
Abstract
Epigenetics studies heritable genomic modifications that occur with the participation of epigenetic modifying enzymes but without alterations of the nucleotide structure. Small-molecule inhibitors of these epigenetic modifying enzymes are known as epigenetic drugs (epi-drugs), which can cause programmed death of tumor cells by affecting the cell cycle, angiogenesis, proliferation, and migration. Epi-drugs include histone methylation inhibitors, histone demethylation inhibitors, histone deacetylation inhibitors, and DNA methylation inhibitors. Currently, epi-drugs undergo extensive development, research, and application. Although epi-drugs have convincing anti-tumor effects, the patient’s sensitivity to epi-drug application is also a fundamental clinical issue. The development and research of biomarkers for epi-drugs provide a promising direction for screening drug-sensitive patients. Here, we review the predictive biomarkers of 12 epi-drugs as well as the progress of combination therapy with chemotherapeutic drugs or immunotherapy. Further, we discuss the improvement in the development of natural ingredients with low toxicity and low side effects as epi-drugs.
Similar content being viewed by others

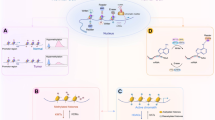

Introduction
Epigenetics studies heritable changes in gene expression are mainly reflected in DNA methylation, histone modification, and chromosome abnormalities and are not due to changes in nucleotide states. Faulty epigenetic reprogramming composes a fundamental part of tumor cell proliferation, escape, intratumoral heterogeneity, and acquisition of therapeutic drug resistance. Accordingly, the genomic alterations and gene transcription abnormalities induced by epigenetic aberrations can have reflective effects in multiple pathways, such as EMT, Hippo signaling, p53 pathway, AMPK signaling, and cellular senescence, which can lead to the induction and maintenance of various cancers [1,2,3,4,5]. Emerging epigenetic drugs (epi-drugs) target enzymes involved in the regulation of aberrant epigenetic modifications in tumors. Further, epi-drugs are usually small-molecule inhibitors that inhibit key enzyme activity [6, 7]. The enzymes involved in aberrant epigenetic processes include DNA methylation-modifying enzymes and histone-modifying enzymes, such as DNA methyltransferases (DNMTs), histone methyltransferases (HMTs), histone demethylases (HDMs), and histone deacetylases (HDACs). At present, epi-drugs that are DNMT inhibitors and HDAC inhibitors are widely developed in preclinical research and clinical applications. Precision medicine paradigms provide hope for the research and clinical application of new representative epi-drugs, including the continuous development of HMT inhibitors, protein arginine methyltransferases (PRMT) inhibitors, and enhancer of zeste homolog 2 (EZH2) inhibitors [8, 9]. Furthermore, targeted drug administration based on different patient biomarkers provides a basis for precision medicine of epi-drugs. One-size-fits-all drug administration should not be adopted in clinical trials; rather, application according to the intratumoral heterogeneity should be preferred. For example, the HDAC inhibitor abexinostat can be administrated selectively according to the Xist expression of breast cancer patients. The use of Xist expression as a biomarker in this instance can improve drug sensitivity [10].
Epi-drugs are more widely used in hematological tumors than in solid tumors. Moreover, the efficacy of epi-drugs in solid tumors is still limited, which may be due to the high degree of cellular differentiation and intratumoral heterogeneity of solid tumors [11]. The intratumoral heterogeneity is observed in colorectal cancer; there are significant heterogeneity in pattern of DNA methylation in colorectal tumors, which associates with times of relapse-free and overall survival [12]. Tumor heterogeneity broadly means that tumors contain cell subsets with different phenotypes. With the development of single-cell sequencing technology, we have gradually realized that through the screening of different cellular surface markers, biochemical metabolism, and other tumor cell aspects, tumor cells are divided into subsets, and in-depth insight into tumor heterogeneity is achieved [13, 14]. By understanding the different levels of tumor heterogeneity, precision therapy has attracted more and more attention to the medical treatment in clinical oncology. Due to the genetic instability and intratumoral heterogeneity of malignant tumors, it is of great clinical significance to understand the biomarkers targeted by epi-drugs in various types of tumors.
Since reversible epigenetic abnormalities can partially lead to the continuous evolution of cancer cells, it is necessary to consider not only the precise treatment with epi-drugs but also the combination of epi-drugs with chemotherapeutic drugs or immunotherapy. The combination of epi-drugs and other types of drugs still presents numerous challenges and risks; thus, understanding the advantages and risks of drug combinations may help to better grasp and utilize epi-drugs and offer other perspectives for cancer therapy. In this review, we summarize and discuss the newly developed biomarkers and drug combinations of 12 epi-drugs of the three types of histone modification inhibitors and DNA methylation inhibitors. In any effort to discover and develop small molecule drugs, it has become key to detect drug-likeness and the targeted proteins. For this point, we provide a comprehensive figure to summarize the predicted oral efficacy and target proteins of epi-drugs [15] (Figs. 2, 4). In addition, a large number of natural components function as small-molecule inhibitors. This paper also discusses the progress of natural epi-drugs in recent years to provide a basic reference for clinical trial design and drug compatibility.
Predictive biomarkers and combination trials of HMT inhibitors
Histone methylation occurs as a covalent modification of arginine (monomethylation or dimethylation) or lysine (monomethylation, dimethylation, or trimethylation). Histone methylation can participate in cancer-related pathways by inhibiting or activating gene transcription, whereas the abnormal function of HMT affects the occurrence and development of cancer [16,17,18]. Therefore, the development and study of small-molecule HMT inhibitors opened up new horizons for a novel generation of antineoplastic drugs. HMTs are typically divided into arginine methyltransferase and lysine methyltransferase in humans, such as the PRMT family and EZH2, respectively (Figs. 1, 2). In related reports, high levels of PRMT family proteins and EZH2 expression were found in hematological tumors and solid tumors [19, 20], but the development and application of PRMT inhibitors and EZH2 inhibitors still have limitations.

Predictive biomarkers and drug combinations of histone methylation inhibitors. Prominent biomarkers and drug combinations of histone methyltransferase (HMT) and histone demethylase (HDM) inhibitors are detailed here. However, other biomarkers and drug combinations also exist. Histone octamer is the basic unit consisting of the nucleosome core particle. Me represents methylation. HMTs can methylate arginine or lysine of histone, meantime HDMs demethylates methylated histones. The small molecule drugs had been developed to inhibit HMT or HDM to regulate an epigenetic process in cancer cells. The application of these inhibitors acts as anticancer drugs by targeting individual biomarkers through different pathway in a variety of tumors. HMT inhibitors include the PRMT inhibitors GSK3368715 and GSK3326595 and the EZH2 inhibitors tazemetostat, CPI-1205, and GSK2816126. The predicted biomarkers for the two PRMT inhibitors are the MTAP and p53-MDM4 axes, respectively. GSK3326595 combined with immunotherapy can effectively exert synergistic anticancer effects in melanoma. The predicted biomarkers of the three EZH2 inhibitors are INI1, the PRC2 complex, and BRAF. The combination of CPI-1205 and an anti-CTL4 antibody can effectively exert a synergistic anticancer effect in melanoma. LSD1 inhibitors as HDM inhibitors include GSK2879552 and tranylcypromine. The DNA hypomethylation levels and ZEB2 status are predictive biomarkers for the selection of GSK2879552 sensitive patients. In hepatic carcinoma treatment, the combination of GSK2879552 and sorafenib exerts improved anticancer effects. In AML, the combination of tranylcypromine with mTORC1 inhibitors effectively exerts synergistic anticancer effects

Oral bioavailability and target prediction of PRMT inhibitors, EZH2 inhibitors, and LSD1 inhibitors. We used the online tool SwissADME (https://www.sib.swiss/) to predict the oral bioavailability and target prediction of seven epi-drugs including PRMT inhibitors, EZH2 inhibitors, and LSD1 inhibitors. Oral bioavailability is an essential parameter for determining the efficacy and side effects of new and developing medications. The prediction of oral bioavailability is displayed for a rapid appraisal of drug-likeness. Six physicochemical properties are taken into account: lipophilicity, size, polarity, solubility, flexibility and saturation. The colored zone is the suitable physiochemical space for oral bioavailability. The pink area represents the optimal range for each property. Lipophility: -0.7 < XLOGP3 < + 5.0; size: 150 g/mol < MV < 500 g/mol; polarity: 20Å2 < TPSA < 130Å2; insolubility: 0 < Log S (ESOL) < 6; insaturation: 0.25 < fraction Csp3 < 1; and flexibility: 0 < Num. rotatable bonds < 9. We also used the website to predict the most probable macromolecular targets of a small molecule, assumed as bioactive of each inhibitor. The pie chart of each inhibitor displays the summary of predication target classes, including kinase, writer, eraser, family A G protein coupled receptor, phosphodiesterase, protease and so on
PRMT inhibitors
GSK3368715 is an oral S-adenosyl-L-methionine (SAM) noncompetitive type 1 PRMT reversible inhibitor. GSK3368715 inhibits the activity of PRMT1, PRMT3, PRMT4, PRMT6, and PRMT8 at different concentrations [21]. The anticancer activity of GSK3368715 was evaluated in more than 200 cell lines of 12 tumor types and in an in vivo model of diffuse large B cell lymphoma (DLBCL) [22]. GSK3368715 treatment showed a promising inhibitory effect in most solid tumor cells, and the tumor inhibition rate of GSK3368715 in renal cell carcinoma, triple-negative breast cancer, and pancreatic cancer was as high as 75%. In addition, researchers also found that the deletion of the methionine phosphorylase (MTAP) gene leads to the accumulation of metabolite 2-methyladenosine in cells, which enhances the sensitivity of certain tumor cells to GSK3368715. Therefore, the MTAP status provides a theoretical basis as a biomarker for patients treated with GSK3368715 [23]. In terms of clinical trials, the first human study of GSK3368715 in solid tumors, including pancreatic cancer, bladder cancer, non-small cell lung cancer, and recurrent/refractory DLBCL, is currently under recruitment (NCT03666988) (Table 1). This study will evaluate the safety, pharmacokinetics, pharmacodynamics, food effects, and preliminary clinical activity of GSK3368715.
GSK3326595 is a selective reversible inhibitor of PRMT5 under clinical development. In breast cancer and lymphoma cell lines, GSK3326595 activates the p53 pathway by inducing alternative splicing of murine double minute 4 (MDM4), regulating the cell cycle to stimulate apoptosis [24]. The p53 protein is classified as a tumor suppressor, and the p53 pathway can be interacted with many other transduction pathways such as Wnt/β catenin, IGF-1/AKT or p38 MAPK [25]. Thus, the integrity of the p53-MDM4 axis in patients may be a potential biomarker for the clinical administration of GSK3326595 [26]. Another small-molecule inhibitor, the cyclin-dependent kinase 4/6 (CDK4/6) inhibitor palbociclib, has been widely studied in the treatment of melanoma. However, there are still certain melanoma cells with acquired drug resistance to palbociclib. In the development of combination trials, it was found that the combination of palbociclib and GSK3326595 could alter the pre-mRNA splicing of MDM4, attenuating the expression of MDM4 to activate p53, further leading to the inhibition of CDK2, eventually resulting in the loss of the acquired drug resistance of palbociclib [27]. Moreover, during the application of GSK3326595 against melanoma, GSK3326595 treatment augmented the level of MHC1 and had a significant inhibitory effect on melanoma in immunoactivity mice. The combination of GSK3326595 and immunotherapy (with an anti-PD-1 antibody) effectively controlled the growth of melanoma and improved the therapeutic effect of a single drug [24]. Current clinical trials on GSK3326595 recruiting patients include a study of the safety and clinical activity of GSK3326595 in the treatment of acute myeloid leukemia (AML) (NCT03614728) and a study of the clinical activity and dose progression of oral GSK3326595 in selected solid tumors and non-Hodgkin's lymphoma (NCT02783300) (Table 1).
EZH2 inhibitors
Tazemetostat is an oral, SAM competitive EZH2 inhibitor, which selectively inhibits the activity of wild-type and mutant EZH2 [28]. The United States Food and Drug Administration (FDA) has approved tazemetostat as the first epi-drug for sarcoma in adults and children over the age of 16 with metastatic or locally advanced epithelioid sarcoma who are not suitable for radical surgery in 2020 [29,30,31]. In early research, tazemetostat has demonstrated anticancer efficacy in a variety of cancers with functional deletion mutations of the SWI/SNF complex or abnormal activation of EZH2 resulting in histone hypermethylation, such as sarcoma, non-Hodgkin's lymphoma, medulloblastoma, and many solid tumors [32]. As a component of the SWI/SNF complex, INI1 is a powerful tumor suppressor gene. In solid tumors with loss of INI1 function, EZH2 can be abnormally recruited, leading to the activation of multiple oncogenes signaling pathways. Therefore, in vivo and in vitro studies have revealed that treatment with tazemetostat can effectively prevent the proliferation and survival of INI1-negative malignant rhabdoid tumor cells [33, 34]. Tazemetostat plays an active role in different types of tumors. In medulloblastoma, tazemetostat reactivates the expression of BAI1 by regulating EZH2 levels, thus preventing the growth of medulloblastoma cells and prolonging the survival time of the orthotopic xenotransplantation model [32]. There are 23 tazemetostat-related clinical trials. Among them, nine are under recruitment, whereas three trials on combination therapy for lymphoma, solid tumors, and malignant stromal tumors have been completed (NCT03010982, NCT02220842, and NCT02860286) (Table 1). Among the different tumor types that depend on the abnormal growth of EZH2, a better study of tazemetostat administration based on biomarkers may be helpful for clinical drug usage.
CPI-1205, an oral indole EZH2 inhibitor, inhibits tumor growth in B cell lymphoma and several types of solid tumors [35, 36]. The PRC2 complex is involved in the process of histone methylation, and its dysfunction is related to the occurrence and development of malignant tumors and tumor prognosis [37,38,39]. The eutectic structure of the CPI-1205 and PRC2 complex plays an important role in anti-tumor effects [40]. In addition, immunotherapy combined with CPI-1205 treatment exerts an improved anticancer function via the modification of EZH2 expression in melanoma. CPI-1205 administration suppresses EZH2 to trigger the phenotype of Tregs in human T cells, whereas the treatment with ipilimumab (an anti-CTLA-4 monoclonal antibody) augments the expression of EZH2 in peripheral blood T cells. Thus, the combination of CPI-1205 and ipilimumab alleviates the side effects of ipilimumab monotherapy, providing a basis for the targeting of the combination therapy [41]. Currently, there are three clinical trials of CPI-1205, including a phase I clinical trial to evaluate the efficacy of CPI-1205 in B cell lymphoma (NCT02395601) (Table 1). These results provide a credible basis for CPI-1205 as a potential cancer treatment.
GSK2816126 inhibits wildtype and mutant EZH2 by competing with SAM as an effective EZH2 inhibitor. GSK2816126 exhibits outstanding anticancer ability in EZH2 mutated malignant tumors and can be administered intravenously in preclinical experiments [42]. In addition, BRAF abnormalities are also present in EZH2 mutated cancers. The combination of the BRAF inhibitors vemurafenib and GSK2816126 revealed a more significant anticancer effect than vemurafenib monotherapy in melanoma models with both a BRAF V600E mutation and an EZH2 abnormality [43]. Therefore, the status of the BRAF V600E mutation with an EZH2 gene copy number variation can be used as a potential tumor therapeutic target. Phase I clinical trials of GSK2816126 in patients with lymphoma and solid tumors have been completed. GSK2816126 was highly active in inhibiting EZH2 mutated tumor growth with a maximum tolerated dose of 2400 mg by intravenous infusion and a dose-limiting toxicity of hepatic transaminitis (NCT02082977) (Table 1) [44].
Predictive biomarkers and combination trials of HDM inhibitors
Histone methylation occurs at lysine and arginine sites and is a reversible process that can be demethylated by HDMs, mainly by LSD1 and demethylases of the JmjC family [45, 46]. As an important mechanism of epigenetic modification, histone demethylation may be involved in the regulation of chromatin remodeling, embryonic development, cellular senescence, tumor proliferation, survival, and other key biological processes [47,48,49]. The first identified HDM LSD1 is a flavin adenine dinucleotide-dependent monoamine oxidase that can be involved in transcriptional regulation. LSD1 has become a promising epigenetic target for a variety of malignant tumors, and its function mainly depends on the demethylase activity at the C terminus. Inhibition of LSD1 activity disturbs the invasion, proliferation, and survival of cancer cells [50, 51]. Therefore, LSD1 inhibitors developed for this mechanism are currently widely used in anti-tumor research and combined immunotherapy (Fig. 1).
LSDI inhibitors
GSK2879552 is an oral, irreversible cyclopropylamine LSD1 inhibitor that is used as an effective antineoplastic drug in a variety of tumors. The antitumor effect of GSK2879552 was examined in 165 cancer cell types, indicating a significant inhibitory effect on 30% of small cell lung cancers and AML. In vitro and in vivo experiments on sensitive small cell lung cancers showed that the MYCL1 copy number, DNA hypomethylation, and TGFβ pathway are associated with GSK2879552 sensitivity to growth inhibition. Helai P. Mohammad identified 45 differentially methylated probes distributed promoter, intronic, and intergenic regions; hence, SMAD2 binding sequences were enriched at differentially methylated regions in sensitivity to GSK2879552. Therefore, the hallmarks of predicted DNA hypomethylation could serve as biomarkers for tumors sensitive to GSK2879552 [52]. In AML, the GSK2879552 therapeutic strategy extinguishes the interaction of ZEB2-KDM1A targeting cells with high ZEB2 levels, inhibits tumor invasion and growth, and then affects tumor survival. The ZEB2 status may act as a biomarker to cope with hematological or solid tumors driven by ZEB2 for GSK2879552 treatment [53]. GSK2879552 also exerts efficacy in combination with other classical antineoplastic drugs and small-molecule inhibitors against cancer. The combination of GSK2879552 with all-trans retinoic acid is also considered in AML, exhibiting a better synergistic effect on cell proliferation and cytotoxicity, ultimately resulting in caspase-mediated cell death, which may achieve ideal anticancer effects in relapsed and refractory AML patients [54]. Resistance often occurs with the tyrosine kinase inhibitor sorafenib in advanced liver cancer patients, but GSK2879552 and sorafenib mechanistically impede the Wnt/β-catenin pathway to attenuate cancer cell stemness, inhibiting acquired resistance and improving the therapeutic effect of sorafenib [55]. Currently, there are three terminated clinical trials associated with GSK2879552 (Table 1). Among them, a phase I trial of GSK2879552 in small cell lung cancer was terminated, because the disease control rate in patients was too high, although the pharmacokinetic properties of the drug were good (NCT02034123).
Tranylcypromine (TCP) was originally used in clinical applications as an antidepressant targeting monoamine oxidase A and B, which are related to the structure of LSD1. Moreover, a TCP analog has been widely recognized as an effective reversible LSD1 inhibitor [56]. In the treatment of AML, a TCP analog effectively inhibited LSD1 interference with GFI1-mediated PU1 target gene inhibition and induced AML differentiation as an LSD1 inhibitor [57]. Since a combination therapy can provide better clinical efficacy than TCP treatment alone, there are a number of achievements in the research of TCP-related combinations for AML therapy. Multiple components of the mTORC1 signaling act as sensitizers for LSD1 inhibition in AML. Among them, the combination of MTORC1 components or mTORC1 inhibitors with TCP, which promotes the differentiation of cell lines and primary cells and enhances MLL-translocated AML differentiation, can be evaluated in early clinical trials [58]. Corin, a synthetic hybrid derived from the HDAC inhibitors entinostat and a TCP analog, showed strong anticancer activity by comprehensively blocking the CoREST complex in melanoma and skin scale-cell carcinoma. Such two-pronged hybrids demonstrate the preferential targeting of specific epigenetic regulatory the CoREST complex and provide unique therapeutic opportunities [59]. There are 26 clinical trials of TCP, and three tumor-related therapeutic trials mainly focus on AML treatment (NCT02261779, NCT02717884, NCT02273102) (Table 1).
Predictive biomarkers and combination trials of HDAC inhibitors
Acetylation occurs on specific lysines of four histones, and the involvement of two key enzymes in the process is required to be catalyzed by a HAT and an HDAC. The HAT acetylates histone lysine residues to activate gene transcription, whereas the HDAC acts as a protease to inhibit gene transcription by deacetylating acetylated histones [60,61,62]. In cancer cells, the overexpression of HDAC enhances histone deacetylation and tightens loose nucleosomes to constrain the expression of tumor suppressor genes, thereby affecting the proliferation, metastasis, and survival of cancer cells. Therefore, HDAC inhibitors promote histone acetylation, relax nucleosomes, and activate the transcription of genes to induce programmed cell death, such as apoptosis [63,64,65]. This class of small-molecule inhibitors has thus become a new class of anti-tumor drugs, but some clinical trials exhibit numerous side effects of pan-HDAC inhibitors, such as hematologic toxicity, diarrhea, weight loss, taste disturbances, electrolyte changes, disordered clotting, fatigue, and cardiac arrhythmias [66]. These findings are not surprising if only considers the essential role of HDACs as key regulators of genes transcription in any tumor type without considering the distinctive HDAC family members and the characteristic of different tumors. In order to achieve good therapeutic effects on a variety of malignant tumors with low toxicity, and high efficiency, we have to cogitate the selectivity of HDAC inhibitors in diverse cancer types. The following paragraphs focus on biomarker and combination guidance for three FDA-approved HDAC inhibitors, vorinostat, abexinostat, and panobinostat (Figs. 3, 4).

Predictive biomarkers and drug combinations of histone deacetylation (HDAC) inhibitors and DNA methyltransferase (DNMT) inhibitors. Prominent biomarkers and drug combinations of HDAC/DNMT inhibitors are detailed here. However, other biomarkers and HDAC/DNMT inhibitor combinations also exist. Me represents methylation. Ac represents acetylation. Similar to histone methylation, methylation of DNA occurs at cytosine residues and needs the participation of DNMT. Besides, histone acetylation has consistently been linked to a chromatin state and regulated by histone acetylases (HATs) and HDACs. The HDAC inhibitors include vorinostat, abexinostat, and panobinostat. The predicted biomarkers for the three HDAC inhibitors are Mcl-1, xCT, and Xist. Vorinostat combined with PFK 118–310 can effectively exert synergistic anticancer effects in breast cancer. Further, panobinostat combined with daratumumab can effectively exert synergistic anticancer effects in multiply myeloma. The DNMT inhibitors include 5'-azacytidine and decitabine. The 5'-azacytidine predicted biomarkers are the UCK1, DDIT3, and PMAIP1 status. The combination of 5'-azacytidine with a SMO inhibitor or AG-221 exerts improved anticancer effects in acute myeloid leukemia. Cellular hypoxia is a predictive biomarker for the selection of decitabine sensitive patients. In renal cell carcinoma, the combination between decitabine and the oxygen nanocarrier H-NPs exerts synergistic anticancer effects

Oral bioavailability and target prediction of HDAC inhibitors and DNMT inhibitors. We used the online tool SwissADME (https://www.sib.swiss/) to predict the oral bioavailability and target prediction of 5 epi-drugs that represent HDAC inhibitors or DNMT inhibitors. The colored zone is the suitable physiochemical space for oral bioavailability. The pink area represents the optimal range for each property. Lipophility: -0.7 < XLOGP3 < + 5.0; size: 150 g/mol < MV < 500 g/mol; polarity: 20Å2 < TPSA < 130Å2; insolubility: 0 < log S (ESOL) < 6; insaturation: 0.25 < fraction Csp3 < 1; flexibility: 0 < num. rotatable bonds < 9. The pie chart of each inhibitor displays the summary of predication target classes
Vorinostat (also called SAHA), an oral HDAC inhibitor, can effectively inhibit the activity of HDAC1, HDAC2, HDAC3, and HDAC6, stimulating stem cell differentiation to affect cell cycle and cell death, and has been approved by the FDA for the treatment of cutaneous T cell lymphoma [67, 68]. Vorinostat exerts antitumor activity through different targeted regulatory mechanisms, such as the induction of BCL-2-induced apoptosis, reduction of glycolysis in a c-Myc-dependent manner to inhibit ATP levels, or synergistic involvement in redox pathways to induce ferroptosis [69]. The treatment of squamous carcinoma cells with vorinostat regulates the expression of Bcl-2 family members and suppresses Mcl-1, a major and tissue-specific survival factor in squamous cell carcinoma, thereby inducing apoptosis. In addition, FBW7-mutant somatic cells are more sensitive to vorinostat treatment. The Mcl-1 status and FBTW7 mutations may act as biomarkers for the application of vorinostat as the basis for the sensitivity of certain tumors to HDAC inhibitor therapy [70, 71]. Moreover, oxidative stress status is also involved in the directive of vorinostat sensitivity; glutamate-cystine transporter xCT levels present a positive correlation with vorinostat in a variety of cancer cells. When vorinostat is combined with the xCT inhibitor SASP, it causes ROS accumulation and induces ferroptosis; thus, xCT levels are also expected to be a potential predictive biomarker for vorinostat treatment [72, 73]. The combination of vorinostat with the natural component artemisinin succinate (ARS) increases the expression of 5-aminolevulinic acid synthase (ALAS1) and enhances the cytotoxicity of ARS by regulating heme synthesis, which has a synergistic antitumor effect and is more effective than monotherapy in solid tumors [74]. In breast cancer, the combined application of vorinostat and the Wnt-β-linked protein blocker PKF118-310 facilitates the induction of differentiation of cancer stem cells to inhibit the EMT process, reducing the number of breast cancer stem cells through a nanoparticle delivery system [75].
Abexinostat, as a moderate oral, isotaximate-containing HDAC inhibitor, can effectively inhibit the activity of HDAC1, HDAC2, HDAC3, HDAC6, HDAC8, and HDAC10. Moreover, abexinostat has been approved by the FDA for the treatment of fourth-line follicular lymphoma as a non-cell cycle-specific cytotoxic epi-drug [76]. Abexinostat appears to have good clinical efficacy in hematological and solid tumors and exhibits better safety than other HDAC inhibitors of the same class. Abexinostat, due to its prominence for DNA double-strand break repair and homologous recombination (HR), plays an anticancer role by decreasing the expression of HR-related gene RAD15, and reducing the ability for HR repair [77]. Interestingly, we demonstrated that abexinostat can participate in the calcium signaling pathway in breast cancer. Abexinostat dysregulated cellular calcium influx via inhibiting Gαq-PLCβ3-mediated calcium signaling by activating the transcription of RGS2, leading to the inhibition of cell proliferation and EMT progression, eventually resulting in cell death in breast cancer [78]. Abexinostat is an effective anticancer drug for treatment-resistant gallbladder cancer, which suppresses cellular growth and proliferation by inhibiting ErbB2 levels and downregulating miR-21 expression [79]. Abexinostat could meritoriously induce the differentiation of breast cancer stem cells with low expression of the long non-coding RNA Xist, increasing the sensitivity to drug response to further induce apoptosis in a variety of breast cancer cell lines. Therefore, Xist may be a predictive biomarker in abexinostat sensitive patients for the exertion of abexinostat anti-tumor characteristics for accurate treatment [80]. Abexinostat has obtained great research value in combination therapy. In addition, the combination of abexinostat with bortezomib displays a good synergistic effect in lymphoma, and the combination of abexinostat with chemotherapy also significantly inhibits tumor growth and metastasis in soft tissue sarcoma; thus, these combination trials deserve further rigorous evaluation in clinical studies [81, 82].
Panobinostat is an oral, nonselective pan-HDAC inhibitor that exerts severe stress on cancer cells to trigger cell death, whereas healthy cells are unaffected. Therefore, panobinostat was approved by the FDA for drug combination with bortezomib or dexamethasone as a third-line treatment in patients with multiple myeloma (MM) [83,84,85]. It should be noted that panobinostat has a black box warning, indicating that the drug has adverse side effects accompanied by severe diarrhea, fatal cardiac events, arrhythmias, and electrocardiogram (ECG) changes. Likewise, panobinostat is usually considered for combination therapy with other agents to treat malignancies. Panobinostat combined with the anti-CD38 monoclonal antibody danatuximab augments CD38 expression and enhances danatuximab monotherapy with significant anti-tumor activity in MM [86]. MEK inhibition has revealed surprising anti-tumor effect in BRAF-mutant melanoma. RAF and MEK inhibition can improve the MAPK signaling and adapt AKT signaling [87, 88]. Surprisingly, the combination of MEK inhibitors with panobinostat is even more effective than other drug combinations by inhibiting the PI3K/AKT pathway and ET-3-mediated YAP signaling, which in turn effectively inhibits tumor growth in subcutaneous and liver metastasis models [89]. Thus, among many HDAC inhibitors, panobinostat has a high-intensity anticancer effect as a combination drug.
Predictive biomarkers and combination trials of DNMT1 inhibitors
DNA methylation is catalyzed by DNMT to obtain a chemical modification with a methyl group by covalent bonding [90]. This modification process does not change the DNA sequence but can induce chromatin structure, DNA stability, and DNA and protein interaction changes to participate in the regulation of gene expression, embryonic development, and control of cellular function and tumorigenesis [91]. DNMT is divided into three families in mammals: DNMT1, DNMT2, and DNMT3. DNMT1, the most widely studied enzyme in the DNMT family, is a key enzyme in DNA replication repair and methylation maintenance [92, 93]. DNMT1 inhibitors are currently widely studied as epigenetic drugs for cancer treatment, and two DNMT inhibitors, azacytidine and decitabine, have been approved by the FDA for certain cancer treatments (Figs. 3, 4).
Decitabine, an irreversible DNMT1 inhibitor, is also a deoxycytidine analog, antimetabolite, which mainly affects the cell cycle and promotes apoptosis. Decitabine behaves as a suicide substrate for DNA methyltransferases and incorporation into DNA without the requirement of a deoxygenation step [94]. Decitabine is approved by the FDA for the treatment of myelodysplastic syndrome. It is worth noting that the anticancer activity of decitabine has a dual mechanism of dose difference, with cytotoxic effects at high concentrations and demethylation effects at low concentrations, indicating a bigger role in the clinical application of malignant tumors. In small cell renal cancer, decitabine promotes T cell activation, enhancing the cancer cell response to immune checkpoint blockade and immune infiltration by stimulating the expression of the transposable element ERV and cytokine secretion [95]. Organic cation/carnitine transporter 2 (OCT2) acts as an important protein involved in renal excretion, and the loss of OCT2 in renal cell carcinoma may lead to decitabine resistance [96, 97]. By combining decitabine and the hemoglobin-based oxygen nanocarrier H-NPs to regulate the hypoxic properties of renal cancer cells, the loss of decitabine activity can be alleviated, enhancing the OCT2 transcriptional process to increase the sensitivity of cancer cells to decitabine treatment. Therefore, modulating the cellular hypoxic environment upon decitabine treatment may serve as a potential clinical application guide for modulating drug resistance [98]. There are more than 400 clinical trials for decitabine, of which 159 have been completed and 110 are recruiting, mainly focusing on hematological oncology and drug combinations, and only a few solid tumor studies of breast, head, and neck cancer (NCT02957968, NCT04252248, and NCT02961101) (Table 1).
5'-Azacytidine (azacytidine), a pyrimidine nucleoside analog, can inhibit DNMT1 activity, weaken DNA methylation, and reverse epigenetic changes. Azacytidine binds to RNA as an antimetabolite of cytidine and has been approved by the FDA for preleukemic myelodysplastic syndrome [99]. Unlike the action mechanism of decitabine, azacytidine is predominantly incorporated into RNA rather than DNA. DNMT displays the nucleophilic attack by the dynamic site Cys residue and methylation by SAM [100]. A recent study on the resistance to azacytidine in AML revealed that azacytidine treatment regulates uridine-cytidine kinase 1 (UCK1) ubiquitination and phosphorylation around the KLHL2/USP28/ATM axis, affecting cellular proliferation and apoptosis. Moreover, the prediction of low levels of UCK1 may be an important candidate biomarker for azacytidine efficacy [101]. In addition, azacytidine activates the integrated stress response (ISR) pathway to induce the activated expression of DDIT3, the classical target in AML, and PMAIP1 and BBC3. Thus, the combination treatment of azacytidine with venetoclax in patients with PMAIP1 and DDIT3 status as potential biomarkers is currently undergoing a phase III clinical trial (NCT02993523) (Table 1) [102]. When azacytidine is used as an anticancer drug, the combination of targeted signaling pathways and epigenetic pathways can effectively improve drug resistance and enhance sensitivity [103]. TET2 and IDH2 mutations resulting from abnormal DNA methylation often occur in AML. Moreover, after the combination of azacytidine with the IDH2 inhibitor AG-221, it can alleviate abnormal changes in DNA methylation, causing a decrease in the number of leukemia cells, which in turn is consistent with anti-leukemia activity to enhance sensitivity [104,105,106]. In elderly patients with AML, an increased rate of drug response to azacytidine treatment is observed. Nonetheless, the sensitizing potential of azacytidine in elderly patients is not associated with common myeloid mutations. Genetic targets in the common deletion region of chromosomes 5 and 7 affect azacytidine sensitivity in elderly patients, such as the silencing of SMO. Consequently, SMO inhibitors combined with azacytidine effectively curb the hedgehog pathway and improve the anti-tumor activity of azacytidine [107].
Natural components as epi-drugs
Natural drugs originate from pharmacologically active ingredients in animals, plants, or minerals, such as international hot natural drugs, the anticancer drug paclitaxel and its derivatives, and the antimalarial drug artemisinin [108]. Due to concerns about the negative effects of chemicals on health and life, the research and development of natural ingredients as drugs have been vigorously carried out in recent years. Further, ordinances on natural drugs have started to relax, and the USA has been unwinding the restrictions on botanicals by modifying the relevant provisions of the FDA. Gradually, natural components have been more intensively studied by researchers. Although the mechanism by which epi-drugs exert epigenetic modifications as enzyme inhibitors has been frequently reported, no drugs have been developed for clinical use. The natural phytoconstituents resveratrol and curcumin act as DNMT inhibitors to exert anticancer effects in certain solid tumors by inhibiting the activity and expression of DNMT and the hypermethylated tumor suppressor gene RASSF1A [109,110,111,112]. There are many related studies on natural inhibitors of histone-modifying enzymes. For example, curcumin, sulforaphane, tanshindiols, rottlerin, and other plant extracts exhibit EZH2 inhibitory activity, which in turn inhibits histone methylation and attenuates tumor proliferation and invasion to induce cellular apoptosis [113,114,115,116]. The anticancer potency of olive oil in MM was investigated; olive oil inhibited the expression of HDAC1, HDAC2, HDAC3, HDAC4, and HDAC6; arrested the cell cycle; and induced caspase 8-dependent apoptosis by regulating Sp1 without affecting the DNMT activity [117]. Moreover, the extracts of natural products also can act as the epigenetic regulator, such as ibotenic acid extracted from fungal species and baicalein exhibit HDAC inhibitory activity to impede the progression of cancer [118, 119] (Table 2). Lycopene, a potent antioxidant, extracted from tomatoes, carrot, watermelon has been identified as a potential DNA methylating agent to reduce genomic instability in breast cancers [120, 121]. As a natural, easy obtainment, high value and potential in use plant pigment, lycopene appears epigenetic activity in various types of cancer. Thus, the in-depth epigenetic mechanism of lycopene is worthy of further excavation and exploration.
Although the development of natural epi-drugs has opened new avenues to reduce the side effects of cancer therapy, the target diversity of natural drug components poses great difficulties for clinical medication guidance. For example, curcumin has the efficacy of EZH2 and DNMT inhibitors. Thus, the pathway of curcumin’s anti-tumor activity is complex, and predicting its biomarker localization in suitable patients is difficult. Moreover, there are too many kinds of natural components, and the systematic screening of natural drugs for their inhibitory effects on epigenetic enzymes still faces great challenges. Even if many natural ingredients with suspected epi-drug properties are screened out, the comprehensive mechanistic interpretation of natural ingredients requires tremendous energy. Still, as long as the road to cancer treatment continues, the pace of research on natural epi-drugs with low side effects will not stop. Future clinical applications for natural drugs are awaited.
Perspectives
Epi-drugs achieve anticancer effects mainly by targeting and inhibiting epigenetic modifying enzymes and adjusting abnormal epigenetic changes in tumors. Thus, epi-drugs participate in the interruption of the cell cycle and the activation of tumor suppressor pathways, which in turn induce programmed cell death in tumorigenesis. However, as targeted modulators, epi-drugs do not only target directly inhibiting enzymes; rather, it should also be determined whether the sensitivity to epi-drugs in different patients is based on biomarkers. Epi-drugs cannot exert their potential antitumor activity at the applicable dose in insensitive to the patients. In addition, the greatest obstacle faced during the development of epi-drugs or other types of drugs is the occurrence of adverse side effects. In the development of epi-drugs, it is necessary not only to find potential biomarkers, but also to ensure that epi-drugs exert their maximum efficacy at a minimum dose and to avoid the generation of toxic side effects. Among the above 12 epi-drugs, six epi-drugs have been approved by the FDA for clinical treatment, including mainly EZH2 inhibitors, HDAC inhibitors, and DNMT inhibitors. We display the overview of clinical trials of 12 epi-drugs in certain cancer types to reference the application on epi-drugs in clinical use, including aspects as target cancer type selection, or in-depth study of the anti-tumor mechanism (Fig. 5). Nevertheless, no PRMT inhibitors and LSD1 inhibitors have currently achieved an FDA approval. Further, there are still some contradictions regarding LSD1 inhibitors. Thus, there is controversy regarding the function of LSD1 as an oncogene or a tumor suppressor. The function of LSD1 inhibitors as anticancer epi-drugs has been recognized, but it needs to be further studied whether their targets exert anticancer effects by inhibiting LSD1.

Targeting of certain cancers with 12 epi-drugs, including examples of clinical trial. The graph depicts the clinical trials of these PRMT, EZH2, LSD1, HDAC and DNMT inhibitors based on searches of the ClinicalTrials.gov database (https://clinicaltrials.gov/). Epi-drugs play an active role in hematological and solid tumors. However, epi-drugs that are mainly EZH2 inhibitors and PRMT inhibitors are more widely used in hematological tumors than in solid tumors. Moreover, the efficacy of EZH2 inhibitors and PRMT inhibitors in solid tumors is still limited. The effects of LSD1 inhibitors, HDAC inhibitors, and DNMT inhibitors on hematological tumors have also been analyzed in several clinical trials. However, clinical studies on their role in solid tumors such as lung cancer, breast cancer, and renal cancer have currently been successively carried out
In addition to the epi-drugs mentioned above that directly target on the epigenetic proteins, there is also a class of drugs that deserve our attention. These kinds of drugs are not typically regarded as "epigenetic" drugs. Although these drugs do not directly act on the epigenetic regulators, they still established epigenetic function. For example, a standard chemotherapy in glioblastoma treatment, temozolomide, is discovered to have strong influence on enhance of DNA methylation resulting in global gene silencing [122, 123]. For the long-term point of view, the CRISPR/Cas approach gives a different perspective to target silence the locus-specific epigenetic genes, which offer puissant possibilities correct epigenetic mutations in tumorigenesis and drug resistance [124]. The epigenetic editing technique can be used to fuse the catalytic domain of epigenetic enzyme and modulate the dysfunction of epigenetic signature at precise positioning of tissues [125]. Based on the CRISPR/Cas approach, epigenetic editing might present new insight into the use of epi-drugs and diminish side effects to a greater extent.
Although cancer monotherapy can minimize the toxic and side effects of epi-drugs, combination trials targeting multiple targets can have a therapeutic effect in malignant tumors due to the tumor heterogeneity. For example, panobinostat is approved by the FDA as a third-line drug for the treatment of MM, and its combination can produce a synergistic effect in the treatment of cancer. Thousands of clinical trials of epi-drugs are currently being carried out, many of which focus on the combination of epi-drugs with immunotherapy or traditional chemotherapeutic drugs. Combination therapy has undergone incremental development, and combination therapy may provide a basic reference for clinical medical administration. Although clinical trials in the direction of combination therapy are currently progressing well, there are only a few clinical trials for available predictive biomarkers of epi-drugs. For less studied biomarkers, clinical trials are relatively conservative. Researchers should not only study biomarkers in depth but also apply the studied biomarkers in the clinical practice and truly achieve precise drug treatment in the future.
Availability of data and materials
All data are included in the paper.
Abbreviations
- Epi-drugs:
-
Epigenetic drugs
- DNMTs:
-
DNA methyltransferases
- HMTs:
-
Histone methyltransferases
- HDMs:
-
Histone demethylases
- HDACs:
-
Histone deacetylases
- HDACs:
-
Histone deacetylases
- PRMT:
-
Protein arginine methyltransferases
- EZH2:
-
Enhancer of zeste homolog 2
- MTAP:
-
Methionine phosphorylase
- DLBCL:
-
Diffuse large B cell lymphoma
- FTIH:
-
First time in humans
- NHL:
-
Non-Hodgkin’s lymphoma
- MDM4:
-
Murine double minute 4
- MDS:
-
Myelodysplastic syndrome
- AML:
-
Acute myeloid leukemia
- SCLC:
-
Small cell lung carcinoma
- NSCLC:
-
Non-small cell lung carcinoma
- SAM:
-
S-Adenosyl methionine
- FDA:
-
United States Food and Drug Administration
- ARS:
-
Artemisinin succinate
- ALAS1:
-
5-Aminolevulinic acid synthase
- HR:
-
Homologous recombination
- MM:
-
Multiple myeloma
- ECG:
-
Electrocardiogram
- UCK1:
-
Uridine-cytidine kinase 1
- ISR:
-
Integrated stress response
- OCT2:
-
Organic cation/carnitine transporter 2
- STS:
-
Soft tissue sarcoma
References
Cui H, Hu Y, Guo D, Zhang A, Gu Y, Zhang S, et al. DNA methyltransferase 3A isoform b contributes to repressing E-cadherin through cooperation of DNA methylation and H3K27/H3K9 methylation in EMT-related metastasis of gastric cancer. Oncogene. 2018;37:4358–71.
Wang T, Qin ZY, Wen LZ, Guo Y, Liu Q, Lei ZJ, et al. Epigenetic restriction of Hippo signaling by MORC2 underlies stemness of hepatocellular carcinoma cells. Cell Death Differ. 2018;25:2086–100.
Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, et al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–5.
Chen W, Cooper TK, Zahnow CA, Overholtzer M, Zhao Z, Ladanyi M, et al. Epigenetic and genetic loss of Hic1 function accentuates the role of p53 in tumorigenesis. Cancer Cell. 2004;6:387–98.
Neumeister P, Albanese C, Balent B, Greally J, Pestell RG. Senescence and epigenetic dysregulation in cancer. Int J Biochem Cell Biol. 2002;34:1475–90.
Morel D, Jeffery D, Aspeslagh S, Almouzni G, Postel-Vinay S. Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat Rev Clin Oncol. 2020;17:91–107.
Berdasco M, Esteller MJ. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010;19:698–711.
Morel D, Almouzni G, Soria J. Postel-Vinay SJAooojotESfMO. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. 2017;28:254–69.
Duan R, Du W, Guo WJJoh, oncology. EZH2: a novel target for cancer treatment. 2020; 13: 104.
Salvador MA, Wicinski J, Cabaud O, Toiron Y, Finetti P, Josselin E, et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low xist expression. Clin Cancer Res Off J Am Assoc Cancer Res. 2013;19:6520–31.
Marjanovic N, Hofree M, Chan J, Canner D, Wu K, Trakala M, et al. Emergence of a high-plasticity cell state during lung cancer evolution. 2020.
Martinez-Cardus A, Moran S, Musulen E, Moutinho C, Manzano JL, Martinez-Balibrea E, et al. Epigenetic homogeneity within colorectal tumors predicts shorter relapse-free and overall survival times for patients with locoregional cancer. Gastroenterology. 2016;151:961–72.
Chen Y, Yin J, Li W, Li H, Chen D, Zhang C, et al. Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. 2020.
Mack S. Bertrand K. Sub-group, Sub-type, and Cell-type Heterogeneity of Ependymoma. 2020;38:15–7.
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717.
Histone Methylation Connects DNA Repair to Metabolism in Cancer Cells. J Cancer discovery. 2020.
Zhang W, Qu J, Liu G. Belmonte JJNrMcb. The ageing epigenome and its rejuvenation. 2020;21:137–50.
Jung G, Hernández-Illán E, Moreira L, Balaguer F. Goel AJNrG, hepatology. Epigenetics of colorectal cancer: biomarker and therapeutic potential. 2020;17:111–30.
León T, Rapoz-D’Silva T, Bertoli C, Rahman S, Magnussen M, Philip B, et al. EZH2-deficient T-cell acute lymphoblastic leukemia is sensitized to CHK1 inhibition through enhanced replication stress. Cancer Discov. 2020;10:998–1017.
Karakashev S, Fukumoto T, Zhao B, Lin J, Wu S, Fatkhutdinov N, et al. EZH2 inhibition sensitizes CARM1-high, homologous recombination proficient ovarian cancers to PARP inhibition. 2020; 37: 157–67.e6.
Li N, Yu J, Luo A, Tang Y, Liu W, Wang S, et al. LncRNA and mRNA signatures associated with neoadjuvant chemoradiotherapy downstaging effects in rectal cancer. J Cell Biochem. 2019;120:5207–17.
Srour N, Mersaoui SR. M-TAP Dance: Targeting PRMT1 and PRMT5 family members to push cancer cells over the edge. Cancer Cell. 2019;36:3–5.
Fedoriw A, Rajapurkar S, O’Brien S, Gerhart S, Mitchell L, Adams N, et al. Anti-tumor activity of the Type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP Loss. Cancer Cell. 2019;36(100–14):e25.
Kim H, Kim H, Feng Y, Li Y, Tamiya H, Tocci S, et al. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci Transl Med. 2020;12:5683.
Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–908.
Gerhart SV, Kellner WA, Christine T, Pappalardi MB, Zhang XP, Rocio MDO, et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci Rep. 2018;8:9711.
Abuhammad S, Cullinane C, Martin C, Bacolas Z, ences KESJPotNAo. Regulation of PRMT5–MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. 2019; 116.
Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. 2013; 110: 7922–7.
Drugs SMHJ. Tazemetostat: First Approval. 2020; 80.
Marsh S. Jimeno AJDot. Tazemetostat for the treatment of multiple types of hematological malignancies and solid tumors. 2020;56:377–87.
Rothbart S, Baylin SJC. Epigenetic Therapy for Epithelioid. Sarcoma. 2020;181:211.
Zhang H, Zhu D, Zhang Z, Kaluz S, Oncogene EGVMJ. EZH2 targeting reduces medulloblastoma growth through epigenetic reactivation of the BAI1/p53 tumor suppressor pathway. 2019; 39.
Sullivan LM, Folpe AL, Pawel BR, Judkins AR, Biegel JA. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod Pathol. 2013;26:385–92.
Hornick JL, Dal Cin P, Fletcher CDMJAJoSP. Loss of INI1 Expression is Characteristic of Both Conventional and Proximal-type Epithelioid Sarcoma. 2009; 33: 542–50.
Fioravanti R, Stazi G, Zwergel C, Valente S, Mai AJCr. Six Years (2012–2018) of Researches on Catalytic EZH2 Inhibitors: The Boom of the 2-Pyridone Compounds. 2018; 18: 1818–32.
Gulati N, Béguelin W, Giulino-Roth LJL, lymphoma. Enhancer of zeste homolog 2 (EZH2) inhibitors. 2018; 59: 1574–85.
Long Y, Hwang T, Gooding A, Goodrich K, Rinn J, Cech TJNg. RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. 2020.
Burr M, Sparbier C, Chan K, Chan Y, Kersbergen A, Lam E, et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. 2019; 36: 385–401.e8.
van Mierlo G, Dirks R, De Clerck L, Brinkman A, Huth M, Kloet S, et al. Integrative Proteomic Profiling Reveals PRC2-Dependent Epigenetic Crosstalk Maintains Ground-State Pluripotency. 2019; 24: 123–37.e8.
Vaswani RG, Gehling VS, Dakin LA, Cook A, Nasveschuk CG, Duplessis M, et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable. 2016: 9928–41.
Goswami S, Apostolou I, Zhang J, Skepner J, Sharma PJJoCI. Modulation of EZH2 expression in T cells improves efficacy of anti–CTLA-4 therapy. 2018; 128.
McCabe M, Ott H, Ganji G, Korenchuk S, Thompson C, Van Aller G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. 2012; 492: 108–12.
Yu H, Ma M, Yan J, Xu L, Yu J, Dai J, et al. Identification of coexistence of BRAF V600E mutation and EZH2 gain specifically in melanoma as a promising target for combination therapy. 2017; 15: 243.
Yap TA, Winter JN, Giulino-Roth L, Longley J, Research PWJ. Phase 1 study of the novel enhancer of zeste homolog 2 (EZH2) inhibitor GSK2816126 in patients with advanced hematological and solid tumors. J Cell Cycle. 2019: clincanres.4121.2018.
Anastas J, Zee B, Kalin J, Kim M, Guo R, Alexandrescu S, et al. Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. 2019; 36: 528–44.e10.
Van Rechem C, Black J, Boukhali M, Aryee M, Gräslund S, Haas W, et al. Lysine demethylase KDM4A associates with translation machinery and regulates protein synthesis. 2015; 5: 255–63.
Hori Y. Kikuchi KJAocr. Chemical Tools with Fluorescence Switches for Verifying Epigenetic Modifications. 2019;52:2849–57.
Jambhekar A, Dhall A. Shi YJNrMcb. Roles and regulation of histone methylation in animal development. 2019;20:625–41.
Chakraborty A, Laukka T, Myllykoski M, Ringel A, Booker M, Tolstorukov M, et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. 2019; 363: 1217–22.
Stewart C, Byers L. Altering the Course of Small Cell Lung Cancer: Targeting Cancer Stem Cells via LSD1 Inhibition. J Cell Cycle. 2015; 28: 4–6.
Shi L, Cui S, Engel J, Tanabe OJNm. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. 2013; 19: 291–4.
Mohammad H, Smitheman K, Kamat C, Soong D, Federowicz K, Van Aller G, et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. 2015; 28: 57–69.
Goossens S, Peirs S, Van Loocke W, Wang J, Takawy M, Matthijssens F, et al. Oncogenic ZEB2 activation drives sensitivity toward KDM1A inhibition in T-cell acute lymphoblastic leukemia. 2017; 129: 981–90.
Smitheman KN, Severson TM, Rajapurkar SR, McCabe MT, Karpinich N, Foley J, et al. Lysine specific demethylase 1 inactivation enhances differentiation and promotes cytotoxic response when combined with all-trans retinoic acid in acute myeloid leukemia across subtypes. 2018.
Huang M, Chen C, Geng J, Han D, Wang T, Xie T, et al. Targeting KDM1A attenuates Wnt/β-catenin signaling pathway to eliminate sorafenib-resistant stem-like cells in hepatocellular carcinoma. 2017; 398: 12.
Ji YY, Lin SD, Wang YJ, Su MB, Yu LFJEJoMC. Tying up tranylcypromine: Novel selective histone lysine specific demethylase 1 (LSD1) inhibitors. 2017; 141: 101.
Barth J, Abou-El-Ardat K, Dalic D, Kurrle N, Maier A, Mohr S, et al. LSD1 inhibition by tranylcypromine derivatives interferes with GFI1-mediated repression of PU.1 target genes and induces differentiation in AML. 2019; 33: 1411–26.
Deb G, Wingelhofer B, Amaral FMR, Maiques-Diaz A, Leukemia TCPSJ. Pre-clinical activity of combined LSD1 and mTORC1 inhibition in MLL-translocated acute myeloid leukaemia. 2019.
Kalin JH, Wu M, Gomez AV, Song Y, Cole PAJNC. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. 2018;9:53.
Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. 2019;574:575–80.
Lee J, Carrer A, Shah S, Snyder N, Wei S, Venneti S, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20:306–19.
Bose D, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, Berger SJC. RNA Binding to CBP Stimulates Histone Acetylation and Transcription. 2017; 168: 135–49.e22.
Romero D. HDAC inhibitors tested in phase III trial. Nat Rev Clin Oncol 2019; 16: 465.
Ott C. Wu C. HDAC Inhibitors Finally Open Up: Chromatin Accessibility Signatures of CTCL J Cell Cycle. 2017;32:1–3.
Lane A. Chabner BJJocoojotASoCO. Histone deacetylase inhibitors in cancer therapy. 2009;27:5459–68.
Bruserud O, Stapnes C, Ersvaer E, Gjertsen BT, Ryningen A. Histone deacetylase inhibitors in cancer treatment: a review of the clinical toxicity and the modulation of gene expression in cancer cell. Curr Pharm Biotechnol. 2007;8:388–400.
Kim Y, Bagot M, Pinter-Brown L, Rook A, Porcu P, Horwitz S, et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. 2018; 19: 1192–204.
Krug L, Kindler H, Calvert H, Manegold C, Tsao A, Fennell D, et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): a phase 3, double-blind, randomised, placebo-controlled trial. 2015; 16: 447–56.
Nguyen T, Zhang Y, Shang E, Shu C, Torrini C, Zhao J, et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. 2020;130:3699–716.
He L, Torres-Lockhart K, Forster N, Ramakrishnan S, Greninger P, Garnett MJ, et al. Mcl-1 and FBW7 Control a Dominant Survival Pathway Underlying HDAC and Bcl-2 Inhibitor Synergy in Squamous Cell Carcinoma. 2012.
Song X, Shen L, Tong J, Kuang C, Zeng S, Schoen R, et al. Mcl-1 inhibition overcomes intrinsic and acquired regorafenib resistance in colorectal cancer. 2020; 10: 8098–110.
Miyamoto K, Watanabe M, Boku S, Sukeno M, Morita M, Kondo H, et al. xCT Inhibition Increases Sensitivity to Vorinostat in a ROS-Dependent Manner. 2020; 12.
Conti L, Bolli E, Di Lorenzo A, Franceschi V, Macchi F, Riccardo F, et al. Immunotargeting of the xCT Cystine/Glutamate Antiporter Potentiates the Efficacy of HER2-Targeted Immunotherapies in Breast Cancer. 2020.
Chen C-P, Chen K, Feng Z, Wen X, B HSJAPS. Synergistic antitumor activity of artesunate and HDAC inhibitors through elevating heme synthesis via synergistic upregulation of ALAS1 expression. 2019; 9: 937–51.
Shamsian A, Sepand M, Javaheri Kachousangi M, Dara T, Ostad S, Atyabi F, et al. Targeting Tumorigenicity of Breast Cancer Stem Cells Using SAHA/Wnt-b Catenin Antagonist Loaded Onto Protein Corona of Gold Nanoparticles. 2020;15:4063–78.
Buggy J, Cao Z, Bass K, Verner E, Balasubramanian S, Liu L, et al. CRA-024781: a novel synthetic inhibitor of histone deacetylase enzymes with antitumor activity in vitro and in vivo. 2006; 5: 1309–17.
Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJJPotNAoS. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. 2007.
Yang T, Wang P, Yin X, Zhang J, Huo M, Gao J, et al. The histone deacetylase inhibitor PCI-24781 impairs calcium influx and inhibits proliferation and metastasis in breast cancer. Theranostics. 2021;11:2058–76.
Kitamura T, Connolly K, Ruffino L, Ajiki T, Lueckgen A, Digiovanni J, et al. The therapeutic effect of histone deacetylase inhibitor PCI-24781 on gallbladder carcinoma in BK5.erbB2 mice. 2012; 57: 84–91.
Salvador MA, Wicinski J, Cabaud O, Toiron Y, Finetti P, Josselin E, et al. The Histone Deacetylase Inhibitor Abexinostat Induces Cancer Stem Cells Differentiation in Breast Cancer with Low Xist Expression. 2013;19:6520–31.
Bhalla S, Balasubramanian S, David K, Sirisawad M, Buggy J, Mauro L, et al. PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NF-kappaB mechanisms and is synergistic with bortezomib in lymphoma cells. 2009; 15: 3354–65.
Lopez G, Liu J, Ren W, Wei W, Wang S, Lahat G, et al. Combining PCI-24781, a Novel Histone Deacetylase Inhibitor, with Chemotherapy for the Treatment of Soft Tissue Sarcoma. 2009; 15: 3472–83.
San-Miguel J, Hungria V, Yoon S, Beksac M, Dimopoulos M, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. 2014; 15: 1195–206.
San-Miguel J, Richardson P, Günther A, Sezer O, Siegel D, Bladé J, et al. Phase Ib study of panobinostat and bortezomib in relapsed or relapsed and refractory multiple myeloma. 2013;31:3696–703.
Panobinostat approved for multiple myeloma. %J Cancer discovery. 2015; 5: OF4.
García-Guerrero E, Gogishvili T, Danhof S, Schreder M, Hudecek MJB. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. 2017; 129: 3386.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703.
Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14.
Faião-Flores F, Emmons MF, Durante MA, Kinose F, Research KSMS. HDAC Inhibition Enhances the In Vivo Efficacy of MEK Inhibitor Therapy in Uveal Melanoma. J Cell Cycle. 2019; 25: clincanres.3382.2018.
Wang Q, Yu G, Ming X, Xia W, Xu X, Zhang Y, et al. Imprecise DNMT1 activity coupled with neighbor-guided correction enables robust yet flexible epigenetic inheritance. 2020.
Ming X, Zhang Z, Zou Z, Lv C, Dong Q, He Q, et al. Kinetics and mechanisms of mitotic inheritance of DNA methylation and their roles in aging-associated methylome deterioration. 2020.
Chen Z. Zhang YJArob. Role of Mammalian DNA Methyltransferases in Development. 2020;89:135–58.
Rošić S, Amouroux R, Requena C, Gomes A, Emperle M, Beltran T, et al. Evolutionary analysis indicates that DNA alkylation damage is a byproduct of cytosine DNA methyltransferase activity. 2018;50:452–9.
Dhillon S. Decitabine/cedazuridine: first approval. Drugs. 2020;80:1373–8.
Cubas AAd, Dunker W, Zaninovich A, Hongo RA, Rathmell WK. DNA hypomethylation promotes transposable element expression and activation of immune signaling in renal cell cancer. 2020.
Koepsell H. Organic cation transporters in health and disease. Pharmacol Rev. 2020;72:253–319.
Aguilar A. Kidney cancer: OCT2 demethylation cracks open oxaliplatin resistance. Nat Rev Nephrol. 2016;12:581.
Chen L, Wang Z, Xu Q, Liu Y, Chen L, Guo S, et al. The failure of DAC to induce OCT2 expression and its remission by hemoglobin-based nanocarriers under hypoxia in renal cell carcinoma. Theranostics. 2020;10:3562–78.
Ramos F. Pre-emptive azacitidine for relapse prevention in acute myeloid leukaemia. Lancet Oncol. 2018;19:1557–8.
Christman JK. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–95.
Zhang H, Huang H, Feng X, Song H, Theranostics XQJ. Deubiquitinase USP28 inhibits ubiquitin ligase KLHL2-mediated uridine-cytidine kinase 1 degradation and confers sensitivity to 5'-azacytidine-resistant human leukemia cells. 2020; 10: 1046–59.
Jin S, Cojocari D, Purkal JJ, Popovic RDCP. 5-Azacitidine induces NOXA to prime AML cells for venetoclax-mediated apoptosis. Clin Cancer Res. 2020;26:3371.
Das M. Venetoclax with decitabine or azacitidine for AML. Lancet Oncol. 2018;19:672.
Shih A, Meydan C, Shank K, Garrett-Bakelman F, Ward P, Intlekofer A, et al. IDH2Combination targeted therapy to disrupt aberrant oncogenic signaling and reverse epigenetic dysfunction in - and -mutant acute myeloid leukemia. Cancer Discov. 2017;7:494–505.
Stein E, Fathi A, DiNardo C, Pollyea D, Roboz G, Collins R, et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: a phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Hematol. 2020;7:e309–19.
Ito K, Lee J, Chrysanthou S, Zhao Y, Josephs K, Sato H, et al. Non-catalytic roles of Tet2 are essential to regulate hematopoietic stem and progenitor cell homeostasis. Cell Rep. 2019;28(2480–90):e4.
Tibes R, Al-Kali A, Oliver G, Delman D, Hansen N, Bhagavatula K, et al. The Hedgehog pathway as targetable vulnerability with 5-azacytidine in myelodysplastic syndrome and acute myeloid leukemia. J Hematol Oncol. 2015;8:114.
Ghogare A. Greer AJCr. Using Singlet Oxygen to Synthesize Natural Products and Drugs. 2016;116:9994–10034.
Amp WZ, Sauter ERJN. Cancer. Trans-Resveratrol Alters Mammary Promoter Hypermethylation in Women at Increased Risk for Breast Cancer. 2012;64:393–400.
Li Y, Jiang P, Apei F, et al. Curcumin reactivates silenced tumor suppressor gene RAR beta by reducing DNA methylation. Phytother Res. 2015;29:1237.
Campbell FC, Collett GP. Chemopreventive properties of curcumin. J Future Oncol. 2005;1:405–14.
Shu L, Khor T, Lee J, Boyanapalli S, Huang Y, Wu T, et al. Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. AAPS J. 2011;13:606–14.
Wu G-Q, Chai K-Q, Zhu X-M, Jiang H, Wang X, Xue Q, et al. Anti-cancer effects of curcumin on lung cancer through the inhibition of EZH2 and NOTCH1. Oncotarget. 2016;7:26535.
Fisher ML, Adhikary G, Dan G, Kaetzel DM, Eckert RL. The Ezh2 polycomb group protein drives an aggressive phenotype in melanoma cancer stem cells and is a target of diet derived sulforaphane. J Mol Carcinog 2016.
Woo J, Kim HY, Byun BJ, Chae CH, Lee JY, Ryu SY, et al. Biological evaluation of tanshindiols as EZH2 histone methyltransferase inhibitors. Bioorg Med Chem Lett J. 2014;24:2486–92.
Zheng N, Wang L, Hou Y, Zhou X, He Y, Wang Z. Rottlerin inhibits cell growth and invasion via down-regulation of EZH2 in prostate cancer. J Cell Cycle. 2018;17:2460.
Juli G, Oliverio M, Bellizzi D, Cantafio MEG. Anti-tumor activity and epigenetic impact of the polyphenol oleacein in multiple myeloma. Cancers. 2019;11:990.
Maruca A, Rocca R, Catalano R, Mesiti F, Costa G, Lanzillotta D, et al. Natural products extracted from fungal species as new potential anti-cancer drugs: a structure-based drug repurposing approach targeting HDAC7. Molecules. 2020;25:5524.
Yu X, Li H, Hu P, Qing Y, Wang X, Zhu M, et al. Natural HDAC-1/8 inhibitor baicalein exerts therapeutic effect in CBF-AML. Clin Transl Med. 2020;10:e54.
King-Batoon A, Leszczynska JM, Klein CB. Modulation of gene methylation by genistein or lycopene in breast cancer cells. Environ Mol Mutagen. 2008;49:36–45.
Chalabi N, Satih S, Delort L, Bignon YJ, Bernard-Gallon DJ. Expression profiling by whole-genome microarray hybridization reveals differential gene expression in breast cancer cell lines after lycopene exposure. Biochim Biophys Acta. 2007;1769:124–30.
Belter A, Barciszewski J, Barciszewska AM. Revealing the epigenetic effect of temozolomide on glioblastoma cell lines in therapeutic conditions. PLoS ONE. 2020;15:e0229534.
Barciszewska AM, Gurda D, Glodowicz P, Nowak S, Naskret-Barciszewska MZ. A new epigenetic mechanism of temozolomide action in glioma cells. PLoS ONE. 2015;10:e0136669.
Geel TM, Ruiters MHJ, Cool RH, Halby L, Voshart DC, Andrade Ruiz L, et al. The past and presence of gene targeting: from chemicals and DNA via proteins to RNA. Philos Trans R Soc Lond B Biol Sci. 2018;373:20170077.
Ganesan A, Arimondo PB, Rots MG, Jeronimo C, Berdasco M. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenetics. 2019;11:174.
Funding
This work was supported by the Major State Basic Research Development Program of China (grant number 2016YFA0102400 to Y.W.) and National Natural Science Foundation of China (No. 41931291 to Y.W.).
Author information
Authors and Affiliations
Contributions
Yan Wang contributed to study design. Tianshu Yang contributed to manuscript editing. Yan Wang and Yunkai Yang contributed to manuscript review. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors give consent for the publication of the manuscript in Cancer Letters.
Competing interests
The authors declare that they have no conflicts of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, T., Yang, Y. & Wang, Y. Predictive biomarkers and potential drug combinations of epi-drugs in cancer therapy. Clin Epigenet 13, 113 (2021). https://doi.org/10.1186/s13148-021-01098-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-021-01098-2