
Abstract
Background
DNA methylation is a key epigenetic modification that can directly affect gene regulation. DNA methylation is highly influenced by environmental factors such as cigarette smoking, which is causally related to chronic obstructive pulmonary disease (COPD) and lung cancer. To date, there have been few large-scale, combined analyses of DNA methylation and gene expression and their interrelations with lung diseases.
Results
We performed an epigenome-wide association study of whole blood gene expression in ~ 6000 individuals from four cohorts. We discovered and replicated numerous CpGs associated with the expression of cis genes within 500 kb of each CpG, with 148 to 1,741 cis CpG-transcript pairs identified across cohorts. We found that the closer a CpG resided to a transcription start site, the larger its effect size, and that 36% of cis CpG-transcript pairs share the same causal genetic variant. Mendelian randomization analyses revealed that hypomethylation and lower expression of CHRNA5, which encodes a smoking-related nicotinic receptor, are causally linked to increased risk of COPD and lung cancer. This putatively causal relationship was further validated in lung tissue data.
Conclusions
Our results provide a large and comprehensive association study of whole blood DNA methylation with gene expression. Expression platform differences rather than population differences are critical to the replication of cis CpG-transcript pairs. The low reproducibility of trans CpG-transcript pairs suggests that DNA methylation regulates nearby rather than remote gene expression. The putatively causal roles of methylation and expression of CHRNA5 in relation to COPD and lung cancer provide evidence for a mechanistic link between patterns of smoking-related epigenetic variation and lung diseases, and highlight potential therapeutic targets for lung diseases and smoking cessation.
Similar content being viewed by others


Background
The effects of environmental exposures on downstream phenotypes are mediated in part by DNA methylation [1]. DNA methylation was long thought to inhibit gene expression [2]. Recent studies, however, have revealed a more complex picture. DNA methylation levels have been shown to be inversely correlated with gene expression across the genome and throughout multiple cell types, but site-specific analyses have revealed positive correlations of DNA methylation with the expression of some genes [3]. CpG sites, locations in the genome where a cytosine is followed by a guanine nucleotide, are often methylated, and CpG sites whose methylation is associated with altered gene expression are referred to as expression quantitative trait methylation sites (eQTMs). CpG sites that are positively correlated with gene expression may act by different mechanisms compared with CpGs that are inversely correlated with expression. As a general feature across different cell types, CpG sites that are inversely correlated with gene expression are significantly more likely to be found in transcriptional repressor CCCTC-binding factor (CTCF) binding sites, enhancers, and promoters, particularly non-CpG island (CGI) promoters, whereas positively correlated CpG sites are more likely to be found in gene bodies [4].
DNA methylation has been studied genome-wide in relation to a wide range of phenotypes, with numerous associations having been reported, including with cancer, autoimmune disease, diabetes, cardiovascular disease, and neurological diseases [5,6,7,8,9,10]. Association, however, does not prove causation. Most disease-associated methylation changes have been found to be a consequence of the traits studied and are not causal of disease [11, 12]. Moreover, DNA methylation is determined by a complex interplay of genetic and environmental factors. In particular, current and prior cigarette smoking have a profound influence on the methylation levels of thousands of CpGs [13]. Mendelian randomization (MR) has been proposed as a means to infer causal relations between DNA methylation and disease outcomes [14]. This approach uses a genetic proxy as an instrument to represent DNA methylation and to evaluate the likelihood of a causal association between DNA methylation and disease.
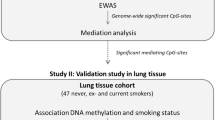
To address a knowledge gap regarding how cigarette smoking affects DNA methylation and gene expression and leads to smoking-related disease outcomes, we performed an epigenome-wide association study (EWAS) of whole blood gene expression in ~ 6000 individuals from four cohort studies and identified thousands of CpG-transcript pairs. Our study had the following aims: (i) identify CpGs associated with gene expression (eQTMs), (ii) explore the functional annotations of eQTMs, (iii) conduct colocalization analyses to investigate how genetic influences on DNA methylation contribute to altered gene expression, and (iv) use MR to infer causal effects of CpGs and expressed genes on smoking-related lung diseases (Fig. 1).

Flowchart of the study design and major findings
Results
eQTMs from discovery and replication data sets
To systematically assess the association between DNA methylation and variation in gene expression, we analyzed DNA methylation and genome-wide transcription in whole blood from 4,170 participants in the Framingham Heart Study (FHS). Clinical characteristics of the study sample are summarized in Additional file 2: Table S1. After adjusting for biological and technical covariates (see Methods for details), we identified 16,416 cis CpG-transcript pairs (CpG site and the associated transcript [eGene] located within 500 kb) and 198,960 trans CpG-transcript pairs (CpG site residing > 500 kb from the eGene) with statistically significant associations. We found that 3% (12,177) of all 401,189 CpG sites tested (see Methods for exclusions of CpGs) were cis eQTMs and were associated with expression levels of 15% (2704) of all 17,873 transcripts studied (P ≤ 1E−8, Bonferroni correction, Additional file 2: Table S2). Another 6% (24,992) of all 401,189 CpG sites tested were trans eQTMs and were associated with 10% (1713) of all transcripts (P ≤ 1E−12, Bonferroni correction, Additional file 2: Table S3).
We sought independent external replication of all significant CpG-transcript pairs from three cohorts with whole blood DNA methylation data and Illumina array-based gene expression: KORA (Kooperative Gesundheitsforschung in der Region Augsburg—Cooperative Health Research in the Region of Augsburg, n = 783) study, InCHIANTI (Invecchiare in Chianti, n = 500) study, and BLSA (Baltimore Longitudinal Study of Aging, n = 150). Following meta-analysis of the three replication studies, 10% of the 16,416 cis CpG-transcript pairs from the discovery sample replicated (at P < 0.05/16,416), with 98% of pairs from discovery showing consistent directions of effect in the meta-analysis of replication cohorts (1672 Affymetrix probes matched up with 1881 Illumina probes, Additional file 2: Table S4). None of the trans CpG-transcript pairs discovered in the FHS replicated in the meta-analysis of the other three cohorts after Bonferroni correction (at P < 0.05/198,960) and fewer than 6% replicated (pairwise comparisons) between the Illumina platform cohorts. Therefore, we focused this report on cis CpG-transcript pairs. To account for the correlation of CpG sites within the same genomic region, we conducted a conditional analysis to identify non-redundant CpGs for a given transcript (see Methods). After adjusting for nearby CpG sites, we identified 3,379 non-redundant cis CpG-transcript pairs (at P < 1E−8) that were the focus of subsequent analyses (Additional file 2: Table S5). Due to the large platform differences and the limited sample size of the Illumina cohorts, our subsequent analyses were performed using the 3,379 non-redundant cis CpG-transcript pairs from FHS discovery. The meta-analyzed results from all four cohorts are provided in Additional file 2: Tables S6.
Functional annotations of eQTMs
The proportion of inter-individual variation in eGene expression explained by cis eQTMs ranged from just under 1% to 75%, with a median R2 of 2%. We found that most CpGs reside in close proximity to their associated transcripts (76% are within 100 kb of the transcription start or end site) and the shorter the distance between a CpG and its paired transcript, the larger the effect size (Pearson correlation r = −0.1, P = 1.2E−08, Fig. 2).

Distance between the CpG site and the transcriptional start or end site vs the proportion of expression variance explained by the CpG. The correlation of distance of the CpG from the transcriptional start site with the proportion of variation in expression explained by the CpG. X-axis is the distance (measured by base pair) between the CpG locus and the transcriptional start/end site of the gene. Y-axis is the proportion of expression variance explained by the CpG (measured by R2)
We found that cis eQTMs are significantly enriched in CpG island shores (regions within a short distance from the CpG islands, P < 1E−4, Chi-square test) but not islands (regions with a high frequency of CpG sites, Fig. 3), and no significant enrichment was found in enhancers. We conducted further annotation using eFORGE (experimentally derived Functional Element Overlap analysis of ReGions from EWAS) [15] to view tissue-specific regulatory components of cis eQTMs across 21 cell lines. We found that cis eQTMs are significantly enriched in blood cell lines (monocytes, T cells, and natural killer cells, among others), indicating a highly tissue-specific pattern (Fig. 4).

Distribution of cis eQTMs in the human genome. Annotation of the cis eQTMs from the ENCODE. The percentage of annotation in different functional categories was compared with 450 K CpGs in the array and enrichment was tested using the Chi-square test, which revealed cis eQTMs were significantly enriched in CpG island shores, but not in islands (P < 1E−4)

Enrichment of DNase 1 hypersensitive sites (DHSs) in cis eQTMs across multiple tissues. Tissue-specific regulatory enrichment of cis eQTMs across 21 cell lines using eFORGE
Among the 3,379 unique, non-redundant cis CpG-transcript pairs, we identified 2,264 (67%) with negative and 1115 (33%) with positive associations. To explore whether the negatively and positively associated eQTMs reflect different functions in relation to regulation of gene expression, we performed separate Gene Ontology enrichment analyses [16] for each type of association (Additional file 2: Table S7). Although some genes were enriched in common biological processes such as immune response, of note, the 271 genes that were positively associated with DNA methylation were enriched in negative regulation of biological processes (Fisher's exact test P = 1.8E−05), whereas the 594 genes that were negatively associated with DNA methylation were enriched in positive regulation of biological processes (Fisher's exact test P = 8.1E−10).
Colocalization of cis eQTMs and eQTLs
DNA methylation can change gene expression without altering DNA sequence. Recent findings suggest that a large portion of this epigenetic regulation is also under genetic control [17]. To identify cis CpG-transcript pairs regulated by genetic variants, we conducted a Bayesian test of colocalization using the coloc package in R to test whether a CpG site and its corresponding transcript within the same genomic region shared the same sentinel variant [18] (see Methods). Among the 3,379 non-redundant cis CpG-transcript pairs from discovery, 2177 shared at least one SNP for both the corresponding cis eQTL variants (gene expression was associated with a SNP) [19] and cis mQTL variants (DNA methylation was associated with a SNP) in FHS participants at FDR < 0.05. Using all SNPs shared by CpG sites and their corresponding transcripts, we conducted a colocalization test for each pair to determine the probability that the two association signals (mQTL and eQTL) were due to the same cis variant (see Methods). For 780 (36%) out of 2,177 cis CpG-transcript pairs with shared SNPs, we observed a probability of > 80% that an mQTL variant colocalized with an eQTL variant (Additional file 2: Table S8).
Cigarette smoking and DNA methylation
Many studies have confirmed that environmental exposures can induce epigenetic changes, i.e., alter DNA methylation. To further investigate the epigenetic mechanisms linking exposures to health outcomes, we explored the DNA methylation signatures of cigarette smoking and linked them to alterations in nearby gene expression using the cis CpG-transcript pairs from discovery. We previously reported that 2,622 CpG sites were differentially methylated in current versus never smokers [13]. Intersecting these CpG sites with all 16,416 cis CpG-transcript pairs from discovery (see Methods, Additional file 2: Table S2), we identified 160 CpGs that differed between current versus never smokers that also are cis eQTMs (Fisher's exact test, P = 3.3E−16, Additional file 2: Table S9). To explore whether these smoking-related cis eQTM sites are under parallel genetic control along with nearby gene expression, we conducted colocalization analysis of cis mQTL variants and cis eQTL variants for the corresponding CpG-transcript pairs. Among the 109 cis CpG-transcript pairs that shared at least one SNP (i.e., an eQTL variant matched a mQTL variant), we identified colocalization (probability > 80%) for 22 cis CpG-transcript pairs. Among the 22 cis CpG-transcript pairs with colocalizing genetic signals for CpG mQTLs and transcript eQTLs, DNA methylation levels of 14 CpGs were decreased in current versus never smokers (11 of these CpGs were associated with increased gene expression and three with decreased gene expression) and methylation levels of eight CpGs were increased in current smokers (five of these CpGs were associated with increased gene expression and three with decreased gene expression) (Table 1 and Additional file 2: Table S10).
Cigarette smoking is a strong environmental and lifestyle risk factor that is linked to many diseases [20]. To investigate the hypothesis that smoking confers disease risk by altering DNA methylation with resultant effects on expression of key cis genes, we intersected the mQTL and eQTL variants associated with cis CpG-transcript pairs with SNPs associated with smoking-related diseases from published GWAS [20]. We identified six SNPs that regulate smoking-related CpG sites and that also have been reported to be associated with chronic obstructive pulmonary disease (COPD) and lung cancer—two prominent smoking-related diseases (Table 2). Because these CpG sites also were associated with nearby gene expression (i.e., they are cis eQTMs), these results suggest that smoking may promote disease by altering DNA methylation of key CpGs and thereby regulate expression of nearby genes. For example, CpGs in CHRNA5 have been found to be related to smoking [21]. We identified an intronic variant, rs17486278, whose C allele was associated with lower DNA methylation of a smoking-associated CpG in CHRNA5 (cg22563815), with reduced expression of CHRNA5, and with increased risk of COPD based on GWAS [22] (Fig. 5).

Genetic effects on smoking-related CpG sites and gene expression and the risk of COPD. Relations between genetic variant (rs17486278), DNA methylation (cg22563815) and gene expression (CHRNA5). Cg225638115 was found to be differentially methylated between current smokers and never smokers. This CpG is associated with the expression of CHRNA5. The CpG and CHRNA5 both are under genetic control of rs17486278, which has been found to be associated with COPD in previous GWAS
We identified smoking-related CpGs at three gene loci (within 1 Mb of CpG site) that also harbor GWAS signals for COPD or lung cancer (Table 2). To further explore epigenetic links between cigarette smoking and smoking-related lung diseases, we conducted MR [23] using four CpGs at these three gene loci (Table 2)—cg19696491 and cg22563815 for CHRNA5, cg03234777 for AMICA1, and cg26850624 for AHRR—with the methylation of the CpG as the exposure, cis mQTLs of these CpGs as the instrumental variables, and lung cancer or COPD as the outcomes [24]. At P < 0.05, we found that lower methylation of two CpG sites at the CHRNA5 locus was associated with increased risk of lung cancers (adenocarcinoma and squamous cell) and COPD, and lower methylation of a CpG in AMICA1 was associated with higher risk for lung cancer (adenocarcinoma but not squamous cell cancer; Table 3). We found no causal association between methylation of AHRR and lung cancer or COPD. The inferred causal relations between DNA methylation and lung cancer were further validated using mQTLs from lung tissue [25] and GWAS from UK Biobank [26] (Table 3). To explore the effects of gene expression of CHRNA5, AMICA1, and AHRR on lung diseases, we conducted MR using expression of these three genes as the exposure, cis eQTLs of these genes from FHS whole blood [19] as the instrumental variables, and lung disease traits as the outcomes. At P < 0.05, we found that lower expression of CHRNA5 was associated with higher risk of lung cancer (Table 4), but not for COPD. The inferred causal relation between gene expression and lung cancer was further tested using cis eQTLs in lung tissue from GTEx [27] as the instrumental variable, expression of CHRNA5 as the exposure, and lung cancer as the outcome. This analysis revealed consistent causal effects of CHRNA5 on lung cancer.
Smoking has profound effects on DNA methylation, and the CHRNA5 locus has been reported to be related to nicotine addiction [28]. To further explore the genetic and environmental effects on CHRNA5, we conducted bidirectional MR analyses of methylation of CHRNA5 in relation to cigarette smoking (see Methods). In the first test (CpG → smoking), we used methylation of CpGs at the CHRNA5 locus (cg19696491, cg22563815) as the exposures, cis mQTLs for these CpGs as the instrumental variables, and pack-years of smoking as the outcome. At P < 0.05, we found that reduced methylation of cg19696491/cg22563815 at CHRNA5 significantly increases smoking exposure in a causal manner. In the second test (smoking → CpG), we used pack-years of smoking as the exposure, pruned GWAS SNPs for smoking from UK Biobank as the instrumental variable, and methylation of CHRNA5 as the outcome. At P < 0.05, we found that greater pack-years of smoking significantly decreases methylation of cg19696491/cg22563815 at CHRNA5 (Table 5). Bidirectional MR revealed that the association of CHRNA5 with risk of lung cancer is causally influenced by both genetic and environment effects (Fig. 6).

Genetic and environmental effects on methylation of CHRNA5 in relation to the risk of lung disease. The possible mechanism of methylation of CHRNA5 affecting the risk of lung disease is through both genetic control and environment influence. The MR test inferred the causal role of methylation of CHRNA5 on lung cancer and COPD. In another bidirectional MR test, the methylation of CHRNA5 and pack-years of smoking played causal roles with each other
Discussion
We conducted a comprehensive assessment of the association of DNA methylation sites with gene expression and generated a resource of thousands cis CpG-transcript pairs that can be used to explore environmentally mediated epigenetic effects on disease. We conducted independent external replication of these findings. We found that the closer a CpG resided to a transcription start site, the larger its effect size is, and that the association of methylation with expression can be positive or negative. Moreover, using colocalization analyses, we found that 36% of cis CpG-transcript pairs share the same causal variant (i.e., the mQTL variant for the CpG matches the eQTL variant for the transcript), indicating that in addition to previously described environmental influences on DNA methylation, genetic effects also play an important role in epigenetic regulation. Using these genetic effects as instrumental variables in MR analyses, we identified a putatively causal role of DNA methylation of CHRNA5 in COPD and lung cancer.
CHRNA5, the gene that encodes the acetylcholine receptor subunit alpha 5, has been reported to be associated with tobacco addiction and lung cancer[29]. The possible molecular mechanism has been established by a chrna5 knockout-mouse model, which is analogous to individuals with reduced α5 receptor function[30]. Dramatically increased nicotine consumption was observed in mice with a null mutation in chrna5. This effect was reversed in knockout mice by re-expressing α5 subunits in the medial habenula of the mouse brain. These findings suggest that nicotine activates α5-containing nicotinic acetylcholine receptors (nAChRs) to suppress nicotine intake. Our MR results further demonstrate that in humans, genetic variants in CHRNA5 affect smoking and lung cancer risk through effects on DNA methylation and gene expression of CHRNA5.
The low reproducibility rate of cis CpG-transcript pairs may be due to two factors. First, the smaller sample size of the replication panel limited the power to replicate. To address this issue, we repeated the discovery-replication experiment in the opposite direction, with discovery from the meta-analysis of KORA, InCHIANTI, and BLSA (4446 cis CpG-transcript pairs) and replication in the FHS. This resulted in 57% of discovery cis CpG-transcript pairs from the meta-analysis of the three cohorts replicated in the FHS (at P < 0.05/4446), which confirmed the first assumption about replication of cis CpG-transcript pairs. Second, technical differences in the gene expression platforms (Affymetrix array in FHS versus Illumina array in the other cohorts) may restrict replication. Barnes et al. reported that only 37% of genes had expression levels that were significantly correlated when measuring the same sample using an Affymetrix array versus Illumina gene expression array [31]. To address this, we explored the consistency of results among the cohorts that used an Illumina array for expression profiling and found that 50% (491/987) of the cis CpG-transcript pairs from BLSA (the smallest sample size) replicated in the other two cohorts that used the same expression array, but none of them replicated in the FHS (Affymetrix expression array). Similar replication rates were observed among pairwise comparisons among Illumina cohorts, suggesting that platform rather than population differences is critical. On the other hand, the low replication of trans CpG-transcript pairs may be due to a lack of power to detect trans associations, substantial platform differences (as was the case for cis pairs), or because DNA methylation regulates only nearby rather than remote gene expression.
Tobacco exposure is a powerful environmental modifier of DNA methylation [13] and a major risk factor for cancer, cardiovascular disease, COPD, and many other diseases [5,6,7,8]. While it is reasonable to hypothesize that environmental factors affect DNA methylation with downstream effects on gene expression that in turn impact disease risk, these relationships are seldom tested formally. In-depth exploration of the inter-relations between genetic variation, DNA methylation, and gene expression is needed to identify mechanisms underlying environmental effects on disease. To that end, we integrated cis CpG-transcript pairs with their corresponding mQTLs and eQTLs, which enabled us to identify genetic variants that jointly regulate DNA methylation and gene expression. Finally, we integrated colocalization analysis with large GWAS databases to explore the relations between cigarette smoking and lung diseases. Although many smoking-related CpG sites were not associated with altered gene expression, we identified three genes (CHRNA5, AMICA1, and AHRR) that exhibited interconnected smoking-DNA methylation–gene expression relationships (Table 2). Using MR, we inferred a causal role of lower methylation and lower expression of CHRNA5 with increased risk for lung cancer (Table 5). For example, carriers of the risk allele for nicotine addiction (rs17486278) have lower methylation of cg19696491/cg22563815 at CHRNA5, which increases smoking exposure and the resultant risk of lung cancer. Individuals who do not carry the risk allele, however, may also be at increased risk for lung disease by virtue of smoking-related altered methylation of CHRNA5, which in turn increases lung cancer risk. If these findings are tested and validated in the clinical setting, it is possible that they can be used as biomarkers to identify high-risk subgroups (e.g., carriers of the risk allele, those with hypomethylation or reduced expression of CHRNA5) or as therapeutic targets for nicotine addiction treatment.
Causal inference analysis using MR is a powerful tool to distinguish causal from non-causal associations. Our previous cross-sectional study [17] reported that current cigarette smoking was associated with increased methylation of cg19696491 (CHRNA5), which is opposite to the MR results in the present investigation. When we limited analyses to individuals with fewer than 60 pack-years of smoking, however, we found that pack-years was inversely correlated with methylation of CHRNA5 (P = 0.0006, Additional file 1: Figure S1), which is consistent with our MR results. We further conducted a longitudinal analysis of DNA methylation changes following smoking cessation among smokers in the FHS who quit during follow-up and found that methylation of cg1969649 (CHRNA5) was significantly higher (Beta = 0.01, P = 0.036) following cessation than when these individuals smoked.
There are several limitations to our study. First, the discovery and replication cohorts used different gene expression platforms, which impaired our ability to replicate results from discovery. Second, DNA methylation and gene expression were profiled in whole blood, which may not reflect tissue-specific effects of DNA methylation on gene expression. Given the tissue-specific nature of eQTMs, our findings should be confirmed in additional disease-relevant tissues and cell types and validated in future studies. Finally, although colocalization has been proposed as a methodology for describing shared genetic influences [32], it relies on a key assumption of no more than two sentinel SNPs at a given locus, which may be inaccurate in some cases.
Our study is among the first investigations of the role of cigarette smoking on DNA methylation and gene expression and how these effects may promote smoking-related diseases. Taken together, our results show that whereas DNA methylation is an important epigenetic mechanism associated with gene expression, genetic variants play important dual roles in the regulation of DNA methylation and gene expression. We demonstrate that genetic variants associated with CpG-transcript pairs (i.e., mQTLs and eQTLs) can be integrated with smoking-related GWAS variants to improve our understanding of the interplay between environmental effects and lung diseases, facilitating the prioritization of candidate genes implicated in the pathogenesis of disease.
Conclusions
By integrating genetic and epigenetic data, we found that altered DNA methylation and gene expression of CHRNA5 have putatively causal effects on lung diseases. Using a bidirectional MR approach, we found evidence that DNA methylation and cigarette smoking have mutual effects on CHRNA5 that in turn influence risk for lung disease. Our findings highlight CHRNA5 as a potential therapeutic target for lung diseases and also for smoking cessation. The present study illustrated the potential clinical utility of identifying high-risk individuals by virtue of genetic and epigenetic biomarkers; broader application might be achieved in other tumor types in relation to other environmentally mediated disease processes.
Methods
Discovery: Framingham Heart Study (FHS)
The FHS is a community-based prospective study, which consists of three generations of participants starting in 1948. The 4170 participants in this study included FHS Offspring cohort (Exam 7; 1998–2001) and Third Generation cohort (Exam 1; 2002–2005) participants. Gene expression: Whole blood was collected in PAXgene™ tubes (PreAnalytiX, Hombrechtikon, Switzerland) and frozen at − 80 °C. RNA was extracted using the whole blood RNA System Kit (Qiagen, Venlo, Netherlands) and mRNA expression profiling was assessed using the Affymetrix Human Exon 1.0 ST GeneChip platform (Affymetrix Inc, Santa Clara, CA), which contains more than 5.5 million probes targeting the expression of 17,873 genes. The Robust Multi-array Average (RMA) package[33] was used to normalize the gene expression values and remove any technical or spurious background variation. Linear regression models were used to adjust for technical covariates (batch, first principal component, and all probeset mean).
DNA methylation: DNA methylation status was assayed using the Infinium HumanMethylation450 BeadChip (Illumina Inc., San Diego, CA). A total of 2,648 samples from FHS offspring cohort were run in two laboratory batches at the Johns Hopkins Center for Inherited Disease Research (laboratory batch #1) and the University of Minnesota Biomedical Genomics Center (laboratory batch #2). A total of 1,522 samples from the FHS Third generation cohort (laboratory batch #3) were run by Illumina (San Diego, CA, USA). QC and normalization were performed using the DASEN methodology implemented in the wateRmelon R package, and the final β values of each CpG for downstream analysis were output as previously described [13]. DNA methylation probes that contained polymorphic SNPs were removed [34]. Because of X-chromosome inactivation, only probes on autosomal chromosomes were analyzed. A total of 401,189 CpGs were retained for further analysis.
Replication cohorts
KORA: The KORA (Kooperative Gesundheitsforschung in der Region Augsburg—Cooperative Health Research in the Region of Augsburg) study is a series of independent population-based epidemiological surveys and follow-up studies of participants living in the region of Augsburg, Southern Germany. In the present study, we included 707 participants (356 males and 351 females aged 62–81 years) of the KORA F4 study for whom DNA methylation and gene expression data were available. KORA F4 (2006–2008, N = 3080) is a follow-up study of the KORA S4 survey (1999/2001, N = 4261). The applied standardized examinations have been described in detail elsewhere [35]. The KORA study has been conducted according to the principles expressed in the Declaration of Helsinki. Written informed consent has been given by each participant. The study was reviewed and approved by the local ethics committee (Bayerische Landesärztekammer).
Gene expression: In the KORA F4 study, gene expression was assessed using the Illumina HumanHT-12_v3 expression BeadChip, as described previously [36]. The gene expression data were quantile normalized and log2 transformed. The gene expression data are available for download at ArrayExpress (E-MTAB-1708).
DNA methylation: Genome-wide DNA methylation in KORA F4 was assessed using the Illumina HumanMethylation450 BeadChip as described elsewhere [37]. In brief, bisulfite converted genomic samples were amplified. After enzymatic fragmentation and application of the samples, the arrays were fluorescently stained and scanned using an Illumina HiScan SQ scanner. Data quality was assessed using GenomeStudio (version 2010.3). The methylation data were preprocessed primarily following the CPACOR pipeline[38]. Background correction was performed using the R package minfi, version 1.6.0 [39] and signals with detection P values ≥ 0.01 or with less than three functional beads were set to missing. Observations with less than 95% of CpG sites providing reliable signals were excluded. Finally, data were quantile normalized as described by [38], using the R package limma, version 3.16.5 [40]. Beta values representing the percentage of DNA methylation of a cytosine were calculated as the ratio of the methylated signal over the sum of the methylated and unmethylated signals. Following exclusion of cross-reactive probes [41], there were 442,279 CpG sites for investigation. Missing methylation values were imputed using a k-nearest neighbors approach (k = 8). Annotations are based on UCSC Genome Browser on Human Feb. 2009 (GRCh37/hg19) Assembly (https://genome.ucsc.edu/).
InCHIANTI
The InCHIANTI study [42] is a population-based, prospective study of human aging in the Tuscany area of Italy. A total of 1,455 participants were enrolled at baseline (1998–2000), with follow-up waves every 3 years. Extensive interviews, questionnaires, medical examinations, physical tests and blood samples were taken at every wave. Ethical approval was granted by the Instituto Nazionale Riposo e Cura Anziani institutional review board in Italy, and participants gave informed consent to participate.
Gene Expression: Peripheral blood specimens were collected at wave 4 (year 9, 2008–9) from 712 individuals, using the PAXgene technology to preserve levels of mRNA transcripts as they were at the point of collection[43]. RNA was extracted from peripheral blood samples using the PAXgene Blood mRNA kit (Qiagen, Crawley, UK) according to the manufacturer’s instructions. RNA was biotinylated and amplified using the Illumina® TotalPrep™ -96 RNA Amplification Kit and directly hybridized with HumanHT-12_v3 Expression BeadChips that include 48,803 probes. Image data were collected on an Illumina iScan and analyzed using the Illumina and Beadstudio software (Illumina, San Diego, California, USA) as previously described[44]. All microarray experiments and analyses complied with MIAME guidelines.
DNA Methylation: CpG methylation data were generated for a subset of the InCHIANTI participants. Samples taken at baseline (year 0) and during follow-up wave 3 (year 9, the ‘gene expression’ wave) were analyzed using the Illumina Infinium HumanMethylation450 BeadChip. Briefly, genomic DNA was bisulfite converted using Zymo EZ-96 DNA Methylation Kit, followed by CpG analysis using the aforementioned Illumina 450 k array. Quality control of the samples included exclusion based on sex-discrepancy and call-rate thresholds. Normalization of the arrays was performed using the ‘wateRmelon’[45] R package (specifically the DASEN method), which includes quantile normalization between probe types and arrays. Samples having 5% of sites with a detection P value > 0.01 were removed. After exclusions, 506 samples having robust data at two waves (9 years apart) were available for analysis. For more detailed methods, see Holly et al. [46].
BLSA
The Baltimore Longitudinal Study of Aging (BLSA) study is a population-based study aimed to evaluate contributors of healthy aging in the older population residing predominantly in the Baltimore-Washington DC area [47]. Starting in 1958, participants have been examined every one to four years depending on their age. There are ~ 1000 active participants enrolled in the study including 150 who have DNA methylation and gene expression data and were included in this investigation. The BLSA has continuing approval from the Institutional Review Board (IRB) of Medstar Research Institute.
Gene Expression: Gene expression profiling was conducted using the same processes as the InCHIANTI study. In brief, peripheral blood samples were collected for the purposes of gene expression profiling between April 2008 and September 2012. RNA samples were extracted with PAXGene blood mRNA kits (Qiagen), and gene expression was assessed using the Illumina HumanHT-12 v4 expression BeadChip. Gene expression data were log2-transformed (values less than or equal to 0 were imputed as 1) and quantile normalized for analysis.
DNA methylation: DNA methylation was conducted using a process consistent with that of the InCHIANTI study. Briefly, genomic DNA was extracted from whole blood using Gentra Puregene DNA purification system (Qiagen Inc., Germantown, MD). This was followed by bisulfite conversion using EZ DNA methylation kit (Zymo Research Corp., Irvine, CA), and genome-wide methylation was measured using the Illumina Infinium HumanMethylation450 BeadChip (Illumina Inc., San Diego, CA) following the manufacturer’s protocol. Quality control of the samples included exclusion based on sex-discrepancy and call-rate thresholds. Normalization of the arrays was performed using the ‘wateRmelon’[45] R package (specifically the DASEN method), which includes quantile normalization between probe-types and arrays. Samples having 5% of sites with a detection P value > 0.01 were removed.
Statistical analysis
eQTM analysis: First, we computed the residuals of the DNA methylation values using a linear mixed effect model adjusting for the following covariates: age, sex, Houseman’s white blood cell type proportions [48], DNA methylation-specific technical variables (e.g., chip, row, column). Then we computed 25 surrogate variables (SVs) for the residuals and computed the residuals of the residuals. Second, we performed the same clean-up protocol on the gene expression dataset, adjusting for age, sex, Houseman’s white blood cell type proportions [48], and gene expression-specific technical variables (e.g., batch effect, RNA integrity number). We used surrogate variable analysis (SVA) to identify unknown confounders [48]. We chose the number of surrogate variables (SVs) by comparing the internal replication rate of CpG-transcript pairs using FHS data (splitting the full set into discovery and replication samples). We examined replication with 0, 25, and 50 SVs and found that 25 SVs maximized the internal replication rate, thus we used 25 SVs to compute residuals. We then applied this protocol to each cohort. Due to differences in laboratory assays, we allowed each cohort to specify their own technical covariates to minimize technical artefacts. Cis was defined as a 500-kb window around the transcript unit.
Conditional eQTM analysis: For each transcript, we performed a conditional analysis by adding the CpG site that is most associated (lowest P value) with the transcript in the previous analysis as an independent variable. The same linear model of the previous analysis was used with the added conditional CpG term. Beta coefficients, standard errors, t values, and P values were then collected.
Meta-analysis: Because only results at P < 1e−4 were stored in some cohorts, regular meta-analysis approaches could not be used because they require complete availability of beta and standard error values for all cohorts. If only results for which the values are available are ignored, then meta-analysis results may be an underestimation. To remedy this situation, we used a method called MetaNSUE [49] to properly estimate the beta coefficients, standard errors, and P values of unstored/missing results and avoid underestimation. The method MetaNSUE does not distinguish random and fixed effects, but it accounts for the between-study heterogeneity and potential covariates by way of its maximum likelihood technique. This method is implemented in the R package MetaNSUE.
Colocalization analysis: For each CpG-transcript pair, the colocalization analysis involved a two-step procedure. Using FHS cis mQTL results, we first identified SNPs associated with CpG sites in 1 Mb region (upstream and downstream). Using FHS cis eQTL results [19], we then identified SNPs associated with transcripts in 1 Mb region. To estimate the probability that cis eQTLs and cis mQTLs residing in the same genomic location shared the same causal variant, we conducted a Bayesian test for colocalization of all pairs using all shared SNPs by the coloc package in R [18]. This method requires specifying a prior probability for a SNP being associated with gene expression only (p1), methylation level only (p2), and with both traits (p12). We applied the default P values, with p1 and p2 set to 1E−4, assuming that 1 in 10,000 SNPs are causal for either trait, and p12 was set to 1E−5.
Smoking-related CpGs: 2,622 CpG sites that were differentially methylated in current versus never smokers were derived from our previous publication [13]. Current smokers were defined as people who reported smoking at least one cigarette per day within 12 months prior to the blood draw, former smokers were defined as people who previously smoked at least one cigarette per day, but stopped more than 12 months prior to the blood draw, and never smokers were defined as people who never smoked. Pack-years was calculated based on self-reported average number of cigarettes per day smoked divided by 20 multiplied by the number of years of smoking, with zero assigned to never smokers. Because the smoking-related CpG lists did not account for the correlation among CpGs, we leverage the 2,622 smoking-related CpGs with 16,416 cis CpG-transcript pairs from all 450 K CpGs to maximize the overlap.
Mendelian Randomization: MR uses genetic variation as a natural experiment that mimic randomized control trials to infer causal relations between an exposure and an outcome using genetic data from observational studies and GWAS (Additional file 1: Figure S2). MR has three assumptions: (1) that the instrumental variable is robustly associated with the exposure, (2) that the instrumental variable acts independently of confounders, and (3) that the instrumental variable only influences the outcome via its effect on the exposure. Using SNPs significantly associated with DNA methylation or gene expression as genetic instruments for MR satisfies assumptions 1 and 2. Using only cis-mQTLs and cis-eQTLs as instrumental variables satisfies assumption 3. MR was conducted in TwoSampleMR package[23] using DNA methylation or gene expression as exposure, separately. Two-sample (SNP-outcome association is from published GWAS and SNP-exposure association is from FHS mQTLs or eQTLs) MR was used to identify putatively causal CpG sites or genes for lung cancer. SNPs and lung cancer associations were based on the published GWAS. Instrumental variables (IV) for each CpG or gene were composed of independent cis mQTLs or cis eQTLs pruned by LD at r2 < 0.001. For CpGs or genes with only one independent SNP after LD pruning, causal effect estimates were determined using the Wald ratio test. When multiple non-redundant SNPs were present, we conducted multi-SNP MR using inverse-variance weighted estimates. Bidirectional MR was first conducted using DNA methylation as exposure and pack-years of smoking as outcome and then vice versa. Summary statistics for SNP-pack years of smoking associations were obtained from UK Biobank GWAS [50]. Pruned SNPs (LD r2 < 0.001) were used as instrumental variables, and the associations between SNPs and methylation level were calculated in FHS.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- eQTMs:
-
Expression quantitative trait methylation sites
- CTCF:
-
Transcriptional repressor CCCTC-binding factor
- MR:
-
Mendelian randomization
- EWAS:
-
Epigenome-wide association study
- FHS:
-
Framingham Heart Study
- eGene:
-
CpG site and the associated transcript
- KORA:
-
Kooperative Gesundheitsforschung in der Region Augsburg—Cooperative Health Research in the Region of Augsburg
- InCHIANTI:
-
Invecchiare in Chianti
- BLSA:
-
Baltimore Longitudinal Study of Aging
- eFORGE:
-
Experimentally derived Functional Element Overlap analysis of ReGions from EWAS
- mQTL:
-
DNA methylation was associated with a SNP
- eQTL:
-
Gene expression was associated with a SNP
- COPD:
-
Chronic obstructive pulmonary disease
- GTEx:
-
Genotype-Tissue Expression
- GWAS:
-
Genome-wide association studies
References
Hannon E, Knox O, Sugden K, Burrage J, Wong CCY, Belsky DW, Corcoran DL, Arseneault L, Moffitt TE, Caspi A, et al. Characterizing genetic and environmental influences on variable DNA methylation using monozygotic and dizygotic twins. PLoS Genet. 2018;14(8):e1007544.
Razin A, Cedar H. DNA methylation and gene expression. Microbiol Rev. 1991;55(3):451–8.
Gutierrez-Arcelus M, Lappalainen T, Montgomery SB, Buil A, Ongen H, Yurovsky A, Bryois J, Giger T, Romano L, Planchon A, et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife. 2013;2:e00523.
Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, Stamatoyannopoulos JA. Role of DNA methylation in modulating transcription factor occupancy. Cell Rep. 2015;12(7):1184–95.
Hanley MP, Hahn MA, Li AX, Wu X, Lin J, Wang J, Choi AH, Ouyang Z, Fong Y, Pfeifer GP, et al. Genome-wide DNA methylation profiling reveals cancer-associated changes within early colonic neoplasia. Oncogene. 2017;36(35):5035–44.
Jeffries MA, Dozmorov M, Tang Y, Merrill JT, Wren JD, Sawalha AH. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics. 2011;6(5):593–601.
Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, Albin RL, Hu H, Rozek LS. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J Alzheimer’s Dis JAD. 2012;29(3):571–88.
Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, Tsai PC, Ried JS, Zhang W, Yang Y, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541(7635):81–6.
Kriebel J, Herder C, Rathmann W, Wahl S, Kunze S, Molnos S, Volkova N, Schramm K, Carstensen-Kirberg M, Waldenberger M, et al. Association between DNA methylation in whole blood and measures of glucose metabolism: KORA F4 study. PLoS ONE. 2016;11(3):e0152314.
Chambers JC, Loh M, Lehne B, Drong A, Kriebel J, Motta V, Wahl S, Elliott HR, Rota F, Scott WR, et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol. 2015;3(7):526–34.
Mendelson MM, Marioni RE, Joehanes R, Liu C, Hedman AK, Aslibekyan S, Demerath EW, Guan W, Zhi D, Yao C, et al. Association of body mass index with DNA methylation and gene expression in blood cells and relations to cardiometabolic disease: a Mendelian randomization approach. PLoS Med. 2017;14(1):e1002215.
Johnson AA, Akman K, Calimport SR, Wuttke D, Stolzing A, de Magalhaes JP. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012;15(5):483–94.
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CE, Aslibekyan S, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9(5):436–47.
Relton CL, Davey Smith G. Two-step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. Int J Epidemiol. 2012;41(1):161–76.
Breeze CE, Paul DS, van Dongen J, Butcher LM, Ambrose JC, Barrett JE, Lowe R, Rakyan VK, Iotchkova V, Frontini M, et al. eFORGE: a tool for identifying cell type-specific signal in epigenomic data. Cell Rep. 2016;17(8):2137–50.
Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8(8):1551–66.
Taylor DL, Jackson AU, Narisu N, Hemani G, Erdos MR, Chines PS, Swift A, Idol J, Didion JP, Welch RP, et al. Integrative analysis of gene expression, DNA methylation, physiological traits, and genetic variation in human skeletal muscle. Proc Natl Acad Sci USA. 2019;116(22):10883–8.
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383.
Joehanes R, Zhang X, Huan T, Yao C, Ying SX, Nguyen QT, Demirkale CY, Feolo ML, Sharopova NR, Sturcke A, et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol. 2017;18(1):16.
Schane RE, Ling PM, Glantz SA. Health effects of light and intermittent smoking: a review. Circulation. 2010;121(13):1518–22.
Tsaprouni LG, Yang TP, Bell J, Dick KJ, Kanoni S, Nisbet J, Vinuela A, Grundberg E, Nelson CP, Meduri E, et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics. 2014;9(10):1382–96.
Hobbs BD, de Jong K, Lamontagne M, Bosse Y, Shrine N, Artigas MS, Wain LV, Hall IP, Jackson VE, Wyss AB, et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet. 2017;49(3):426–32.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R et al: The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018, 7.
Wang Y, McKay JD, Rafnar T, Wang Z, Timofeeva MN, Broderick P, Zong X, Laplana M, Wei Y, Han Y, et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat Genet. 2014;46(7):736–41.
Morrow JD, Glass K, Cho MH, Hersh CP, Pinto-Plata V, Celli B, Marchetti N, Criner G, Bueno R, Washko G, et al. Human lung DNA methylation quantitative trait loci colocalize with chronic obstructive pulmonary disease genome-wide association loci. Am J Respir Crit Care Med. 2018;197(10):1275–84.
nealelab: 2018.
Consortium GT. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45(6):580–5.
Berrettini WH, Doyle GA. The CHRNA5-A3-B4 gene cluster in nicotine addiction. Mol Psychiatry. 2012;17(9):856–66.
Lassi G, Taylor AE, Timpson NJ, Kenny PJ, Mather RJ, Eisen T, Munafo MR. The CHRNA5-A3-B4 gene cluster and smoking: from discovery to therapeutics. Trends Neurosci. 2016;39(12):851–61.
Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471(7340):597–601.
Barnes M, Freudenberg J, Thompson S, Aronow B, Pavlidis P. Experimental comparison and cross-validation of the Affymetrix and Illumina gene expression analysis platforms. Nucleic Acids Res. 2005;33(18):5914–23.
Giambartolomei C, Zhenli Liu J, Zhang W, Hauberg M, Shi H, Boocock J, Pickrell J, Jaffe AE, CommonMind C, Pasaniuc B, et al. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics. 2018;34(15):2538–45.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64.
Daca-Roszak P, Pfeifer A, Zebracka-Gala J, Rusinek D, Szybinska A, Jarzab B, Witt M, Zietkiewicz E. Impact of SNPs on methylation readouts by Illumina Infinium HumanMethylation450 BeadChip Array: implications for comparative population studies. BMC Genom. 2015;16:1003.
Holle R, Happich M, Lowel H, Wichmann HE, Group MKS. KORA—a research platform for population based health research. Gesundheitswesen. 2005;67(Suppl 1):S19-25.
Schramm K, Marzi C, Schurmann C, Carstensen M, Reinmaa E, Biffar R, Eckstein G, Gieger C, Grabe HJ, Homuth G, et al. Mapping the genetic architecture of gene regulation in whole blood. PLoS ONE. 2014;9(4):e93844.
Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, Weidinger S, Lattka E, Adamski J, Peters A, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE. 2013;8(5):e63812.
Lehne B, Drong AW, Loh M, Zhang W, Scott WR, Tan ST, Afzal U, Scott J, Jarvelin MR, Elliott P, et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 2015;16:37.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–9.
Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21(9):2067–75.
Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–9.
Ferrucci L, Bandinelli S, Benvenuti E, Di Iorio A, Macchi C, Harris TB, Guralnik JM. Subsystems contributing to the decline in ability to walk: bridging the gap between epidemiology and geriatric practice in the InCHIANTI study. J Am Geriatr Soc. 2000;48(12):1618–25.
Debey-Pascher S, Eggle D, Schultze JL. RNA stabilization of peripheral blood and profiling by bead chip analysis. Methods Mol Biol. 2009;496:175–210.
Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6(5):e1000952.
Pidsley R. CC YW, Volta M, Lunnon K, Mill J, Schalkwyk LC: A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genom. 2013;14:293.
Holly AC, Pilling LC, Hernandez D, Lee BP, Singleton A, Ferrucci L, Melzer D, Harries LW. Splicing factor 3B1 hypomethylation is associated with altered SF3B1 transcript expression in older humans. Mech Ageing Dev. 2014;135:50–6.
Ferrucci L. The Baltimore Longitudinal Study of Aging (BLSA): a 50-year-long journey and plans for the future. J Gerontol A Biol Sci Med Sci. 2008;63(12):1416–9.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012;13:86.
Radua J, Schmidt A, Borgwardt S, Heinz A, Schlagenhauf F, McGuire P, Fusar-Poli P. Ventral striatal activation during reward processing in psychosis: a neurofunctional meta-analysis. JAMA Psychiatry. 2015;72(12):1243–51.
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779.
Acknowledgements
The Framingham Heart Study is funded by National Institutes of Health contract N01-HC-25195. The DNA methylation laboratory work for this investigation was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD (D. Levy, Principal Investigator). The analytical component of this project was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, and the Center for Information Technology, National Institutes of Health, Bethesda, MD. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
Funding
Open Access funding provided by the National Institutes of Health (NIH). The KORA study was initiated and financed by the Helmholtz Zentrum München – German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Furthermore, KORA research has been supported within the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ. The German Diabetes Study (GDS) was initiated and financed by the German Diabetes Center, which is funded by the German Federal Ministry of Health (Berlin, Germany), the Ministry of Culture and Science of the state North Rhine-Westphalia (Düsseldorf, Germany), and grants from the German Federal Ministry of Education and Research (Berlin, Germany) to the German Center for Diabetes Research e.V. (DZD). The funders had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All participants from the FHS, KORA, InCHIANTI, and BLSA gave informed consent for participation in this study and for the collection of plasma and DNA for analysis. The FHS study protocol was approved by Boston Medical Center. The KORA study protocol was approved by the Ethics Committee of the Bavarian Medical Association, Germany. The InCHIANTI ethical approval was granted by the Instituto Nazionale Riposo e Cura Anziani institutional review board in Italy, and participants gave informed consent to participate. The BLSA has approval from the Institutional Review Board (IRB) of Medstar Research Institute.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplemental Figures.
Additional file 2.
Supplemental Tables.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yao, C., Joehanes, R., Wilson, R. et al. Epigenome-wide association study of whole blood gene expression in Framingham Heart Study participants provides molecular insight into the potential role of CHRNA5 in cigarette smoking-related lung diseases. Clin Epigenet 13, 60 (2021). https://doi.org/10.1186/s13148-021-01041-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-021-01041-5