrome by a small neurogenic molecule, NSI-189. Neuropharmacology. 2019;144:337–44.
Gu B, Zhu M, Glass MR, Rougié M, Nikolova VD, Moy SS, Carney PR, Philpot BD. Cannabidiol attenuates seizures and EEG abnormalities in Angelman syndrome model mice. J Clin Invest. 2019;129:5462–7.
Cruz E, Descalzi G, Steinmetz A, Scharfman HE, Katzman A, Alberini CM. CIM6P/IGF-2 receptor ligands reverse deficits in Angelman syndrome model mice. Autism Res. 2021;14:29–45.
Sun AX, Yuan Q, Fukuda M, Yu W, Yan H, Lim GGY, Nai MH, D’Agostino GA, Tran HD, Itahana Y, Wang D, Lokman H, Itahana K, Lim SWL, Tang J, Chang YY, Zhang M, Cook SA, Rackham OJL, Lim CT, Tan EK, Ng HH, Lim KL, Jiang YH, Je HS. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science. 2019;366:1486–92.
Chung L, Bey AL, Towers AJ, Cao X, Kim IH, Jiang YH. Lovastatin suppresses hyperexcitability and seizure in Angelman syndrome model. Neurobiol Dis. 2018;110:12–9.
Kumar V, Joshi T, Vatsa N, Singh BK, Jana NR. Simvastatin restores HDAC1/2 activity and improves behavioral deficits in Angelman syndrome model mouse. Front Mol Neurosci. 2019;12:289.
Wang J, Lou SS, Wang T, Wu RJ, Li G, Zhao M, Lu B, Li YY, Zhang J, Cheng X, Shen Y, Wang X, Zhu ZC, Li MJ, Takumi T, Yang H, Yu X, Liao L, Xiong ZQ. UBE3A-mediated PTPA ubiquitination and degradation regulate PP2A activity and dendritic spine morphology. Proc Natl Acad Sci U S A. 2019;116:12500–5.
Guzzetti S, Calzari L, Buccarello L, Cesari V, Toschi I, Cattaldo S, Mauro A, Pregnolato F, Mazzola SM, Russo S. Taurine administration recovers motor and learning deficits in an Angelman syndrome mouse model. Int J Mol Sci. 2018;19:1088.
Güemes M, Shah P, Roženková K, Gilbert C, Morgan K, Hussain K. Severe hyperinsulinaemic hypoglycaemia in Beckwith–Wiedemann syndrome due to paternal uniparental disomy of 11p15.5 managed with sirolimus therapy. Horm Res Paediatr. 2016;85:353–7.
Lemoine A, Harbison MD, Salem J, Tounian P, Netchine I, Dubern B. Effect of cyproheptadine on weight and growth velocity in children with Silver–Russell syndrome. J Pediatr Gastroenterol Nutr. 2018;66:306–11.
Srivastava T, Krudys J, Mardis NJ, Sebestyen-VanSickle J, Alon US. Cinacalcet as adjunctive therapy in pseudohypoparathyroidism type 1b. Pediatr Nephrol. 2016;31:795–800.
Garcin L, Kariyawasam D, Busiah K, Fauret-Amsellem AL, Le Bourgeois F, Vaivre-Douret L, Cavé H, Polak M, Beltrand J. Successful off-label sulfonylurea treatment of neonatal diabetes mellitus due to chromosome 6 abnormalities. Pediatr Diabetes. 2018;19:663–9.
Carmody D, Beca FA, Bell CD, Hwang JL, Dickens JT, Devine NA, Mackay DJ, Temple IK, Hays LR, Naylor RN, Philipson LH, Greeley SA. Role of noninsulin therapies alone or in combination in chromosome 6q24-related transient neonatal diabetes: sulfonylurea improves but does not always normalize insulin secretion. Diabetes Care. 2015;38:e86–7.
Yorifuji T, Hashimoto Y, Kawakita R, Hosokawa Y, Fujimaru R, Hatake K, Tamagawa N, Nakajima H, Fujii M. Relapsing 6q24-related transient neonatal diabetes mellitus successfully treated with a dipeptidyl peptidase-4 inhibitor: a case report. Pediatr Diabetes. 2014;15:606–10.
Søvik O, Aagenaes O, Eide S, Mackay D, Temple IK, Molven A, Njølstad PR. Familial occurrence of neonatal diabetes with duplications in chromosome 6q24: treatment with sulfonylurea and 40-yr follow-up. Pediatr Diabetes. 2012;13:155–62.
Reichova A, Schaller F, Bukatova S, Bacova Z, Muscatelli F, Bakos J. The impact of oxytocin on neurite outgrowth and synaptic proteins in Magel2-deficient mice. Dev Neurobiol. 2021;81:366–88.
Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578:229–36.
Coller BS. Ethics of human genome editing. Annu Rev Med. 2019;70:289–305.
Wu Z, Parry M, Hou XY, Liu MH, Wang H, Cain R, Pei ZF, Chen YC, Guo ZY, Abhijeet S, Chen G. Gene therapy conversion of striatal astrocytes into GABAergic neurons in mouse models of Huntington’s disease. Nat Commun. 2020;11:1105.
Thompson AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil JA, Hongeng S, Magrin E, Schiller GJ, Payen E, Semeraro M, Moshous D, Lefrere F, Puy H, Bourget P, Magnani A, Caccavelli L, Diana JS, Suarez F, Monpoux F, Brousse V, Poirot C, Brouzes C, Meritet JF, Pondarré C, Beuzard Y, Chrétien S, Lefebvre T, Teachey DT, Anurathapan U, Ho PJ, von Kalle C, Kletzel M, Vichinsky E, Soni S, Veres G, Negre O, Ross RW, Davidson D, Petrusich A, Sandler L, Asmal M, Hermine O, De Montalembert M, Hacein-Bey-Abina S, Blanche S, Leboulch P, Cavazzana M. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378:1479–93.
Le Meur G, Lebranchu P, Billaud F, Adjali O, Schmitt S, Bézieau S, Péréon Y, Valabregue R, Ivan C, Darmon C, Moullier P, Rolling F, Weber M. Safety and long-term efficacy of AAV4 gene therapy in patients with RPE65 Leber congenital amaurosis. Mol Ther. 2018;26:256–68.
George LA, Ragni MV, Rasko JEJ, Raffini LJ, Samelson-Jones BJ, Ozelo M, Hazbon M, Runowski AR, Wellman JA, Wachtel K, Chen Y, Anguela XM, Kuranda K, Mingozzi F, High KA. Long-term follow-up of the first in human intravascular delivery of AAV for gene transfer: AAV2-hFIX16 for severe hemophilia B. Mol Ther. 2020;28:2073–82.
Zhu J, Liu JQ, Shi M, Cheng X, Ding M, Zhang JC, Davis JP, Varikuti S, Satoskar AR, Lu L, Pan X, Zheng P, Liu Y, Bai XF. IL-27 gene therapy induces depletion of Tregs and enhances the efficacy of cancer immunotherapy. JCI Insight. 2018;3:1088.
Liu W, Kim GB, Krump NA, Zhou Y, Riley JL, You J. Selective reactivation of STING signaling to target Merkel cell carcinoma. Proc Natl Acad Sci U S A. 2020;117:13730–9.
Daily JL, Nash K, Jinwal U, Golde T, Rogers J, Peters MM, Burdine RD, Dickey C, Banko JL, Weeber EJ. Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS ONE. 2011;6: e27221.
Judson MC, Shyng C, Simon JM, Davis CR, Punt AM, Salmon MT, Miller NW, Ritola KD, Elgersma Y, Amaral DG, Gray SJ, Philpot BD. Dual-isoform hUBE3A gene transfer improves behavioral and seizure outcomes in Angelman syndrome model mice. JCI Insight. 2021;6:20.
Masiuk KE, Brown D, Laborada J, Hollis RP, Urbinati F, Kohn DB. Improving gene therapy efficiency through the enrichment of human hematopoietic stem cells. Mol Ther. 2017;25:2163–75.
Ferrua F, Cicalese MP, Galimberti S, Giannelli S, Dionisio F, Barzaghi F, Migliavacca M, Bernardo ME, Calbi V, Assanelli AA, Facchini M, Fossati C, Albertazzi E, Scaramuzza S, Brigida I, Scala S, Basso-Ricci L, Pajno R, Casiraghi M, Canarutto D, Salerio FA, Albert MH, Bartoli A, Wolf HM, Fiori R, Silvani P, Gattillo S, Villa A, Biasco L, Dott C, Culme-Seymour EJ, van Rossem K, Atkinson G, Valsecchi MG, Roncarolo MG, Ciceri F, Naldini L, Aiuti A. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott–Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019;6:e239–53.
Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, Caccavelli L, Neven B, Bourget P, El Nemer W, Bartolucci P, Weber L, Puy H, Meritet JF, Grevent D, Beuzard Y, Chretien S, Lefebvre T, Ross RW, Negre O, Veres G, Sandler L, Soni S, de Montalembert M, Blanche S, Leboulch P, Cavazzana M. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376:848–55.
Tucci F, Galimberti S, Naldini L, Valsecchi MG, Aiuti A. A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders. Nat Commun. 2022;13:1315.
Adhikari A, Copping NA, Beegle J, Cameron DL, Deng P, O’Geen H, Segal DJ, Fink KD, Silverman JL, Anderson JS. Functional rescue in an Angelman syndrome model following treatment with lentivector transduced hematopoietic stem cells. Hum Mol Genet. 2021;30:1067–83.
Varkuti BH, Kepiro M, Liu Z, Vick K, Avchalumov Y, Pacifico R, MacMullen CM, Kamenecka TM, Puthanveettil SV, Davis RL. Neuron-based high-content assay and screen for CNS active mitotherapeutics. Sci Adv. 2020;6:eaaw8702.
Zhang R, Zhu Z, Lv H, Li F, Sun S, Li J, Lee CS. Immune checkpoint blockade mediated by a small-molecule nanoinhibitor targeting the PD-1/PD-L1 pathway synergizes with photodynamic therapy to elicit antitumor immunity and antimetastatic effects on breast cancer. Small. 2019;15: e1903881.
Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, Fekkes P, Karr C, Klimek V, Lai G, Lee L, Kumar P, Lee SC, Liu X, Mackenzie C, Meeske C, Mizui Y, Padron E, Park E, Pazolli E, Peng S, Prajapati S, Taylor J, Teng T, Wang J, Warmuth M, Yao H, Yu L, Zhu P, Abdel-Wahab O, Smith PG, Buonamici S. H3B–8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. 2018;24:497–504.
Scherer M, Santana AG, Robinson K, Zhou S, Overkleeft HS, Clarke L, Withers SG. Lipid-mimicking phosphorus-based glycosidase inactivators as pharmacological chaperones for the treatment of Gaucher’s disease. Chem Sci. 2021;12:13909–13.
Martin JK 2nd, Sheehan JP, Bratton BP, Moore GM, Mateus A, Li SH, Kim H, Rabinowitz JD, Typas A, Savitski MM, Wilson MZ, Gitai Z. A dual-mechanism antibiotic kills Gram-negative bacteria and avoids drug resistance. Cell. 2020;181:1518-32.e14.
Ward RA, Fawell S, Floc’h N, Flemington V, McKerrecher D, Smith PD. Challenges and opportunities in cancer drug resistance. Chem Rev. 2021;121:3297–351.
Huang HS, Allen JA, Mabb AM, King IF, Miriyala J, Taylor-Blake B, Sciaky N, Dutton JW, Lee HM, Chen X, Jin J, Bridges AS, Zylka MJ, Roth BL, Philpot BD. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2012;481:185.
King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, Chamberlain SJ, Philpot BD, Zylka MJ. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501:58.
Crooke ST, Baker BF, Crooke RM, Liang XH. Antisense technology: an overview and prospectus. Nat Rev Drug Discov. 2021;20:427–53.
Yu AM, Tu MJ. Deliver the promise: RNAs as a new class of molecular entities for therapy and vaccination. Pharmacol Ther. 2021;107967.
Jafar-Nejad P, Powers B, Soriano A, Zhao H, Norris DA, Matson J, DeBrosse-Serra B, Watson J, Narayanan P, Chun SJ, Mazur C, Kordasiewicz H, Swayze EE, Rigo F. The atlas of RNase H antisense oligonucleotide distribution and activity in the CNS of rodents and non-human primates following central administration. Nucleic Acids Res. 2021;49:657–73.
Scoles DR, Meera P, Schneider MD, Paul S, Dansithong W, Figueroa KP, Hung G, Rigo F, Bennett CF, Otis TS, Pulst SM. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017;544:362–6.
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–12.
Milazzo C, Mientjes EJ, Wallaard I, Rasmussen SV, Erichsen KD, Kakunuri T, van der Sman ASE, Kremer T, Miller MT, Hoener MC, Elgersma Y. Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. Jci Insight. 2021;6:15.
Silva-Santos S, van Woerden GM, Bruinsma CF, Mientjes E, Jolfaei MA, Distel B, Kushner SA, Elgersma Y. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Invest. 2015;125:2069–76.
Sonzogni M, Zhai P, Mientjes EJ, van Woerden GM, Elgersma Y. Assessing the requirements of prenatal UBE3A expression for rescue of behavioral phenotypes in a mouse model for Angelman syndrome. Mol Autism. 2020;11:70.
Eguchi A, Wleklinski MJ, Spurgat MC, Heiderscheit EA, Kropornicka AS, Vu CK, Bhimsaria D, Swanson SA, Stewart R, Ramanathan P, Kamp TJ, Slukvin I, Thomson JA, Dutton JR, Ansari AZ. Reprogramming cell fate with a genome-scale library of artificial transcription factors. Proc Natl Acad Sci U S A. 2016;113:E8257–66.
Tak YE, Horng JE, Perry NT, Schultz HT, Iyer S, Yao Q, Zou LS, Aryee MJ, Pinello L, Joung JK. Augmenting and directing long-range CRISPR-mediated activation in human cells. Nat Methods. 2021;18:1075–81.
Bailus BJ, Pyles B, McAlister MM, O’Geen H, Lockwood SH, Adams AN, Nguyen JT, Yu A, Berman RF, Segal DJ. Protein delivery of an artificial transcription factor restores widespread Ube3a expression in an Angelman syndrome mouse brain. Mol Ther. 2016;24:548–55.
Pickar-Oliver A, Gersbach CA. The next generation of CRISPR–Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20:490–507.
Gaj T, Ojala DS, Ekman FK, Byrne LC, Limsirichai P, Schaffer DV. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci Adv. 2017;3:eaar3952.
Ekman FK, Ojala DS, Adil MM, Lopez PA, Schaffer DV, Gaj T. CRISPR–Cas9-mediated genome editing increases lifespan and improves motor deficits in a Huntington’s disease mouse model. Mol Ther Nucleic Acids. 2019;17:829–39.
György B, Lööv C, Zaborowski MP, Takeda S, Kleinstiver BP, Commins C, Kastanenka K, Mu D, Volak A, Giedraitis V, Lannfelt L, Maguire CA, Joung JK, Hyman BT, Breakefield XO, Ingelsson M. CRISPR/Cas9 mediated disruption of the Swedish APP allele as a therapeutic approach for early-onset Alzheimer’s disease. Mol Ther Nucleic Acids. 2018;11:429–40.
Wolter JM, Mao HQ, Fragola G, Simon JM, Krantz JL, Bazick HO, Oztemiz B, Stein JL, Zylka MJ. Cas9 gene therapy for Angelman syndrome trapsUbe3a-ATSlong non-coding RNA. Nature. 2020;587:281.
Schmid RS, Deng XF, Panikker P, Msackyi M, Breton C, Wilson JM. CRISPR/Cas9 directed to the Ube3a antisense transcript improves Angelman syndrome phenotype in mice. J Clin Invest. 2021;131:5.
Pollyea DA, Pratz K, Letai A, Jonas BA, Wei AH, Pullarkat V, Konopleva M, Thirman MJ, Arellano M, Becker PS, Chyla B, Hong WJ, Jiang Q, Potluri J, DiNardo CD. Venetoclax with azacitidine or decitabine in patients with newly diagnosed acute myeloid leukemia: Long term follow-up from a phase 1b study. Am J Hematol. 2021;96:208–17.
Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S, Batlevi CL, Campbell P, Ribrag V, Damaj GL, Dickinson M, Jurczak W, Kazmierczak M, Opat S, Radford J, Schmitt A, Yang J, Whalen J, Agarwal S, Adib D, Salles G. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020;21:1433–42.
Rodriguez CP, Wu QV, Voutsinas J, Fromm JR, Jiang X, Pillarisetty VG, Lee SM, Santana-Davila R, Goulart B, Baik CS, Chow LQM, Eaton K, Martins R. A phase II trial of pembrolizumab and vorinostat in recurrent metastatic head and neck squamous cell carcinomas and salivary gland cancer. Clin Cancer Res. 2020;26:837–45.
Alamer E, Zhong C, Liu Z, Niu Q, Long F, Guo L, Gelman BB, Soong L, Zhou J, Hu H. Epigenetic suppression of HIV in myeloid cells by the BRD4-selective small molecule modulator ZL0580. J Virol. 2020;94:e01880.
Ray KK, Nicholls SJ, Buhr KA, Ginsberg HN, Johansson JO, Kalantar-Zadeh K, Kulikowski E, Toth PP, Wong N, Sweeney M, Schwartz GG. Effect of apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes: a randomized clinical trial. JAMA. 2020;323:1565–73.
Liu Y, Lin H, Jiang L, Shang Q, Yin L, Lin JD, Wu WS, Rui L. Hepatic Slug epigenetically promotes liver lipogenesis, fatty liver disease, and type 2 diabetes. J Clin Invest. 2020;130:2992–3004.
Gräff J, Joseph NF, Horn ME, Samiei A, Meng J, Seo J, Rei D, Bero AW, Phan TX, Wagner F, Holson E, Xu J, Sun J, Neve RL, Mach RH, Haggarty SJ, Tsai LH. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell. 2014;156:261–76.
Qin L, Ma K, Wang ZJ, Hu Z, Matas E, Wei J, Yan Z. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat Neurosci. 2018;21:564–75.
Polex-Wolf J, Lam BY, Larder R, Tadross J, Rimmington D, Bosch F, Cenzano VJ, Ayuso E, Ma MK, Rainbow K, Coll AP, O’Rahilly S, Yeo GS. Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader–Willi syndrome. J Clin Invest. 2018;128:960–9.
Cruvinel E, Budinetz T, Germain N, Chamberlain S, Lalande M, Martins-Taylor K. Reactivation of maternal SNORD116 cluster via SETDB1 knockdown in Prader–Willi syndrome iPSCs. Hum Mol Genet. 2014;23:4674–85.
Langouët M, Glatt-Deeley HR, Chung MS, Dupont-Thibert CM, Mathieux E, Banda EC, Stoddard CE, Crandall L, Lalande M. Zinc finger protein 274 regulates imprinted expression of transcripts in Prader–Willi syndrome neurons. Hum Mol Genet. 2018;27:505–15.
Kim Y, Lee HM, Xiong Y, Sciaky N, Hulbert SW, Cao XY, Everitt JI, Jin J, Roth BL, Jiang YH. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader–Willi syndrome. Nat Med. 2017;23:213–22.
Nuñez JK, Chen J, Pommier GC, Cogan JZ, Replogle JM, Adriaens C, Ramadoss GN, Shi Q, Hung KL, Samelson AJ, Pogson AN, Kim JYS, Chung A, Leonetti MD, Chang HY, Kampmann M, Bernstein BE, Hovestadt V, Gilbert LA, Weissman JS. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 2021;184:2503-19.e17.
Moreno AM, Alemán F, Catroli GF, Hunt M, Hu M, Dailamy A, Pla A, Woller SA, Palmer N, Parekh U, McDonald D, Roberts AJ, Goodwill V, Dryden I, Hevner RF, Delay L, Gonçalves Dos Santos G, Yaksh TL, Mali P. Long-lasting analgesia via targeted in situ repression of Na(V)1.7 in mice. Sci Transl Med. 2021;13:9056.
Lu Z, Liu Z, Mao W, Wang X, Zheng X, Chen S, Cao B, Huang S, Zhang X, Zhou T, Zhang Y, Huang X, Sun Q, Li JD. Locus-specific DNA methylation of Mecp2 promoter leads to autism-like phenotypes in mice. Cell Death Dis. 2020;11:85.
Carpenter MD, Hu Q, Bond AM, Lombroso SI, Czarnecki KS, Lim CJ, Song H, Wimmer ME, Pierce RC, Heller EA. Nr4a1 suppresses cocaine-induced behavior via epigenetic regulation of homeostatic target genes. Nat Commun. 2020;11:504.
Liu XS, Wu H, Ji X, Stelzer Y, Wu XB, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233.
Krishnan V, Stoppel DC, Nong Y, Johnson MA, Nadler MJ, Ozkaynak E, Teng BL, Nagakura I, Mohammad F, Silva MA, Peterson S, Cruz TJ, Kasper EM, Arnaout R, Anderson MP. Autism gene Ube3a and seizures impair sociability by repressing VTA Cbln1. Nature. 2017;543:507–12.
Xu X, Li C, Gao X, Xia K, Guo H, Li Y, Hao Z, Zhang L, Gao D, Xu C, Xu H, Xiong ZQ, Qiu Z, Mei L, Xie X, Ruan K, Hu R. Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res. 2018;28:48–68.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (81371215 and 81670786) and The Key R & D Projects of Zhejiang Provincial Department of Science and Technology (2021 C03094). Figures in the article are created with BioRender.com.
Funding
This work was supported by National Natural Science Foundation of China (81371215 and 81670786) and The Key R & D Projects of Zhejiang Provincial Department of Science and Technology (2021 C03094).
Author information
Authors and Affiliations
Contributions
YC and YQ completed the conceptualization and original draft preparation; YC, YQ, XZ, XW, CH, and FX reviewed the manuscript; and CZ supervised and revised the work critically for important intellectual content. All authors read and approved the final manuscript. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors gave their consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chao, Y., Qin, Y., Zou, X. et al. Promising therapeutic aspects in human genetic imprinting disorders. Clin Epigenet 14, 146 (2022). https://doi.org/10.1186/s13148-022-01369-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-022-01369-6