
Abstract
Background
Prenatal inflammation has been proposed as an important mediating factor in several adverse pregnancy outcomes. C-reactive protein (CRP) is an inflammatory cytokine easily measured in blood. It has clinical value due to its reliability as a biomarker for systemic inflammation and can indicate cellular injury and disease severity. Elevated levels of CRP in adulthood are associated with alterations in DNA methylation. However, no studies have prospectively investigated the relationship between maternal CRP levels and newborn DNA methylation measured by microarray in cord blood with reasonable epigenome-wide coverage. Importantly, the timing of inflammation exposure during pregnancy may also result in different effects. Thus, our objective was to evaluate this prospective association of CRP levels measured during multiple periods of pregnancy and in cord blood at delivery which was available in one cohort (i.e., Effects of Aspirin in Gestation and Reproduction trial), and also to conduct a meta-analysis with available data at one point in pregnancy from three other cohorts from the Pregnancy And Childhood Epigenetics consortium (PACE). Secondarily, the impact of maternal randomization to low dose aspirin prior to pregnancy on methylation was assessed.
Results
Maternal CRP levels were not associated with newborn DNA methylation regardless of gestational age of measurement (i.e., CRP at approximately 8, 20, and 36 weeks among 358 newborns in EAGeR). There also was no association in the meta-analyses (all p > 0.5) with a larger sample size (n = 1603) from all participating PACE cohorts with available CRP data from first trimester (< 18 weeks gestation). Randomization to aspirin was not associated with DNA methylation. On the other hand, newborn CRP levels were significantly associated with DNA methylation in the EAGeR trial, with 33 CpGs identified (FDR corrected p < 0.05) when both CRP and methylation were measured at the same time point in cord blood. The top 7 CpGs most strongly associated with CRP resided in inflammation and vascular-related genes.
Conclusions
Maternal CRP levels measured during each trimester were not associated with cord blood DNA methylation. Rather, DNA methylation was associated with CRP levels measured in cord blood, particularly in gene regions predominately associated with angiogenic and inflammatory pathways.
Trial registration
Clinicaltrials.gov, NCT00467363, Registered April 30, 2007, http://www.clinicaltrials.gov/ct2/show/NCT00467363
Similar content being viewed by others

Background
Inflammation is a non-specific response to insults to the body. A five-fold increase in inflammation, as measured by CRP levels, occurs over the course of normal pregnancy [1], and the fetal impact is unknown. Elevated inflammation beyond the physiological increase during pregnancy has been implicated as a mediator in the associations of a myriad of prenatal exposures (e.g., infectious disease [2], maternal obesity [3, 4], maternal stress [5], and air pollution [6]) with offspring health. Moreover, maternal inflammation is linked with preterm birth [7, 8], preeclampsia [9, 10], and childhood asthma/allergies [11,12,13,14]. Also, women with chronic inflammatory diseases such as rheumatoid arthritis have lower weight babies [15]. Developmental programming studies suggest that prenatal exposures could alter long-term offspring health through epigenetic changes, often effected via DNA methylation. DNA methylation is one epigenetic modification that has become frequently studied due to the availability of reliable and affordable technologies.
C-reactive protein (CRP) is a cytokine routinely used to measure inflammation. CRP is an acute phase reactant produced by the liver, and serum levels can increase 3000-fold within a short amount of time as part of innate immunity [16]. Its plasma half-life is about 18 h but in the acute phase may only be 75 min [16]. Rapid increase in CRP is associated with infections rather than prenatal exposures or systemic inflammation. While many cytokines including interleukins and interferons are part of the inflammatory response, CRP is most often studied due to reliable assay and clinical use as a global marker of systemic chronic inflammation. In adults, circulating CRP has been cross-sectionally associated with altered DNA methylation patterns in white blood cells in a meta-analysis of epigenome-wide studies with data from 8863 Europeans and 4111 African Americans [17]. Moreover, evidence from a longitudinal study of over 100 adults shows DNA methylation changes specific to CRP levels measured 10 years prior, suggesting lasting influence of inflammation on DNA methylation [18]. However, it is unclear whether CRP throughout pregnancy has similar effects on newborn methylation, and whether the timing of the exposure at different gestational ages, including at delivery, matters. Longitudinal analyses of CRP during pregnancy, therefore, could be informative in understanding the role of inflammation on newborn methylation. Decreasing maternal inflammation could also be another way to demonstrate a causal role, and low dose aspirin use may decrease maternal CRP [19], which may in turn alter newborn methylation.
We aimed to comprehensively identify newborn DNA methylation differences associated with prenatal CRP exposure. In the Effects of Aspirin in Gestation and Reproduction (EAGeR) study, high-sensitivity CRP levels were measured longitudinally in maternal serum at three time points during pregnancy and in cord blood at delivery. Newborn DNA methylation was assessed in cord blood obtained at delivery. In addition, as part of the Pregnancy And Childhood Epigenetic (PACE) consortium [20], maternal CRP levels measured in the first trimester and DNA methylation in cord blood were available from three international pregnancy cohorts.
Results
Women in EAGeR had an average pre-pregnancy BMI of 25.2 kg/m2, predominantly did not smoke, and 43% had household income levels above $100,000 (Table 1). Levels of maternal and cord blood CRP are provided in Table 2. No significant associations between maternal CRP concentrations during pregnancy were found regardless of gestational age at measure in EAGeR (i.e., in first, second, or third trimester) in crude models adjusted for covariates (i.e., Model 1: maternal age, smoking, income, pre-pregnancy BMI, plate) or in two models additionally adjusting for cell type distribution using two different cord blood reference panels (i.e., Model 2, Bakulski [21]; Model 3, Gervin [22] (Supplemental Tables A–C in the online repository, Figshare)). In sensitivity analysis, we repeated all analyses without exclusion of CRP >10 mg/dl so as to not exclude acute infections but found no associations aside from one CpG with third trimester CRP levels which was not in a known gene region (cg14503868; beta = − 0.02, FDR p = 0.01; located in Chr 9, position 128504605, closest to PBX3) (See Supplemental Tables A4, B4, C4 on Figshare).
Similarly, in the meta-analysis with three PACE cohorts for first trimester CRP measures, no significant associations were found. Table 3 lists the top 5 associations (all FDR p > 0.05) in the meta-analysis and the direction of association from each cohort. Supplemental Table 1 provides characteristics of the participants in the other cohorts, and Supplemental Table 2 provides information on the genomic inflation of the various models run from each study.
One association with newborn methylation was identified in additional analyses performed in EAGeR to evaluate whether cumulative inflammation over pregnancy (as estimated by the area under the curve of CRP measured in gestational weeks 8–36) was associated with DNA methylation in offspring (Supplemental Tables D1-3 on Figshare). The CpG (cg09180262) was significant only for Model 3 with small effect size (− 0.0002 per log unit CRP). We also assessed “the extremes” by comparing newborn methylation in infants born to women consistently in the highest tertile of CRP (n = 64) for all 3 trimesters of pregnancy compared to newborns of women consistently at the lowest tertile (n = 62). In this analysis, DNA methylation levels at 5 CpGs were associated with CRP, the most highly associated CpG being in the intron of the major histocompatibility complex, class II, DQ beta 1 (HLA-DQB1). See Supplemental Table 3 for annotated results of the top 6 CpGs and Supplemental Tables D–E on Figshare for full results.
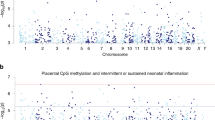
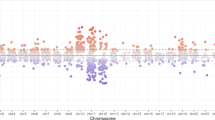
In the analysis comparing newborn CRP measured in cord blood at the same time as DNA methylation, 108 significant associations were observed in crude models (Model 1) without cell-type adjustment initially in EAGeR. After Model 2 adjustment using the Bakulski reference [21] to estimate cell type distribution, 71 significant associations were found. This number was further reduced in Model 3 after adjustment for cell type as estimated by Gervin et al. [22], and 33 significant associations were identified (Table 4). Supplemental Figure 2 shows the overlap in the associations found between the 3 models. Of all the CpGs identified, 20 were FDR-significant in all three models regardless of cell type adjustment, whereas 6 associations (as marked by asterisks in Table 4) were significant only in Model 3. Effect sizes of Model 3 associations ranged from 2 to 15% differences in methylation per log unit change in CRP at delivery, and the top 8 CpGs were Bonferroni significant (p < 6 × 10−8). There were almost equal numbers of CpGs that had higher methylation (18 CpGs) as lower methylation (15 CpGs) in relation to increasing concentrations of CRP at delivery. The top CpG (cg13138089, p = 2 × 10−14) is in the CpG island for the gene ECEL1P2 (Endothelin Converting Enzyme Like 1 Pseudogene 2), which is relevant for endothelial function. Apart from the 3 CpGs on the X chromosome, 3 associated with non-coding RNAs, and 4 with no known genes within 5 kb, the gene locations and functions of the remaining 23 CpGs were reviewed (Supplemental Text). Among the 33 significant CpGs, no significant cell type specific signals were identified by eFORGE. However, enrichment for transcription factors related to immune cell lineages preserved across evolutionary lines was noted from several databases (Supplemental Figure 3 and Supplemental Table 4).
Lastly, approximately half of the EAGeR participants were randomized to low-dose aspirin as expected (Table 1). Randomization to low dose aspirin was not associated with differences in newborn methylation (Supplemental Figure 1), except for DNA methylation at one CpG (cg2002882, chromosome 13, position 79169823, beta = 0.009, FDR-p = 0.04).
Discussion
We report, to our knowledge, the first study to examine the longitudinal association between prenatal inflammation as repeatedly measured by CRP and newborn DNA methylation at delivery (Supplemental Figure 4). Associations relevant to inflammation/angiogenesis were identified in newborns at delivery suggesting that CRP, as an acute phase reactant, associates with methylation of circulating cells when measured at the same time point, reiterating CRP’s role in inflammatory and angiogenic pathways although the temporality of these associations remains unclear. Few associations in genes related to histocompatibility proteins/antigens were identified with persistent high CRP.
CRP during pregnancy
The general lack of association between circulating maternal CRP levels during pregnancy and newborn DNA methylation may be related to several reasons. First, due to its short half-life, CRP may represent transient acute inflammation rather than chronic inflammation. However, few associations emerged even when we modeled longitudinal CRP levels in two different ways to capture women with relatively and chronically higher levels throughout pregnancy. These analyses were done by modeling cumulative CRP using an area under the curve model with repeated measures and by categorizing women consistently in the top tertile of CRP from first through third trimester. For the latter, we observed a few differences in DNA methylation located at genes related to immune function after cell type adjustment. HLA-DQB1 codes for a class II major histocompatibility complex protein, PUM3 codes for a minor histocompatibility antigen, and MYO1G codes for a minor histocompatibility antigen precursor. In other studies evaluating prenatal exposures on offspring methylation, HLA-DQB1 methylation differed between siblings at 4–6 years of age before and after maternal weight loss from bariatric surgery [24], and MYO1G methylation was associated with environmental chemical exposure [25]. However, these findings require replication given the selectiveness of the sample.
As CRP does not cross the placental barrier [26], its indirect influence on neonatal health may be modified by downstream factors including immune responses. And while the majority of previous studies have found maternal CRP to be associated with smaller newborn size [27, 28], it is inconsistently associated with other conditions [2, 29]. Studies linking maternal CRP with childhood asthma/allergies [12, 14] have also been inconsistent. These observations, in combination with the natural rise of CRP during pregnancy [30], make it difficult to evaluate normative rather than “elevated” CRP in the context of pregnancy. We modeled linear associations with the understanding that higher inflammation is indicative of cellular damage, but in the context of pregnancy, it is possible that some non-linear relationships exist for the lowest and the highest CRP levels. Second, CRP levels have been shown not to correlate with other cytokines measured early in pregnancy [31]. Thus, other biomarkers that may provide a fuller picture of prenatal inflammation may be needed to further investigate the role of inflammation. Third, cord blood CRP levels were 10- to 20-fold lower than maternal CRP levels (Table 2), despite evidence that maternal CRP increases dramatically during labor and delivery [32]. Placental production of CRP is largely directed into maternal circulation [33]. Thus, natural protective mechanisms may cause maternal CRP to not reflect local inflammation in the developing fetus and explains why it is not associated with newborn methylation. Last, the sample size of EAGeR (n = 391) has limited power to detect differences. Nevertheless, a meta-analysis of over 1600 newborns and the findings for CRP at delivery in EAGeR suggest otherwise. Taken together, maternal CRP levels during pregnancy are generally not associated with alterations in DNA methylation patterns to the offspring as measured at delivery in cord blood.
Randomization to low-dose aspirin was also largely not associated with differences in newborn methylation. The single association identified on chromosome 13 (cg20028827) after accounting for multiple testing (FDR p = 0.04) was located ~ 3.5 kb upstream of the promoter of POU4F1 (chr13:79173227-79177695). It is in a DNase hypersensitive site (chr13:79169325-79170815) with peaks for transcription factors EZH2 and SUZ12, among others. The levels of H2K4Me1 are slightly higher but with no apparent enhancer overlap. However, the CpG is within the intron of several ncRNA variants of the OBI1-AS1 (previously known as the PUF1-AS1 or RNF219-AS1). A log unit increase in CRP levels was associated with a < 1% increase in methylation levels (adjusted beta = 0.009). POU4F1 codes for neural transcriptors, particular to ganglions, by playing a role in development of calcium channels [34]. However, its methylation has only been examined with regard to breast tissue tumors (where it was found to be hypermethylated and its expression silenced) [35].
CRP at delivery
While no associations with maternal CRP were identified, newborn CRP levels were associated with differences in cord blood methylation. Interestingly, 13 of the CpGs were located in or near genes associated with inflammatory or angiogenic pathways (ECEL1P2, SPEG, ARHGEF17, OLFML2A, HSPB6, HSPE1, IFITM3, ZADH2, DEPDC1, TRIM6, SERPINB10, JMJD1C, and RGS5). Of note, the top 7 genes, 5 of which exceeded Bonferroni significance, are among these genes. eFORGE results also confirm these methylation changes corresponded to transcription factors involved in development of immune cell lineages. These observations further support their biological relevance in relation to CRP. Previous studies that have measured inflammation and methylation in adults have been systematically reviewed [36]. We compared the cross-sectional associations found in the current analysis at birth with those of previous epigenome-wide association studies among adults but were unable to find overlap between statistically significant CpGs identified by other studies and our results [17, 18]. All but one study used the 450K panel, such that most of the identified CpGs in the current analysis are unique. We add to the list of CpGs identified, which include sites that are novel to the EPIC microarray. Myte et al. also used the EPIC microarray but had a limited sample size of < 200 adults [18]. Cord blood samples unlike those from older children or adults normally contain nucleated red blood cells, which may also explain differences among studies [37]. Moreover, immune system development continues after delivery as the newborn comes into contact with microbes, whereas adult immune systems have matured with respect to a range of environmental exposures and their own microbiome [38].
Limitations
EAGeR included women with 1–2 previous pregnancy losses. We cannot exclude that genetic causes of pregnancy loss [39], including a role for CRP [40], may play a role. This may limit the generalizability of the EAGeR results. However, the PACE birth cohorts contributing to the first trimester analysis were recruited from the general population and echoed the same findings with regards to maternal CRP. The small sample size of EAGeR and the meta-analysis may have limited power to detect weaker associations. Given that inflammation plays an integral role in recruitment of white blood cells, using a mix of cells as in buffy coat or any blood sample without prior selection of cell type for DNA methylation measurement requires estimation of cell type based on previous reference panels [21, 22]. Remaining estimation errors cannot be excluded. Also, the specificity of the associations identified to CRP is unclear. Our results are based on cord blood derived methylation and unclear if generalizable to other tissues. CRP is just one acute phase protein (APP), and various cytokines/chemokines either leading to APP production are increased by APPs (e.g., interleukins, TNF-a) in turn recruits immune cells. Thus, associations might have been due to correlated cytokine production and upstream factors. Lastly, the Illumina platform covers a large number of CpG sites but only a small fraction of the genome. It might not provide sufficient coverage to evaluate differences in global methylation as other techniques [41]. Nevertheless, site specific differences identified in this approach revealed specific genes and biological pathways related to inflammation.
Conclusion
In conclusion, DNA methylation levels as measured in cord blood do not correlate with prenatal levels of CRP over pregnancy. Rather, methylation levels are consistent with concurrently measured CRP. The differentially methylated CpG sites suggest pathways that are modulated by inflammatory cytokines. Future research in larger studies and using different biomarkers of inflammation apart from CRP are needed for understanding the role of prenatal inflammation in fetal and neonatal developmental programming. Other epigenetic mechanisms such as histone acetylation and non-coding RNAs may also provide insights into the role of prenatal inflammation.
Methods
Study design
The Effects of Aspirin in Gestation and Reproduction (EAGeR) trial (2007–2011; NCT00467363) randomized women prior to conception who previously experienced 1–2 prior pregnancy losses to 81 mg low-dose aspirin and 400 μg folic acid, or placebo plus 400 μg folic acid [42]. Randomization to low-dose aspirin did not significantly alter live birth rates [42]. Its design and results are detailed elsewhere [42]. The current analysis was nested among 391 newborns with DNA methylation data available from the Utah site [43]. The University of Utah IRB approved the study (Salt Lake City, Utah IRB #1002521), and all participants provided written informed consent prior to enrollment. Among the 391 newborns with methylation data, we excluded 12 newborns of non-white self-reported ethnicity (to avoid population stratification in meta-analysis across cohorts) and 9 for missing CRP measures.
Laboratory methods
Women in EAGeR underwent multiple blood draws; first at baseline prior to pregnancy and at approximately 8, 20, and 36 weeks gestation during pregnancy. In addition, among 428 deliveries from the Utah site, 10 mL cord blood was collected in a plasma collection tube with ethylenediaminetetraacetic acid (EDTA) [43]. CRP was quantified from maternal serum samples and cord blood plasma by means of an immunoturbidimetric assay (Roche COBAS 6000, Roche Diagnostics, IN) with a limit of detection (LOD) of 0.15 mg/L. Cord blood DNA underwent bisulphite conversion with standardized kits (e.g. Zymo EZ DNA MethylationTM kit, Zymo, Irvine, CA), followed by measurements of DNA methylation using the Infinium MethylationEPIC BeadChip microarray [43].
Statistical analysis
Data cleaning for EAGeR
Methylation data were processed using the minfi package in R [44], including the identification of failed probes and scaling with Illumina control probes to determine methylation values. The beta value (β) was determined for each of the CpG sites by the fluorescent signals (β = Max (M, 0) / [Max (M, 0) + Max (U, 0) + 100), where M and U are the intensity of methylated and unmethylated allele [45]. β values approaching 1 are completely methylated, and those close to 0 are unmethylated. Background and dye-bias corrections were applied. Quantile normalization was used to normalize beta values between two types of probes [46]. The purpose of this step is to eliminate potential probe type bias (type I vs II probes). Cell type mixture was then estimated on the full set of normalized data (FlowSorted.CordBlood.450K package) [21]. Principal component analysis (PCA) was performed to further detect outliers and samples mismatched in sex compared against information from electronic medical records. Five samples with sex mismatch were further excluded. We extracted the detection p value for each methylation measure (per site per sample) and filtered data that failed detection p value (p > 0.01). Beta values were replaced as missing if either detection p value > 0.01 or bead counts < 3. We removed samples and CpG sites with low passing rate (< 97%) based on detection p value and bead counts. After probe removal, 821,665 CpG probes remained.
Modeling exposure for EAGeR
To exclude acute inflammation from infection, maternal and cord plasma samples with CRP above 10 mg/L were excluded from analysis (15 at preconception, 39 at 8 weeks, 54 at 20 weeks, 28 at 36 weeks, 1 at delivery from EAGeR). Among the 391 neonates with DNA methylation data, 358 remained for analyses with CRP measured at any time during pregnancy. CRP was natural log transformed for normality. Longitudinal measures were also used in combination to examine inflammation in two ways. First, the cumulative concentration of CRP (log transformed) across the first, second, and third trimesters was approximated using linear mixed models. The linear mixed model was fitted with a random intercept and random centered week of CRP measurements. The model estimates also incorporated fixed effects including an intercept and a linear and quadratic term of centered week of CRP measurements, based on the regression curve of the original CRP concentrations. For each subject, parameters from the model were used to define the predicted CRP curves. The area below the curves was divided into 1000 parts. The area of each part was then calculated as a rectangle, since they are very small, and summed to obtain the cumulative CRP concentrations over pregnancy [47]. In addition, methylation was also examined by comparing two extreme groups of women that is women in the top tertile of CRP across all trimesters were compared with women in the bottom tertile across all trimesters. Cumulative and tertile analyses did not exclude maternal CRP levels above 10 mg/L to better characterize inflammation across the whole of pregnancy for the same woman. While the primary analysis excluded CRP levels above 10 to rule out outliers that would have strong influence on estimates based on linear regression models, it would have excluded too many women for cumulative or tertile analyses who just had one of the 3 with an “elevated” measure. The cumulative and tertile analyses were more robust to outlying CRP.
Modeling outcomes for EAGeR
Trimming of the outlying methylation values at IQR ± 3 × IQR (IQR = inter-quartile range) was performed after values were normalized. Complete case analyses were performed without imputation. Linear mixed effects models were used to test associations between methylation beta values at each CpG site and measured CRP at each time point and as cumulative or by extreme tertiles with adjustment for covariates. Batch effects (as covariates of chip and row) were accounted for through random effects. FDR-correction was applied to account for multiple testing.
Covariates for EAGeR
Covariates included maternal age (continuous, by self-report), smoking (yes/no by self-report), income (5 categories from ≥ $100,000, $75,000–99,999, $40,000–69,999, $20,000–19,999, ≤ $19,999 by self-report), pre-pregnancy BMI (continuous, by direct measure prior to pregnancy), plate (for batch effects), and with or without cell count distribution (in separate models). Rather than adjustment for self-reported ethnicity, 12 non-white participants were excluded from EAGeR analyses. Three models were run; Model 1 adjusting for covariates and two models additionally adjusting for cell type distribution using two separate reference panels. Model 2 adjusted for covariates and cell counts based on a cord blood reference using minfi in R [21], including B-cell, CD-4+ T cells, CD-8+ T cells, granulocytes, monocytes, NK-cells, and nucleated RBCs. A major difference previously identified between adult and cord blood cell count distribution in terms of DNA contribution from buffy coat samples is the proportion of nucleated red blood cells [21]. Model 3 used a more recent cord blood reference pooling together larger number of samples with available cell distribution information [22].
Meta-analysis
As part of the PACE consortium [20], three pregnancy cohorts (INMA [48], Generation R [49], PREDO [50]) provided results for the associations between CRP measured in first trimester samples and DNA methylation measured in cord blood at delivery adjusting for the covariates mentioned above including with and without adjustment for cell type using two different references as described above [21, 22]. A meta-analysis of results was conducted with EAGeR as the fourth cohort. Inverse-variance weighted fixed effects meta-analysis was performed among a total of 1603 newborns using the software METAL [51]. Specific analytical and laboratory methods are included in Supplemental materials for each of the cohorts.
Bioinformatics
All FDR-significant CpGs were searched in the UCSC Genome Browser for genes within 5 kb of the location provided by Illumina. Information on distance to the TSS or which region within a gene body the CpG is located was ascertained. Gene names were further updated using the NCBI Gene Database. To further understand functional enrichment, eFORGE (v2.0) was utilized [52].
Availability of data and materials
EAGeR data can be shared upon written request to the corresponding author. Data from the Generation R Study are available upon reasonable request to the director, Vincent Jaddoe (generation r@erasmusmc.nl), and subject to local rules and regulations. Due to ethical issues and consent, the PREDO datasets analyzed during the current study are not publicly available. However, an interested researcher can obtain a de-identified dataset after approval from the PREDO study board. Data requests may be subject to further review by the national register authority and by the ethical committees. Data can be obtained upon reasonable request from the PREDO study board (predo.study@helsinki. fi) or individual researchers.
Abbreviations
- CRP:
-
C-reactive protein
- hsCRP:
-
High-sensitivity C-reactive protein
- DNA:
-
Deoxyribonucleic acid
- EAGeR:
-
Effects of Aspirin in Gestation and Reproduction
- LDA:
-
Low-dose aspirin
- PACE:
-
Pregnancy and Childhood Epigenetic
- RBC:
-
Red blood cell
References
Bertran N, Camps J, Fernandez-Ballart J, Murphy MM, Arija V, Ferre N, et al. Evaluation of a high-sensitivity turbidimetric immunoassay for serum C-reactive protein: application to the study of longitudinal changes throughout normal pregnancy. Clinical chemistry and laboratory medicine : CCLM / FESCC. 2005;43(3):308–13.
Azizia MM, Irvine LM, Coker M, Sanusi FA. The role of C-reactive protein in modern obstetric and gynecological practice. Acta Obstet Gynecol Scand. 2006;85(4):394–401.
Sureshchandra S, Marshall NE, Wilson RM, Barr T, Rais M, Purnell JQ, et al. Inflammatory determinants of pregravid obesity in placenta and peripheral blood. Front Physiol. 2018;9:1089.
Sureshchandra S, Wilson RM, Rais M, Marshall NE, Purnell JQ, Thornburg KL, et al. Maternal pregravid obesity remodels the DNA methylation landscape of cord blood monocytes disrupting their inflammatory program. J Immunol. 2017;199(8):2729–44.
Hantsoo L, Kornfield S, Anguera MC, Epperson CN. Inflammation: a proposed intermediary between maternal stress and offspring neuropsychiatric risk. Biol Psychiatry. 2018.
Li Q, Wang YY, Guo Y, Zhou H, Wang X, Wang Q, et al. Effect of airborne particulate matter of 2.5mum or less on preterm birth: a national birth cohort study in China. Environ Int. 2018.
Pitiphat W, Gillman MW, Joshipura KJ, Williams PL, Douglass CW, Rich-Edwards JW. Plasma C-reactive protein in early pregnancy and preterm delivery. Am J Epidemiol. 2005;162(11):1108–13.
Boggess KA, Lieff S, Murtha AP, Moss K, Jared H, Beck J, et al. Maternal serum C-reactive protein concentration early in pregnancy and subsequent pregnancy loss. Am J Perinatol. 2005;22(6):299–304.
Tjoa ML, van Vugt JM, Go AT, Blankenstein MA, Oudejans CB, van Wijk IJ. Elevated C-reactive protein levels during first trimester of pregnancy are indicative of preeclampsia and intrauterine growth restriction. J Reprod Immunol. 2003;59(1):29–37.
Wolf M, Kettyle E, Sandler L, Ecker JL, Roberts J, Thadhani R. Obesity and preeclampsia: the potential role of inflammation. Obstet Gynecol. 2001;98(5 Pt 1):757–62.
Lapin B, Ownby D, Turyk M, Piorkowski J, Freels S, Chavez N, et al. Relationship between in utero C-reactive protein levels and asthma in at-risk children. Ann Allergy Asthma Immunol. 2015;115(4):282–7.
Sonnenschein-van der Voort AM, Jaddoe VW, Moll HA, Hofman A, van der Valk RJ, de Jongste JC, et al. Influence of maternal and cord blood C-reactive protein on childhood respiratory symptoms and eczema. Pediatr Allergy Immunol. 2013;24(5):469–75.
Fink NR, Chawes B, Bonnelykke K, Thorsen J, Stokholm J, Rasmussen MA, et al. Levels of systemic low-grade inflammation in pregnant mothers and their offspring are correlated. Sci Rep. 2019;9(1):3043.
Morales E, Guerra S, Garcia-Esteban R, Guxens M, Alvarez-Pedrerol M, Bustamante M, et al. Maternal C-reactive protein levels in pregnancy are associated with wheezing and lower respiratory tract infections in the offspring. Am J Obstet Gynecol. 2011;204(2):164 e1–9.
Murphy VE, Smith R, Giles WB, Clifton VL. Endocrine regulation of human fetal growth: the role of the mother, placenta, and fetus. Endocr Rev. 2006;27(2):141–69.
Ansar W. Biology of c reactive protein in health and disease. New York, NY: Springer Berlin Heidelberg; 2015. pages cm p.
Ligthart S, Marzi C, Aslibekyan S, Mendelson MM, Conneely KN, Tanaka T, et al. DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases. Genome Biol. 2016;17(1):255.
Myte R, Sundkvist A, Van Guelpen B, Harlid S. Circulating levels of inflammatory markers and DNA methylation, an analysis of repeated samples from a population based cohort. Epigenetics. 2019;14(7):649–59.
Sjaarda LA, Radin RG, Silver RM, Mitchell E, Mumford SL, Wilcox B, et al. Preconception low-dose aspirin restores diminished pregnancy and live birth rates in women with low-grade inflammation: a secondary analysis of a randomized trial. J Clin Endocrinol Metab. 2017;102(5):1495–504.
Felix JF, Joubert BR, Baccarelli AA, Sharp GC, Almqvist C, Annesi-Maesano I, et al. Cohort profile: Pregnancy And Childhood Epigenetics (PACE) consortium. Int J Epidemiol. 2018;47(1):22–3u.
Bakulski KM, Feinberg JI, Andrews SV, Yang J, Brown S, Stephanie LM, et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics. 2016;11(5):354–62.
Gervin K, Salas LA, Bakulski KM, van Zelm MC, Koestler DC, Wiencke JK, et al. Systematic evaluation and validation of reference and library selection methods for deconvolution of cord blood DNA methylation data. Clin Epigenetics. 2019;11(1):125.
Illumina I. Field guide to methylation methods: Illumina, Inc.; 2016 [updated 2/12/2020. Available from: https://www.illumina.com/content/dam/illumina-marketing/documents/products/other/field_guide_methylation.pdf.
Berglind D, Muller P, Willmer M, Sinha I, Tynelius P, Naslund E, et al. Differential methylation in inflammation and type 2 diabetes genes in siblings born before and after maternal bariatric surgery. Obesity (Silver Spring). 2016;24(1):250–61.
Su KY, Li MC, Lee NW, Ho BC, Cheng CL, Chuang YC, et al. Perinatal polychlorinated biphenyls and polychlorinated dibenzofurans exposure are associated with DNA methylation changes lasting to early adulthood: findings from Yucheng second generation. Environ Res. 2019;170:481–6.
Nielsen FR, Bek KM, Rasmussen PE, Qvist I, Tobiassen M. C-reactive protein during normal pregnancy. Eur J Obstet Gynecol Reprod Biol. 1990;35(1):23–7.
Kuzawa CW, Fried RL, Borja JB, McDade TW. Maternal pregnancy C-reactive protein predicts offspring birth size and body composition in metropolitan Cebu, Philippines. J Dev Orig Health Dis. 2017;8(6):674–81.
Lowe R, Gemma C, Beyan H, Hawa MI, Bazeos A, Leslie RD, et al. Buccals are likely to be a more informative surrogate tissue than blood for epigenome-wide association studies. Epigenetics. 2013;8:4.
Soegaard SH, Rostgaard K, Skogstrand K, Wiemels JL, Schmiegelow K, Hjalgrim H. Neonatal inflammatory markers are associated with childhood B-cell precursor acute lymphoblastic leukemia. Cancer Res. 2018;78(18):5458–63.
Kalva-Borato DC, Ribas JT, Parabocz GC, Borba LM, Maciel MAS, Santos FAD, et al. Biomarkers in non-complicated pregnancy: insights about serum myeloperoxidase and ultrasensitive C-reactive protein. Exp Clin Endocrinol Diabetes. 2019;127(9):585–9.
Stokkeland LMT, Giskeodegard GF, Stridsklev S, Ryan L, Steinkjer B, Tangeras LH, et al. Serum cytokine patterns in first half of pregnancy. Cytokine. 2019;119:188–96.
Logan CA, Thiel L, Bornemann R, Koenig W, Reister F, Brenner H, et al. Delivery mode, duration of labor, and cord blood adiponectin, leptin, and C-reactive protein: results of the population-based Ulm Birth Cohort studies. PLoS One. 2016;11(2):e0149918.
Malek A, Bersinger NA, Di Santo S, Mueller MD, Sager R, Schneider H, et al. C-reactive protein production in term human placental tissue. Placenta. 2006;27(6-7):619–25.
Sherrill HE, Jean P, Driver EC, Sanders TR, Fitzgerald TS, Moser T, et al. Pou4f1 Defines a subgroup of type I spiral ganglion neurons and is necessary for normal inner hair cell presynaptic Ca(2+) signaling. J Neurosci. 2019;39(27):5284–98.
Faryna M, Konermann C, Aulmann S, Bermejo JL, Brugger M, Diederichs S, et al. Genome-wide methylation screen in low-grade breast cancer identifies novel epigenetically altered genes as potential biomarkers for tumor diagnosis. FASEB J. 2012;26(12):4937–50.
Gonzalez-Jaramillo V, Portilla-Fernandez E, Glisic M, Voortman T, Ghanbari M, Bramer W, et al. Epigenetics and inflammatory markers: a systematic review of the current evidence. Int J Inflamm. 2019;2019:6273680.
Hanion-Lundberg KM, Kirby RS, Gandhi S, Broekhuizen FF. Nucleated red blood cells in cord blood of singleton term neonates. Am J Obstet Gynecol. 1997;176(6):1149–54 discussion 54-6.
Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, et al. Stereotypic immune system development in newborn children. Cell. 2018;174(5):1277–92 e14.
Pereza N, Ostojic S, Kapovic M, Peterlin B. Systematic review and meta-analysis of genetic association studies in idiopathic recurrent spontaneous abortion. Fertil Steril. 2017;107(1):150–9 e2.
Ahmed SK, Mahmood N, Malalla ZH, Alsobyani FM, Al-Kiyumi IS, Almawi WY. C-reactive protein gene variants associated with recurrent pregnancy loss independent of CRP serum levels: a case-control study. Gene. 2015;569(1):136–40.
Dwi Putra SE, Reichetzeder C, Hasan AA, Slowinski T, Chu C, Kramer BK, et al. Being born large for gestational age is associated with increased global placental DNA methylation. Sci Rep. 2020;10(1):927.
Schisterman EF, Silver RM, Lesher LL, Faraggi D, Wactawski-Wende J, Townsend JM, et al. Preconception low-dose aspirin and pregnancy outcomes: results from the EAGeR randomised trial. Lancet. 2014;384(9937):29–36.
Yeung EH, Guan W, Mumford SL, Silver RM, Zhang C, Tsai MY, et al. Measured maternal prepregnancy anthropometry and newborn DNA methylation. Epigenomics. 2019;11(2):187–98.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–9.
Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99(6):3740–5.
Touleimat N, Tost J. Complete pipeline for Infinium((R)) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4(3):325–41.
Ghassabian A, Albert PS, Hornig M, Yeung E, Cherkerzian S, Goldstein RB, et al. Gestational cytokine concentrations and neurocognitive development at 7 years. Transl Psychiatry. 2018;8(1):64.
Guxens M, Ballester F, Espada M, Fernandez MF, Grimalt JO, Ibarluzea J, et al. Cohort profile: the INMA--INfancia y Medio Ambiente--(Environment and Childhood) project. Int J Epidemiol. 2012;41(4):930–40.
Kooijman MN, Kruithof CJ, van Duijn CM, Duijts L, Franco OH, van IJzendoorn MH, et al. The Generation R Study: design and cohort update 2017. Eur J Epidemiol. 2016;31(12):1243–64.
Girchenko P, Lahti M, Tuovinen S, Savolainen K, Lahti J, Binder EB, et al. Cohort profile: prediction and prevention of preeclampsia and intrauterine growth restriction (PREDO) study. Int J Epidemiol. 2017;46(5):1380–1g.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–1.
Breeze CE, Reynolds AP, van Dongen J, Dunham I, Lazar J, Neph S, et al. eFORGE v2.0: updated analysis of cell type-specific signal in epigenomic data. Bioinformatics. 2019;35(22):4767–9.
Acknowledgements
INMA researchers would like to thank all the participants for their generous collaboration. A full roster of the INMA Project Investigators can be found at: http://www.proyectoinma.org/presentacioninma/listado-investigadores/en_listado-investigadores.html
Generation R Study: The Generation R Study is conducted by the Erasmus Medical Center in close collaboration with the School of Law and Faculty of Social Sciences of the Erasmus University Rotterdam; the Municipal Health Service Rotterdam area, Rotterdam; the Rotterdam Homecare Foundation, Rotterdam; and the Stichting Trombosedienst & Artsenlaboratorium Rijnmond (STAR-MDC), Rotterdam. We gratefully acknowledge the contribution of children and parents, general practitioners, hospitals, midwives, and pharmacies in Rotterdam. The study protocol was approved by the Medical Ethical Committee of the Erasmus Medical Centre, Rotterdam. Written informed consent was obtained for all participants. The generation and management of the Illumina 450K methylation array data (EWAS data) for the Generation R Study was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Netherlands. We thank Mr. Michael Verbiest, Ms. Mila Jhamai, Ms. Sarah Higgins, Mr. Marijn Verkerk, and Dr. Lisette Stolk for their help in creating the EWAS database. We thank Dr. A. Teumer for his work on the quality control and normalization scripts.
The PREDO study would not have been possible without the dedicated contribution of the PREDO study group members: E Hamäläinen, E Kajantie, H Laivuori, PM Villa, A-K Pesonen, A Aitokallio-Tallberg, A-M Henry, VK Hiilesmaa, T Karipohja, R Meri, S Sainio, T Saisto, S Suomalainen-Konig, V-M Ulander, T Vaitilo (Department of Obstetrics and Gynaecology, University of Helsinki and Helsinki University Central Hospital, Helsinki, Finland), L Keski-Nisula, Maija-Riitta Orden (Kuopio University Hospital, Kuopio Finland), E Koistinen, T Walle, R Solja (Northern Karelia Central Hospital, Joensuu, Finland), M Kurkinen (Päijät-Häme Central Hospital, Lahti, Finland), P.Taipale. P Staven (Iisalmi Hospital, Iisalmi, Finland), and J Uotila (Tampere University Hospital, Tampere, Finland). We thank all the PREDO children and their parents for their enthusiastic participation. We also thank all the research nurses, research assistants, and laboratory personnel involved in the PREDO study.
Funding
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (National Institutes of Health, Bethesda, MD, USA), and the EAGeR trial was specifically funded under contract numbers HHSN267200603423, HHSN267200603424, HHSN267200603426, and HHSN275201300023I-HHSN2750008. SL was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. Main funding of the epigenetic studies in INMA was grants from Instituto de Salud Carlos III (Red INMA G03/176, CB06/02/0041), Spanish Ministry of Health (FIS-PI04/1436, FIS-PI08/1151 including FEDER funds, FIS-PI11/00610, FIS-FEDER-PI06/0867, FIS-FEDER-PI03-1615, MS13/00054, CP18/00018), Generalitat de Catalunya-CIRIT 1999SGR 00241, Fundació La marató de TV3 (090430), EU Commission (261357-MeDALL: Mechanisms of the Development of ALLergy), and European Research Council (268479-BREATHE: BRain dEvelopment and Air polluTion ultrafine particles in scHool childrEn). The general design of the Generation R Study is made possible by financial support from the Erasmus Medical Center, Rotterdam; the Erasmus University Rotterdam; the Netherlands Organization for Health Research and Development; and the Ministry of Health, Welfare and Sport. The EWAS data was funded by a grant from the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO), Netherlands Consortium for Healthy Aging (NCHA; project nr. 050-060-810), by funds from the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, and by a grant from the National Institute of Child and Human Development (R01HD068437). This project received funding from the European Union’s Horizon 2020 research and innovation programme (633595, DynaHEALTH: 733206, LIFECYCLE) and from the European Joint Programming Initiative “A Healthy Diet for a Healthy Life” (JPI HDHL, NutriPROGRAM project, ZonMw the Netherlands no.529051022; and Precise project, ZonMw the Netherlands no. P75416). LD has received funding from the European Joint Programming Initiative. The PREDO study has been funded by the Academy of Finland, EraNet Neuron, EVO (a special state subsidy for health science research), University of Helsinki Research Funds, the Signe and Ane Gyllenberg Foundation, the Emil Aaltonen Foundation, the Finnish Medical Foundation, the Jane and Aatos Erkko Foundation, the Novo Nordisk Foundation, the Päivikki and Sakari Sohlberg Foundation, and the Sigrid Juselius Foundation granted to members of the PREDO study board. Methylation assays were funded by the Academy of Finland.
Author information
Authors and Affiliations
Contributions
EY drafted the initial manuscript. ES conceived, designed, and oversaw the EAGeR trial. ES, SM, and RS collected the EAGeR clinical data. WG, XZ, and LAS contributed to data analysis. JF and SL designed and oversaw PACE. PPB, AM, LD, DC, JL, and EM analyzed results in their respective cohorts. EY, JS, LD, KR, EH, EB, DC, JL, and JF designed and oversaw the epigenetic studies in their respective cohorts. All authors critically revised the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
IRB approved the EAGeR trial (Salt Lake City, Utah IRB #1002521), and all participants provided written informed consent prior to enrollment. The INMA study was conducted with the approval of the hospital ethics committee, and written informed consent was obtained from all women from INMA cohort located in Sabadell. The Generation R Study has been approved by the Medical Ethical Committee of the Erasmus MC, University Medical Center Rotterdam (MEC 198.782/2001/31). Informed consent was obtained for all participants. The PREDO study protocol was approved by the Ethics Committee of Obstetrics and Gynaecology and Women, Children, and Psychiatry of the Helsinki and Uusimaa Hospital District and by the participating hospitals. All participants provided written informed consent. Consent of participating children was provided by parent(s)/guardian(s).
Consent for publication
All authors contributed to the manuscript and approve its submission.
Competing interests
The authors report no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yeung, E.H., Guan, W., Zeng, X. et al. Cord blood DNA methylation reflects cord blood C-reactive protein levels but not maternal levels: a longitudinal study and meta-analysis. Clin Epigenet 12, 60 (2020). https://doi.org/10.1186/s13148-020-00852-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-020-00852-2