
Abstract
According to the “Developmental Origins of Health and Disease” (DOHaD) concept, the early-life environment is a critical period for fetal programming. Given the epidemiological evidence that air pollution exposure during pregnancy adversely affects newborn outcomes such as birth weight and preterm birth, there is a need to pay attention to underlying modes of action to better understand not only these air pollution-induced early health effects but also its later-life consequences. In this review, we give an overview of air pollution-induced placental molecular alterations observed in the ENVIRONAGE birth cohort and evaluate the existing evidence. In general, we showed that prenatal exposure to air pollution is associated with nitrosative stress and epigenetic alterations in the placenta. Adversely affected CpG targets were involved in cellular processes including DNA repair, circadian rhythm, and energy metabolism. For miRNA expression, specific air pollution exposure windows were associated with altered miR-20a, miR-21, miR-146a, and miR-222 expression. Early-life aging markers including telomere length and mitochondrial DNA content are associated with air pollution exposure during pregnancy. Previously, we proposed the air pollution-induced telomere-mitochondrial aging hypothesis with a direct link between telomeres and mitochondria. Here, we extend this view with a potential co-interaction of different biological mechanisms on the level of placental oxidative stress, epigenetics, aging, and energy metabolism. Investigating the placenta is an opportunity for future research as it may help to understand the fundamental biology underpinning the DOHaD concept through the interactions between the underlying modes of action, prenatal environment, and disease risk in later life. To prevent lasting consequences from early-life exposures of air pollution, policy makers should get a basic understanding of biomolecular consequences and transgenerational risks.
Similar content being viewed by others

Air pollution and the Developmental Origins of Health and Disease hypothesis
Air pollution is a global public health issue causing premature death and disease. It comprises different pollutants in gaseous (i.e., carbon oxides, nitrogen oxides, sulfur oxides, and ozone), volatile (i.e., ammonia, polycyclic aromatic hydrocarbons, and quinones), or particulate form (i.e., coarse, fine or ultrafine particles, and black carbon) derived from both natural and anthropogenic sources. In 2015, a component of air pollution, namely particulate matter smaller than 2.5 μm in aerodynamic diameter (PM2.5), was estimated to cause 4.2 million of deaths worldwide of which 202,000 children younger than 5 years [1]. Children are at higher risk of adverse health effects caused by air pollution, even at low levels, because their immune system and lungs are not fully developed, especially during in utero and early life [2].
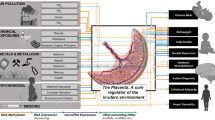
Life in utero is considered a particularly sensitive period during which maternal exposure to unfavorable conditions may not only influence fetal development and induce adverse pregnancy outcomes but also have long-term effects influencing offspring susceptibility to diseases later in adulthood, as postulated by the Developmental Origins of Health and Disease (DOHaD) hypothesis [3, 4]. Substantial evidence associates exposure to air pollution during pregnancy with a range of adverse health outcomes at birth, including increased risk of low birth weight [5,6,7] and prematurity [6, 8], and also in adult life, such as cardiovascular disease [9], respiratory problems [10,11,12], and neurodevelopmental alterations [13] and even cancers [14]. However, the biological chain of events through which exposure to air pollution in utero influences an individual’s later-life health is still poorly known. As the placenta is a crucial organ for fetal development, alterations in the placenta on the molecular level, induced by air pollution, may be important as to the early origins of health and disease. This altered biomolecular functioning of the placenta may contribute to early and even later-life health consequences. In this review, we describe all the available evidence of placental molecular processes associated with prenatal air pollution exposure in the ENVIRONAGE (ENVIRonmental influence ON AGEing in early life) birth cohort, situated in Belgium [15]. The biomolecular processes associated with air pollution exposure can be categorized into nitrosative stress, epigenetic alterations, and aging markers. Alterations in these placental molecular processes may lead to an altered newborn phenotype which may underlie a higher susceptibility for developing diseases later in life.
Can air pollution particles reach the human placenta?
In a recent review, numerous investigations examined whether (nano) particles, in general, can pass the placenta and showed a dependency on size, shape, and surface charge [16]. Furthermore, a study by Valentino et al. [17] strengthened the hypothesis of transplacental particle translocation by showing “nanoparticle-like” aggregates in the cytoplasm of placental trophoblastic cells of rabbits exposed to aerosolized diesel exhaust particles. While these experimental studies show that translocation across the placenta is biologically possible, no such direct evidence in the context of human life exists. Recently, we detected the abundant presence of black carbon (BC) particles in human placenta at both the maternal and fetal side (Fig. 1) [18]. These findings confirm that ambient particles can be translocated directly towards the fetus and represent a potentially novel mechanism explaining the adverse effects from early life onwards, in addition to particle-induced inflammation in the lungs. Furthermore, we showed that urinary carbonaceous particles reflect residential BC exposure and traffic-related exposure [19], showing the translocation of particles from the lung to the system.

Evidence of black carbon particles from ambient air pollution in human placenta. White-light generation by the black carbon particles (white and further indicated using arrowheads) under femtosecond pulsed laser illumination is observed. Second harmonic generation from collagen (red) and two-photon autofluorescence from placental cells (green) are detected simultaneously. Scale bar 40 μm [18]
Air pollution and placental oxidative/nitrosative stress
As air pollution particles may translocate into and cross the placental barrier [18, 20], they may induce placental modifications [21]. Oxidative stress may be one of the key elements of air pollution-induced placental alterations. Air pollution particles are able to generate reactive oxygen/nitrogen species (ROS/RNS) in both a direct and indirect way [22]. Particles may have free radicals present on their surface or may directly generate reactive hydroxyl radicals via a Fenton reaction in the presence of soluble transition metals on the particle surface, such as for instance iron [23]. One of the indirect sources of ROS production is through the PM-induced altered functioning of NADPH oxidases, telomere-mitochondrial dysregulation, and activation of inflammatory cells [22, 24]. These generated ROS can in turn directly induce lipid, protein, and DNA damage. Within the ENVIRONAGE birth cohort, we assessed placental nitrosative stress and mitochondrial 8-deoxyguanosine damage. We observed that each interquartile range (IQR) increment in entire pregnancy PM2.5 exposure resulted in a 35.0% (95% CI 13.9 to 60.0%) increase in placental 3-nitrotyrosine levels, whereas an IQR increase in BC showed a 13.9% (95% CI − 0.21 to 29.9%) increase [25]. Direct oxidative damage in mitochondria was measured by 8-hydroxy-2′-deoxyguanosine (8-OHdG) levels in cord blood and maternal blood samples. Interestingly, we observed that elevated exposure to PM10 during trimester 1 and 2 of pregnancy resulted in an increase in mitochondrial 8-OHdG, while results for PM2.5 were less pronounced [26]. Whether this observation is reflective of placental mitochondrial damage remains to be elucidated. Within this regard, a study of 891 newborns of the Czech Republic showed increased 8-OHdG in placenta in association with exposure to PM2.5 in the first 4 months of pregnancy [27]. These authors also observed that newborns with above-median levels of 8-OHdG had higher probability of intrauterine growth restriction compared with newborns below-median level of oxidative DNA damage.
Taken together, these findings demonstrate that air pollution-related ROS/RNS production may affect the in utero environment. This can be supported by previous studies investigating smoking during pregnancy [28] and environmental toxic metals [29] in association with oxidative stress in the placenta.
Air pollution-induced placental epigenetic alterations
Placental epigenetics is another important target to study early-life effects of air pollution, which includes changes in DNA methylation, histone and noncoding RNA modification, and chromatin remodeling. These processes are able to influence health outcomes during the life course and even across generations [30,31,32]. During pregnancy, especially DNA methylation is an important mechanism as it is involved in “epigenetic reprogramming”. During this process, DNA methylation patterns are erased and re-established, first in gametogenesis and again in early embryogenesis [33]. These epigenetic waves make the early embryonic development a critical period [34]. Within the ENVIRONAGE birth cohort, we have studied placental epigenetic signatures in association to air pollution on different levels, from global DNA methylation, gene-specific DNA methylation to miRNA expression, and we focused both on nuclear and mitochondrial DNA (mtDNA) targets. Our findings from epigenetic alterations induced by air pollution exposure in the ENVIRONAGE birth cohort are summarized in Table 1, in addition to other available evidence.
Placental global DNA methylation
In 2013, we were the first to show in 240 mother-newborn pairs that placental global DNA methylation was inversely associated with first trimester PM2.5 exposure, especially during the critical period of implantation [35]. These findings were confirmed by another study which showed that pregnant mothers living close to major roadways (i.e., a marker of traffic-related air pollution) had lower levels of placental DNA methylation in LINE-1 but not AluYb8, which are surrogate markers of global DNA methylation [36]. Furthermore, in a Chinese study involving 181 mother-newborn pairs (80 fetal growth restriction newborns, 101 normal weight newborns), placental LINE-1 DNA methylation was inversely associated with first trimester PM10 exposure [37]. In contrast, a nested case-control study (n = 100) in Iran showed a positive correlation of global methylation with first trimester PM2.5 and PM10 exposure [38]. It should be mentioned that the Iran study did not use the same technique nor investigated LINE-1 DNA methylation. Furthermore, the EDEN cohort showed a positive association of placental Alu DNA methylation with day before birth PM10 exposure but not with placental LINE-1 DNA methylation [39]. Nevertheless, these studies highlight that air pollution exposure already has an important impact on methylation patterns very early in embryonic development, directly after conception. This may be critical in development as it has been shown in mice that disturbances of DNA methylation in the placenta are associated with abnormal embryonic development [40] and that genetic inactivation of DNA methyltransferases (DNMTs) is lethal to developing mouse embryos [41].
Placental candidate gene methylation
We have carried out different candidate gene methylation studies (Table 1). The rationale of these studies was based on the DOHaD hypothesis, in which we focused on key biological processes that are involved both in growth and development early in life and in age-related diseases later in life. We analyzed in the placentas of 407 newborns the promoter regions of regulatory genes in the circadian pathway (i.e., the central biological clock that maintains the daily cellular rhythm in accordance with the external environment). PM2.5 exposure during the last trimester of pregnancy was positively associated with placental methylation of CLOCK, BMAL1, NPAS2, CRY1-2, and PER1-3 [42]. Previous findings stipulated that dysfunctions in the clock mechanism are prevalent in a variety of diseases, such as cancer, metabolic conditions, and neurological disorders [43, 44].
Further, we evaluated whether promoter regions of key DNA repair genes (including base-excision and nucleotide-excision repair genes) and tumor suppressor genes were differentially methylated in the placenta (n = 463). Higher entire pregnancy PM2.5 exposure was positively associated with methylation of the promoter regions from repair genes (APEX1, OGG1, ERCC4) and from the tumor suppressor p53, whereas promoter methylation of DAPK1 was inversely associated. Similar findings were observed for APEX1 and ERCC1 in association with BC exposure [45]. In addition, we found that increased levels of both PM2.5 and BC were positively associated with higher mutation rates in placental DNA. These findings are in line with a study of Perera and colleagues [46], who showed that air pollution can induce aromatic DNA adducts in cord blood, and with an experimental study of Zhou and colleagues [47], in which hypermethylation of p53 was shown in human bronchial epithelial cells after 10 days of PM2.5 exposure.
We also observed an inverse association between placental LEP promoter methylation (i.e., an energy-regulating hormone involved in fetal growth and development) and PM2.5 exposure during the second trimester of pregnancy [48]. Additionally, this association was strengthened by the determination of the oxidative/nitrosative stress biomarker 3-nitrotyrosine (3-NTp) [48], which showed a similar association as the modeled PM2.5 exposures, that was independent of maternal smoking.
Aside from the ENVIRONAGE birth cohort, further evidence was found by Cai et al. [37] showing that exposure to PM10 during the first two trimesters of pregnancy was positively associated with placental methylation of HSD11B2 (i.e., genes involved in the glucocorticoid metabolism and fetal growth). The observed associations were more pronounced in the fetal growth-restricted newborn subset (n = 80), compared to the normal growth newborns (n = 101).
Finally, we evaluated methylation of two regions of the mtDNA, i.e., D-loop control region and the 12S rRNA. In a study sample of 381 mother-newborn pairs, higher methylation levels of these two mitochondrial genome regions in association with prenatal PM2.5 exposure were observed, with the first trimester as most critical exposure window [49]. A higher methylation of mtDNA in relation to air pollution exposure is in accordance with a study in the blood of steelworkers [50].
Altogether, these candidate-based methylation studies show that mapping placental epigenome modifications attributable to air pollution offers a unique opportunity to unravel biomolecular signatures playing a potential role in the mediation of air pollution influence on postnatal life [51].
Placental miRNA expression
In placental tissue, microRNA expression has been observed both in extracellular vesicles and in tissue biopsies. Several studies showed the ability of microRNA expression patterns to link pregnant women who were at risk of developing preeclampsia [52], preterm birth [53], or fetal growth restriction [54]. Furthermore, microRNA expression patterns in placental tissue have been shown to act in a sexually dimorphic manner in relation to both maternal obesity [55] and placental stress [56].
Until now, most studies focused on chemical exposures including cadmium [57], phthalates [58], arsenic [59], and endocrine disrupting chemicals [60] in association with placental miRNAs. Moreover, multiple studies confirm a role for miRNAs in the response to air pollution exposure in adults [61,62,63]. However, the literature on air pollution exposure in association with placental or, more broadly, early in life microRNA expression in humans is limited.
The first evidence for a role of miRNA expression in response to air pollution exposure in prenatal life came from an animal study [64]. In this study, pregnant rats were exposed to PM2.5 for extended periods of time, causing an increased number of immune cells in mother rats. Expression levels of cerebral cortical miR-6315, miR-3588, and miR-466b-5p were upregulated while a decreased expression of miR-338-5p and let-7e-5p was observed. Further, PM2.5 exposure increased miR-3560 and let-7b-5p in the hippocampus, while miR-99b-5p, miR-92b-5p, and miR-99a-5p were decreased. All of these miRNAs were related to neurobiological processes [64].
We were the first human cohort (ENVIRONAGE) on air pollution exposure and placental miRNA expression in which we investigated six miRNAs (miR-16, miR-20a, miR-21, miR-34a, miR-146a, miR-222) in 210 placenta samples (Table 1). These miRNAs are involved in important cellular processes such as cell cycle, proliferation, apoptosis, inflammation, and angiogenesis. A positive association with first-trimester PM2.5 exposure was identified for placental miR-20a expression, whereas second-trimester exposure was negatively associated with the expression of miR-21a, miR-146a, and miR-222. Furthermore, first-trimester PM2.5 exposure was positively associated with miR-21 expression, whereas it was negatively associated with second-trimester PM2.5 exposure. Tumor suppressor phosphatase and tensin homolog (PTEN) was identified as a common target of the miRNAs significantly associated with PM exposure [65].
One other study used cord blood as biological sample for miRNA expression and smoking status as exposure. They investigated whether miR-155 and miR-233 expression in 450 cord blood and maternal blood samples from the LINA (Lifestyle and Environmental Factors and Their Influence on Newborns Allergy Risk) study was associated with smoking behavior during pregnancy [66]. They found that increased maternal urinary cotinine concentrations (i.e., a marker for short-term smoking exposure) during pregnancy were associated with elevated miR-223 expression in cord blood. Cord blood miR-155 expression was related to lower toluene metabolite S-benzylmercapturic acid concentrations in maternal urine. Moreover, they demonstrated in newborns that a high miR-223 expression in cord blood cells was associated with lower cord blood regulatory T cell numbers.
Air pollution exposure and the aging phenotype
Besides epigenetic alterations in association with air pollution exposure, more downstream placental targets to evaluate the potential impact of air pollution in the DOHaD hypothesis were evaluated in the ENVIRONAGE birth cohort. We have a profound interest in aging-related targets, including telomeres and mitochondria. This is because of the fact that these targets have been widely studied in adult populations and have been associated with age-related diseases. Telomeres shorten throughout the life-span, and this shortening may be influenced by environmental factors, including air pollution [67]. Telomeres play a role in cell senescence and human aging and are indicative of disease risks, and in this regard, short telomeres have independently of chronological age been associated with higher risks for cardiovascular disease [68], type 2 diabetes [69], respiratory diseases [70], and mortality [71]. In addition, mitochondrial dysfunction and mutations play an import role in neurodegenerative diseases [72], cardiovascular diseases [73], and aging [74].
In the ENVIRONAGE birth cohort, we observed for 174 newborns a decrease of 17.4% in placental mtDNA content for a 10-μg/m3 increment in PM10 exposure during the third trimester of pregnancy [75]. Furthermore, in a larger subset of 381 individuals, we observed that an increment of 10-μg/m3 in PM2.5 during the third trimester was associated with a decrease of 23.6% in mtDNA content [49]. In 2017, we showed that placental telomere length (TL) was negatively associated with PM2.5 exposure during weeks 15–27 of gestation [76]. In this study, we applied a distributed lag model which enabled us to investigate weekly exposures during pregnancy in association with placental TL. The estimated effect of a 5-μg/m3 increase in PM2.5 during the second trimester and entire pregnancy was associated with 7.1% and 13.2% shorter placental TL, respectively. Furthermore, shorter placental TL has been observed with increased residential proximity to a major road and a decreased residential greenness [77], and with prenatal cadmium exposure [78].
The importance of placental TL for later-life conditions is rather unclear. However, if placental TL relates to cell senescence, this may influence placental aging, with health consequences on the short- and potential long-term. In this regard, it has been shown that placental senescence is observed in placentas complicated with intrauterine growth restriction or preeclampsia, and indeed, shorter placental TL was observed in these conditions [78]. This directly impacts fetal development and outcomes. In uncomplicated pregnancies, a high variability in placental TL is observed [79], and although these pregnancies may result in a healthy newborn phenotype, later-life consequences may be programmed at the level of telomeres. Indeed, it has been shown that placental TL may predict later-life TL [80], and therefore, changes in placental TL may be predictive for later-life risks in telomere length-associated diseases. However, prospective follow-up studies are needed to confirm whether newborn TL indeed reflects later-life disease risks.
Interplay of oxidative stress, epigenetics, and the air pollution-induced telomere/mitochondrial axis of aging
We previously proposed the air pollution-induced telomere-mitochondrial aging hypothesis [24], with its fundamental basis on findings of a direct link between mitochondria and telomeres [81, 82]. Telomeres are highly sensitive to ROS, and air pollution has shown to increase levels of ROS, which may target telomeres, and shorten them leading to potential dysfunctional telomeres (Fig. 2). Telomere dysfunction in mice showed p53 activation which resulted in suppression of peroxisome proliferator-activated receptor gamma (Pparγ) co-activator 1 alpha and beta (Pgc-1α,β) genes [81]. Repression of Pgc-1α,β leads to a strong decrease in mitochondrial biogenesis and function, subsequently leading to an impaired ATP generation and an increase in ROS production. Alterations in the energy metabolism are a driver of the aging process. Furthermore, DNA damage at telomeres activates several signaling pathways and reduces Sirt1 gene expression, which leads to mitochondrial dysfunction, partially through elevated p53 and reduced PGC [83]. This indicates an intimate relationship and interaction between telomeres and mitochondria.

An extended view of the air pollution-induced telomere-mitochondrial aging hypothesis. Our previous hypothesis showed that the presence of air pollution-induced ROS within cells induces DNA damage which leads to telomere shortening. Both DNA damage and telomere shortening are associated with increased levels of p53, which on its turn leads to increased mitochondrial dysfunction. Furthermore, disturbances in mitochondria can also increase cellular ROS production. We extended this view with epigenetic regulation. A dynamic regulation exists between epigenetic marks and TL. High trimethylated histones at the subtelomeric and telomeric region as well as high subtelomeric DNA methylation by DNMTs are a negative regulator of TL. Additionally, shortening of telomeres leads to a decrease in both histone trimethylation and subtelomeric DNA methylation and global DNA methylation (Alu, LINE-1). Furthermore, microRNAs might be involved through DICER1 regulation that is linked to DNMT expression and on its turn affects methylation processes of the genome and subtelomeric regions. Finally, miRNAs are also under the regulation of both DNA methylation and p53. Both p53 and DICER1 may be under regulation of ROS
However, extending this view with epigenetic regulation of TL and mitochondria may be essential in understanding air pollution-induced placental molecular alterations as shown in the ENVIRONAGE study (Fig. 2). In this regard, clear evidence is available that a dynamic regulation of epigenetic marks and TL is present, because both epigenetic marks may influence TL regulation and homeostasis, but vice versa telomere shortening may alter epigenetic marks. In this regard, it has been shown that telomeric and subtelomeric regions are enriched in trimethylated histones H3K9me3 and H4K20me3 (trimethylation of histone H3 at lysine 9 and of histone H4 at lysine 20), and subtelomeric regions are highly methylated by DNMT1, DNMT3a, and DNMT3b enzymes [84]. This high DNA and histone methylation state has shown to be a negative regulator of TL [84], as cells deficient in DNMTs displayed a strong decrease of subtelomeric DNA methylation and showed elongated telomeres, potentially due to telomerase, and increase telomere recombination [85]. On the other hand, TL may influence the epigenetic landscape. As telomeres shorten, this may lead to a decrease in trimethylation of H3K9 and H4K20 in the telomeric and subtelomeric region as well as a decrease in subtelomeric DNA methylation as shown in telomerase-deficient Terc−/− mice experiments with short telomeres [86]. However, subsequently, this may lead to telomere elongation and maintenance processes as described above. In human population-based studies, lower global DNA methylation (LINE-1 and Alu) has been associated with shorter telomeres [87, 88].
Within the context of air pollution actions on the epigenetic landscape, several theories exist how changes in DNA methylation status can be induced: (1) ROS generated by air pollution-induced oxidative stress can react with DNA, resulting into different DNA lesions, including base modifications, strand breaks, and inter- and intra-strand crosslinks [89]. Due to these DNA alterations, DNMTs are not able to recognize this damaged DNA as a reaction substrate, which leads to a global hypomethylation [90]. Additionally, it has been shown that the repair of damaged DNA by homologous recombination (HR) induces DNA methylation [91], and chromatin, damaged by oxidative stress, recruits DNMT1, which results in DNA methylation changes [92]. One of the most frequently occurring ROS-generated DNA lesions is the oxidation of guanine, resulting in the formation of 8-OHdG [93]. The presence of an 8-OHdG residue inhibits the ability of DNMT to methylate nearby located cytosines [94]. Furthermore, ROS can increase 5mC oxidation via 5hMC, which eventually leads to global hypomethylation [95]. (2) Environmental chemicals may interfere with S-adenosyl methionine (SAM) which results in a methylation reduction by DNMT due to a depletion of available methyl groups from SAM [96, 97]. Furthermore, DNMT gene expression is reduced by long-term environmental exposures [98, 99]. (3) Another suggested mechanism is the so-called transcription factor occupancy theory, in which an interplay between the presence or absence of transcription factors (TF) and the degree of gene-specific DNA methylation exists [96]. In this regard, research by Martin and Fry [100] showed that genes of which the methylation status (evaluated in cord blood or placenta) was associated with prenatal exposures (including arsenic, cadmium, lead, manganese, mercury, and tobacco smoke), shared binding sites for TFs that had a known relationship with these prenatal exposures.
A role of microRNAs in the regulation of DNA methylation and telomeres has also been shown (Fig. 2). In Dicer1-deficient mice, a downregulation of the miR-290 cluster was observed. This downregulation leads to an increase in mRNA levels of Rbl2 (retinoblastoma-like 2 protein) that subsequently inhibits DNMT expression. This decrease in DNMT results in a hypomethylation of the genome and subtelomeric regions, leading to the aforementioned increase in TL and telomere recombination [101]. In this regard, we could evaluate in a small sub-population of the ENVIRONAGE birth cohort that placental TL was associated with miRNA expression. More precisely, we observed that miR-34a, miR-146a, miR-210, and miR-222 expression was positively associated with placental TL in newborn girls [102]. However, in this small sub-population (n = 203), the mediating effect of miRNA expression in the association between air pollution and TL could not be evaluated. Nevertheless, as high exposure to PM2.5 during second trimester was associated with both shorter placental TL and a reduced miR-146a and miR-222 expression, and both miRNAs were associated with longer placental TL in girls, these miRNA targets may warrant further attention. Also, in these miRNA-air pollution associations, ROS may play an important role [103]. First, ROS could act on the biogenesis enzymes of miRNAs, and it has been shown that H2O2-treated JAR trophoblast cells selectively inhibited Dicer activity [104, 105]. Second, ROS may regulate miRNA expression through the alteration of transcription factors, including p53 and NF-κB [103]. p53 is a major tumor suppressor involved in cellular senescence and is assumed to play a pivotal role in our proposed “hypothesis” [24]. Air pollution exposure may alter the expression of p53, and recent studies showed that p53, as a transcription factor, is an important regulator of miRNA expression [106]. Therefore, p53 may be an important target which links air pollution and miRNA regulation with a potential implication as to the aging phenotype and later-life diseases. Finally, miRNA genes are by themselves under the regulation of DNA methylation [103], and therefore, the air pollution/ROS-related DNA methylation regulatory mechanisms may affect miRNA expression, indicating a close interplay between the different epigenetic mechanisms.
Aside from nuclear DNA, it should be noted that mitochondria are the second cellular location to house an abundance of DNA (mtDNA). During recent years, it has been shown that the mitochondrial genome can also undergo epigenetic modifications. Within the ENVIRONAGE birth cohort, we were able to evaluate that indeed an important interplay exists between placental mtDNA content and mtDNA methylation. We observed that the effect of prenatal PM2.5 exposure on placental mtDNA content was mediated for 54% by mitochondrial 12S RNA methylation and for 27% by mitochondrial D-loop methylation [49]. Although studies on mitochondrial epigenetics are still in its infancy, PM2.5 exposure may be a potential candidate with important links to mitochondrial epigenetics [107].
Challenges and opportunities for using the placenta in early-life environmental exposure research
The findings we reviewed in this paper underscore the sensitivity of the biomolecular system to environmental factors during the early period of developmental plasticity. Methylation patterns are re-established during early pregnancy, making this a highly sensitive window of susceptibility to the effects of prenatal air pollution exposure. This may lead to an adaptive response altering placental and fetal development with possibly a long-lasting impact in later life. However, the crucial question remains about the time window in which air pollution exposure influences biomolecular processes during pregnancy as measurements on placenta can only be performed at birth. In this respect, it is noteworthy that in observational studies, the conventional approach of averaging exposures over relatively large time windows (trimesters or the entire pregnancy) can be further refined by using distributed lag models to allow a more detailed investigation of prenatal exposure windows and enable the identification of critical periods during pregnancy for the association with air pollution exposure [76, 108].
Until now, most studies investigating early-life air pollution exposure and placental alterations have focused on DNA methylation, while we were the first investigating microRNAs and even aging-related markers. This can probably be explained by the interpretation capability and the availability of high-throughput laboratory techniques [109]. But even for biomolecular processes, changes need to be interpreted in the context of their biological relevance. For example, although DNA methylation is usually associated with alterations in gene expression [110], it is not known whether small changes in the methylation status of a given promoter necessarily translate into an alteration in gene expression [111, 112]. Furthermore, establishing a cutoff value for differential DNA methylation as biologically relevant is difficult, as this can depend on the type of study, sample size, heterogeneity of the tissue, the method or technique used, or even interpretation of the data. In this respect, larger differences are desirable between cases and controls for a certain disease phenotype, whereas for epidemiological studies, subtle changes in DNA methylation levels can have a functional meaning by revealing biological pathways involved in disease development or to unravel underlying mechanisms of action. In the ENVIRONAGE birth cohort, the reported associations of air pollution exposure with biomolecular processes are generally low in terms of the size of the estimate, although they are significant in terms of nominal p values [113]. To prevail false-positive or false-negative findings, studies should participate in consortia to analyze their findings to improve the generalization of the results [30]. Within this context, the Pregnancy and Childhood Epigenetics cohort (PACE [114], n > 29,000) investigates the cord blood methylome. Similar approaches for placenta epigenetics are currently initiated but must carefully consider differences in the definition of exposures, biological sampling, laboratory techniques, and demographic and lifestyle characteristics of the study population.
Another challenge is that each molecular layer will not only interact with themselves but also display reciprocal relations with other biological networks as discussed in this review. Therefore, studies would benefit from using multi-omics approaches in which different molecular levels are integrated. So far, a few studies in adults included more than one molecular layer of epigenetics or other omics, for example by combining DNA methylation with the transcriptome [115], with inflammatory protein levels [115, 116], or with genetic variation [117].
In addition, the heterogeneity of the biological sample is also challenging when using the placenta. Gene regulation is tissue-, cell-, and context-specific, giving rise to cell-to-cell variation. Cellular composition explains a large part of the observed variability in gene regulation; thus, failing to account for the cellular heterogeneity may result in false-positive outcomes [109]. Measuring the cellular composition would be ideal but is in practice not always feasible. In the past years, efforts have been made to establish algorithms that predict peripheral and cord blood cell composition [118, 119]; however, for placenta, this is not available. Expanding these algorithms to underexplored tissues such as placenta will improve interpretation of results with regard to environmental exposures on placental molecular alterations.
Although molecular alterations are at times an adaptive reaction rather than a cause of disease, we now know that these changes may play an important role in diseases, including cancer, and occur long before disease develops. Epidemiological evidence demonstrated the role of diet and stress in changing the epigenetic program over generations. Examples of this are evidenced in the Dutch hunger winter study [120] and Holocaust survivors and their offspring [121]. These extreme living conditions entailed permanent changes in the epigenetic make-up, and among similar lines, evidence is accumulating that this occurs also for less stringent environmental conditions or exposures. Epigenetic analysis, as demonstrated in our overview, can be used to assess in utero and transgenerational effects. Therefore, epigenetics can lead us to understand the fundamental biology underpinning Barker’s DOHaD hypothesis in terms of interactions between the genome, prenatal environment, and disease risk. For example, Janssen et al. [35] showed placental hypomethylation in association with prenatal PM exposure. Furthermore, the observations of transplacental carcinogenesis by air pollution-induced ALU mutation rate parallels changes in the methylation of genes involved in the DNA repair machinery [45].
While we are only at the beginning to understand transplacental mechanisms, the evidence is mounting that prenatal exposure to ambient air pollution, oxidative stress, epigenetic alterations in DNA repair genes, metabolic genes, and changes in biological aging processes are all molecular processes involved in age-related diseases including cancer. The strength of molecular epidemiology within environmental health is the progress it offers in the understanding of fetal programming and the unraveling of the complex interplay between external and biological factors in order to increase our knowledge about DOHaD in terms of diseases at older age.
Developmental vulnerability should be a priority for environmental public health policies and practices to protect the most susceptible period of human life due to the long-term consequences. Follow-up of child or birth cohorts is crucial to understand the clinical consequences of early-life epigenetic changes on sub-optimal organ development resulting in a decreased reserve capacity of different organ systems and its risk later in life. The strategy aiming at effective protection of pregnant women, unborn children, and infants against lifelong consequences of exposure to combinations of adverse lifestyle factors requires that public health policy makers should get a basic understanding of epigenetic consequences and transgenerational risks.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Cohen AJ, Brauer M, Burnett R, Anderson HR, Frostad J, Estep K, Balakrishnan K, Brunekreef B, Dandona L, Dandona R, Feigin V, Freedman G, Hubbell B, Jobling A, Kan H, Knibbs L, Liu Y, Martin R, Morawska L, Pope CA 3rd, Shin H, Straif K, Shaddick G, Thomas M, van Dingenen R, van Donkelaar A, Vos T, Murray CJL, Forouzanfar MH. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet. 2017;389(10082):1907–18.
Schwartz J. Air pollution and children’s health. Pediatrics. 2004;113(4 Suppl):1037–43.
Barker DJ. Fetal nutrition and cardiovascular disease in later life. Br Med Bull. 1997;53(1):96–108.
Godfrey KM, Barker DJ. Fetal programming and adult health. Public Health Nutr. 2001;4(2b):611–24.
Pedersen M, Giorgis-Allemand L, Bernard C, Aguilera I, Andersen AM, Ballester F, Beelen RM, Chatzi L, Cirach M, Danileviciute A, Dedele A, Eijsden M, Estarlich M, Fernandez-Somoano A, Fernandez MF, Forastiere F, Gehring U, Grazuleviciene R, Gruzieva O, Heude B, Hoek G, de Hoogh K, van den Hooven EH, Haberg SE, Jaddoe VW, Klumper C, Korek M, Kramer U, Lerchundi A, Lepeule J, Nafstad P, Nystad W, Patelarou E, Porta D, Postma D, Raaschou-Nielsen O, Rudnai P, Sunyer J, Stephanou E, Sorensen M, Thiering E, Tuffnell D, Varro MJ, Vrijkotte TG, Wijga A, Wilhelm M, Wright J, Nieuwenhuijsen MJ, Pershagen G, Brunekreef B, Kogevinas M, Slama R. Ambient air pollution and low birthweight: a European cohort study (ESCAPE). Lancet Respir Med. 2013;1(9):695–704.
Li X, Huang S, Jiao A, Yang X, Yun J, Wang Y, Xue X, Chu Y, Liu F, Liu Y, Ren M, Chen X, Li N, Lu Y, Mao Z, Tian L, Xiang H. Association between ambient fine particulate matter and preterm birth or term low birth weight: an updated systematic review and meta-analysis. Environ Pollut. 2017;227:596–605.
Dadvand P, Parker J, Bell ML, Bonzini M, Brauer M, Darrow LA, Gehring U, Glinianaia SV, Gouveia N, Ha EH, Leem JH, van den Hooven EH, Jalaludin B, Jesdale BM, Lepeule J, Morello-Frosch R, Morgan GG, Pesatori AC, Pierik FH, Pless-Mulloli T, Rich DQ, Sathyanarayana S, Seo J, Slama R, Strickland M, Tamburic L, Wartenberg D, Nieuwenhuijsen MJ, Woodruff TJ. Maternal exposure to particulate air pollution and term birth weight: a multi-country evaluation of effect and heterogeneity. Environ Health Perspect. 2013;121(3):267–373.
Klepac P, Locatelli I, Korosec S, Kunzli N, Kukec A. Ambient air pollution and pregnancy outcomes: a comprehensive review and identification of environmental public health challenges. Environ Res. 2018;167:144–59.
Breton CV, Mack WJ, Yao J, Berhane K, Amadeus M, Lurmann F, Gilliland F, McConnell R, Hodis HN, Kunzli N, Avol E. Prenatal air pollution exposure and early cardiovascular phenotypes in young adults. PLoS One. 2016;11(3):e0150825.
Bharadwaj P, Zivin JG, Mullins JT, Neidell M. Early-life exposure to the Great Smog of 1952 and the development of asthma. Am J Respir Crit Care Med. 2016;194(12):1475–82.
Schultz ES, Hallberg J, Bellander T, Bergstrom A, Bottai M, Chiesa F, Gustafsson PM, Gruzieva O, Thunqvist P, Pershagen G, Melen E. Early-life exposure to traffic-related air pollution and lung function in adolescence. Am J Respir Crit Care Med. 2016;193(2):171–7.
Hehua Z, Qing C, Shanyan G, Qijun W, Yuhong Z. The impact of prenatal exposure to air pollution on childhood wheezing and asthma: a systematic review. Environ Res. 2017;159:519–30.
Chiu YH, Hsu HH, Coull BA, Bellinger DC, Kloog I, Schwartz J, Wright RO, Wright RJ. Prenatal particulate air pollution and neurodevelopment in urban children: examining sensitive windows and sex-specific associations. Environ Int. 2016;87:56–65.
Lavigne E, Belair MA, Do MT, Stieb DM, Hystad P, van Donkelaar A, Martin RV, Crouse DL, Crighton E, Chen H, Brook JR, Burnett RT, Weichenthal S, Villeneuve PJ, To T, Cakmak S, Johnson M, Yasseen AS 3rd, Johnson KC, Ofner M, Xie L, Walker M. Maternal exposure to ambient air pollution and risk of early childhood cancers: a population-based study in Ontario, Canada. Environ Int. 2017;100:139–47.
Janssen BG, Madhloum N, Gyselaers W, Bijnens E, Clemente DB, Cox B, Hogervorst J, Luyten L, Martens DS, Peusens M, Plusquin M, Provost EB, Roels HA, Saenen ND, Tsamou M, Vriens A, Winckelmans E, Vrijens K, Nawrot TS. Cohort Profile: The ENVIRonmental influence ON early AGEing (ENVIRONAGE): a birth cohort study. Int J Epidemiol. 2017;46(5):1386-7 m.
Muoth C, Aengenheister L, Kucki M, Wick P, Buerki-Thurnherr T. Nanoparticle transport across the placental barrier: pushing the field forward. Nanomedicine (London, England). 2016;11(8):941–57.
Valentino SA, Tarrade A, Aioun J, Mourier E, Richard C, Dahirel M, Rousseau-Ralliard D, Fournier N, Aubriere MC, Lallemand MS, Camous S, Guinot M, Charlier M, Aujean E, Al Adhami H, Fokkens PH, Agier L, Boere JA, Cassee FR, Slama R, Chavatte-Palmer P. Maternal exposure to diluted diesel engine exhaust alters placental function and induces intergenerational effects in rabbits. Part Fibre Toxicol. 2016;13(1):39.
Bove H, Bongaerts E, Slenders E, Bijnens EM, Saenen ND, Gyselaers W, Van Eyken P, Plusquin M, Roeffaers MBJ, Ameloot M, Nawrot TS. Ambient black carbon particles reach the fetal side of human placenta. Nat Commun (Accepted. 2019). https://doi.org/10.1038/s41467-019-11654-3
Saenen ND, Bove H, Steuwe C, Roeffaers MBJ, Provost EB, Lefebvre W, Vanpoucke C, Ameloot M, Nawrot TS. Children’s urinary environmental carbon load. A novel marker reflecting residential ambient air pollution exposure? Am J Respir Crit Care Med. 2017;196(7):873–81.
Wick P, Malek A, Manser P, Meili D, Maeder-Althaus X, Diener L, Diener PA, Zisch A, Krug HF, von Mandach U. Barrier capacity of human placenta for nanosized materials. Environ Health Perspect. 2010;118(3):432–6.
Carvalho MA, Bernardes LS, Hettfleisch K, Pastro LD, Vieira SE, Saldiva SR, Saldiva PH, Francisco RP. Associations of maternal personal exposure to air pollution on fetal weight and fetoplacental Doppler: a prospective cohort study. Reprod Toxicol (Elmsford, NY). 2016;62:9–17.
Risom L, Moller P, Loft S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat Res. 2005;592(1-2):119–37.
Ghio AJ, Carraway MS, Madden MC. Composition of air pollution particles and oxidative stress in cells, tissues, and living systems. J Toxicol Environ Health B Crit Rev. 2012;15(1):1–21.
Martens DS, Nawrot TS. Air pollution stress and the aging phenotype: the telomere connection. Curr Environ Health Rep. 2016;3(3):258–69.
Saenen ND, Vrijens K, Janssen BG, Madhloum N, Peusens M, Gyselaers W, Vanpoucke C, Lefebvre W, Roels HA, Nawrot TS. Placental nitrosative stress and exposure to ambient air pollution during gestation: a population study. Am J Epidemiol. 2016;184(6):442–9.
Grevendonk L, Janssen BG, Vanpoucke C, Lefebvre W, Hoxha M, Bollati V, Nawrot TS. Mitochondrial oxidative DNA damage and exposure to particulate air pollution in mother-newborn pairs. Environ Health. 2016;15:10.
Rossner P Jr, Tabashidze N, Dostal M, Novakova Z, Chvatalova I, Spatova M, Sram RJ. Genetic, biochemical, and environmental factors associated with pregnancy outcomes in newborns from the Czech Republic. Environ Health Perspect. 2011;119(2):265–71.
Aycicek A, Varma M, Ahmet K, Abdurrahim K, Erel O. Maternal active or passive smoking causes oxidative stress in placental tissue. Eur J Pediatr. 2011;170(5):645–51.
Singh L, Anand M, Singh S, Taneja A. Environmental toxic metals in placenta and their effects on preterm delivery-current opinion. Drug Chem Toxicol. 2018:1–8. https://doi.org/10.1080/01480545.2018.1515216.
Gruzieva O, Xu CJ, Breton CV, Annesi-Maesano I, Anto JM, Auffray C, Ballereau S, Bellander T, Bousquet J, Bustamante M, Charles MA, de Kluizenaar Y, den Dekker HT, Duijts L, Felix JF, Gehring U, Guxens M, Jaddoe VV, Jankipersadsing SA, Merid SK, Kere J, Kumar A, Lemonnier N, Lepeule J, Nystad W, Page CM, Panasevich S, Postma D, Slama R, Sunyer J, Soderhall C, Yao J, London SJ, Pershagen G, Koppelman GH, Melen E. Epigenome-wide meta-analysis of methylation in children related to prenatal NO2 air pollution exposure. Environ Health Perspect. 2017;125(1):104–10.
Plusquin M, Chadeau-Hyam M, Ghantous A, Alfano R, Bustamante M, Chatzi L, Cuenin C, Gulliver J, Herceg Z, Kogevinas M, Nawrot TS, Pizzi C, Porta D, Relton CL, Richiardi L, Robinson O, Sunyer J, Vermeulen R, Vriens A, Vrijheid M, Henderson J, Vineis P. DNA methylome marks of exposure to particulate matter at three time points in early life. Environ Sci Technol. 2018;52(9):5427–37.
Barouki R, Melen E, Herceg Z, Beckers J, Chen J, Karagas M, Puga A, Xia Y, Chadwick L, Yan W, Audouze K, Slama R, Heindel J, Grandjean P, Kawamoto T, Nohara K. Epigenetics as a mechanism linking developmental exposures to long-term toxicity. Environ Int. 2018;114:77–86.
Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8(4):253–62.
Michels KB. Epigenetic Epidemiology. Springer Science+Business Media B.V. 2012. https://doi.org/10.1007/978-94-007-2495-2_21.
Janssen BG, Godderis L, Pieters N, Poels K, Kici Ski M, Cuypers A, Fierens F, Penders J, Plusquin M, Gyselaers W, Nawrot TS. Placental DNA hypomethylation in association with particulate air pollution in early life. Part Fibre Toxicol. 2013;10(1):22.
Kingsley SL, Eliot MN, Whitsel EA, Huang YT, Kelsey KT, Marsit CJ, Wellenius GA. Maternal residential proximity to major roadways, birth weight, and placental DNA methylation. Environ Int. 2016;92-93:43–9.
Cai J, Zhao Y, Liu P, Xia B, Zhu Q, Wang X, Song Q, Kan H, Zhang Y. Exposure to particulate air pollution during early pregnancy is associated with placental DNA methylation. Sci Total Environ. 2017;607-608:1103–8.
Maghbooli Z, Hossein-Nezhad A, Adabi E, Asadollah-Pour E, Sadeghi M, Mohammad-Nabi S, Zakeri Rad L, Malek Hosseini AA, Radmehr M, Faghihi F, Aghaei A, Omidifar A, Aghababei Y, Behzadi H. Air pollution during pregnancy and placental adaptation in the levels of global DNA methylation. PLoS One. 2018;13(7):e0199772.
Abraham E, Rousseaux S, Agier L, Giorgis-Allemand L, Tost J, Galineau J, Hulin A, Siroux V, Vaiman D, Charles MA, Heude B, Forhan A, Schwartz J, Chuffart F, Bourova-Flin E, Khochbin S, Slama R, Lepeule J. Pregnancy exposure to atmospheric pollution and meteorological conditions and placental DNA methylation. Environ Int. 2018;118:334–47.
Yin LJ, Zhang Y, Lv PP, He WH, Wu YT, Liu AX, Ding GL, Dong MY, Qu F, Xu CM, Zhu XM, Huang HF. Insufficient maintenance DNA methylation is associated with abnormal embryonic development. BMC Med. 2012;10:26.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–57.
Nawrot TS, Saenen ND, Schenk J, Janssen BG, Motta V, Tarantini L, Cox B, Lefebvre W, Vanpoucke C, Maggioni C, Bollati V. Placental circadian pathway methylation and in utero exposure to fine particle air pollution. Environ Int. 2018;114:231–41.
Barclay JL, Husse J, Bode B, Naujokat N, Meyer-Kovac J, Schmid SM, Lehnert H, Oster H. Circadian desynchrony promotes metabolic disruption in a mouse model of shiftwork. PLoS One. 2012;7(5):e37150.
Papazyan R, Zhang Y, Lazar MA. Genetic and epigenomic mechanisms of mammalian circadian transcription. Nat Struct Mol Biol. 2016;23(12):1045–52.
Neven KY, Saenen ND, Tarantini L, Janssen BG, Lefebvre W, Vanpoucke C, Bollati V, Nawrot TS. Placental promoter methylation of DNA repair genes and prenatal exposure to particulate air pollution: an ENVIRONAGE cohort study. Lancet Planet Health. 2018;2(4):e174–e83.
Perera F, Hemminki K, Jedrychowski W, Whyatt R, Campbell U, Hsu Y, Santella R, Albertini R, O'Neill JP. In utero DNA damage from environmental pollution is associated with somatic gene mutation in newborns. Cancer Epidemiol Biomark Prev. 2002;11(10 Pt 1):1134–7.
Zhou W, Tian D, He J, Wang Y, Zhang L, Cui L, Jia L, Zhang L, Li L, Shu Y, Yu S, Zhao J, Yuan X, Peng S. Repeated PM2.5 exposure inhibits BEAS-2B cell P53 expression through ROS-Akt-DNMT3B pathway-mediated promoter hypermethylation. Oncotarget. 2016;7(15):20691–703.
Saenen ND, Vrijens K, Janssen BG, Roels HA, Neven KY, Vanden Berghe W, Gyselaers W, Vanpoucke C, Lefebvre W, De Boever P, Nawrot TS. Lower placental leptin promoter methylation in association with fine particulate matter air pollution during pregnancy and placental nitrosative stress at birth in the ENVIRONAGE Cohort. Environ Health Perspect. 2017;125(2):262–8.
Janssen BG, Byun HM, Gyselaers W, Lefebvre W, Baccarelli AA, Nawrot TS. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: an ENVIRONAGE birth cohort study. Epigenetics. 2015;10(6):536–44.
Byun HM, Panni T, Motta V, Hou L, Nordio F, Apostoli P, Bertazzi PA, Baccarelli AA. Effects of airborne pollutants on mitochondrial DNA methylation. Part Fibre Toxicol. 2013;10:18.
Khot VV, Chavan-Gautam P, Mehendale S, Joshi SR. Variable methylation potential in preterm placenta: implication for epigenetic programming of the offspring. Reprod Sci (Thousand Oaks, Calif). 2017;24(6):891–901.
Canfield J, Arlier S, Mong EF, Lockhart J, VanWye J, Guzeloglu-Kayisli O, Schatz F, Magness RR, Lockwood CJ, Tsibris JCM, Kayisli UA, Totary-Jain H. Decreased LIN28B in preeclampsia impairs human trophoblast differentiation and migration. FASEB J. 2019;33(2):2759-2769. https://doi.org/10.1096/fj.201801163R.
Fallen S, Baxter D, Wu X, Kim TK, Shynlova O, Lee MY, Scherler K, Lye S, Hood L, Wang K. Extracellular vesicle RNAs reflect placenta dysfunction and are a biomarker source for preterm labour. J Cell Mol Med. 2018;22(5):2760–73.
Whitehead CL, Teh WT, Walker SP, Leung C, Larmour L, Tong S. Circulating MicroRNAs in maternal blood as potential biomarkers for fetal hypoxia in-utero. PLoS One. 2013;8(11):e78487.
Prince CS, Maloyan A, Myatt L. Maternal obesity alters brain derived neurotrophic factor (BDNF) signaling in the placenta in a sexually dimorphic manner. Placenta. 2017;49:55–63.
Schroeder M, Jakovcevski M, Polacheck T, Drori Y, Luoni A, Roh S, Zaugg J, Ben-Dor S, Albrecht C, Chen A. Placental miR-340 mediates vulnerability to activity based anorexia in mice. Nat Commun. 2018;9(1):1596.
Brooks SA, Fry RC. Cadmium inhibits placental trophoblast cell migration via miRNA regulation of the transforming growth factor beta (TGF-beta) pathway. Food Chem Toxicol. 2017;109(Pt 1:721–6.
LaRocca J, Binder AM, McElrath TF, Michels KB. First-trimester urine concentrations of phthalate metabolites and phenols and placenta miRNA expression in a cohort of U.S. women. Environ Health Perspect. 2016;124(3):380–7.
Rahman ML, Liang L, Valeri L, Su L, Zhu Z, Gao S, Mostofa G, Qamruzzaman Q, Hauser R, Baccarelli A, Christiani DC. Regulation of birthweight by placenta-derived miRNAs: evidence from an arsenic-exposed birth cohort in Bangladesh. Epigenetics. 2018;13(6):573–90.
Sood S, Shekhar S, Santosh W. Dimorphic placental stress: a repercussion of interaction between endocrine disrupting chemicals (EDCs) and fetal sex. Med Hypotheses. 2017;99:73–5.
Bollati V, Marinelli B, Apostoli P, Bonzini M, Nordio F, Hoxha M, Pegoraro V, Motta V, Tarantini L, Cantone L, Schwartz J, Bertazzi PA, Baccarelli A. Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes. Environ Health Perspect. 2010;118(6):763–8.
Espin-Perez A, Krauskopf J, Chadeau-Hyam M, van Veldhoven K, Chung F, Cullinan P, Piepers J, van Herwijnen M, Kubesch N, Carrasco-Turigas G, Nieuwenhuijsen M, Vineis P, Kleinjans JCS, de Kok T. Short-term transcriptome and microRNAs responses to exposure to different air pollutants in two population studies. Environ Pollut. 2018;242(Pt A:182–90.
Chen R, Li H, Cai J, Wang C, Lin Z, Liu C, Niu Y, Zhao Z, Li W, Kan H. Fine particulate air pollution and the expression of microRNAs and circulating cytokines relevant to inflammation, coagulation, and vasoconstriction. Environ Health Perspect. 2018;126(1):017007.
Chao MW, Yang CH, Lin PT, Yang YH, Chuang YC, Chung MC, Tseng CY. Exposure to PM2.5 causes genetic changes in fetal rat cerebral cortex and hippocampus. Environ Toxicol. 2017;32(4):1412–25.
Tsamou M, Vrijens K, Madhloum N, Lefebvre W, Vanpoucke C, Nawrot TS. Air pollution-induced placental epigenetic alterations in early life: a candidate miRNA approach. Epigenetics. 2018;13(2):135–46.
Herberth G, Bauer M, Gasch M, Hinz D, Roder S, Olek S, Kohajda T, Rolle-Kampczyk U, von Bergen M, Sack U, Borte M, Lehmann I. Maternal and cord blood miR-223 expression associates with prenatal tobacco smoke exposure and low regulatory T-cell numbers. J Allergy Clin Immunol. 2014;133(2):543–50.
Martens DS, Nawrot TS. Ageing at the level of telomeres in association to residential landscape and air pollution at home and work: a review of the current evidence. Toxicol Lett. 2018;298:42-52.
Haycock PC, Heydon EE, Kaptoge S, Butterworth AS, Thompson A, Willeit P. Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. Bmj. 2014;349:g4227.
Willeit P, Raschenberger J, Heydon EE, Tsimikas S, Haun M, Mayr A, Weger S, Witztum JL, Butterworth AS, Willeit J, Kronenberg F, Kiechl S. Leucocyte telomere length and risk of type 2 diabetes mellitus: new prospective cohort study and literature-based meta-analysis. PLoS One. 2014;9(11):e112483.
McDonough JE, Martens DS, Tanabe N, Ahangari F, Verleden SE, Maes K, Verleden GM, Kaminski N, Hogg JC, Nawrot TS, Wuyts WA, Vanaudenaerde BM. A role for telomere length and chromosomal damage in idiopathic pulmonary fibrosis. Respir Res. 2018;19(1):132.
Wang Q, Zhan Y, Pedersen NL, Fang F, Hagg S. Telomere length and all-cause mortality: a meta-analysis. Ageing Res Rev. 2018;48:11–20.
Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95.
Moslehi J, DePinho RA, Sahin E. Telomeres and mitochondria in the aging heart. Circ Res. 2012;110(9):1226–37.
Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123(3):951–7.
Janssen BG, Munters E, Pieters N, Smeets K, Cox B, Cuypers A, Fierens F, Penders J, Vangronsveld J, Gyselaers W, Nawrot TS. Placental mitochondrial DNA content and particulate air pollution during in utero life. Environ Health Perspect. 2012;120(9):1346–52.
Martens DS, Cox B, Janssen BG, Clemente DBP, Gasparrini A, Vanpoucke C, Lefebvre W, Roels HA, Plusquin M, Nawrot TS. Prenatal air pollution and newborns’ predisposition to accelerated biological aging. JAMA Pediatr. 2017;171(12):1160–7.
Bijnens E, Zeegers MP, Gielen M, Kicinski M, Hageman GJ, Pachen D, Derom C, Vlietinck R, Nawrot TS. Lower placental telomere length may be attributed to maternal residential traffic exposure; a twin study. Environ Int. 2015;79:1–7.
Lin S, Huo X, Zhang Q, Fan X, Du L, Xu X, Qiu S, Zhang Y, Wang Y, Gu J. Short placental telomere was associated with cadmium pollution in an electronic waste recycling town in China. PLoS One. 2013;8(4):e60815.
Martens DS, Plusquin M, Gyselaers W, De Vivo I, Nawrot TS. Maternal pre-pregnancy body mass index and newborn telomere length. BMC Med. 2016;14(1):148.
Bijnens EM, Zeegers MP, Derom C, Martens DS, Gielen M, Hageman GJ, Plusquin M, Thiery E, Vlietinck R, Nawrot TS. Telomere tracking from birth to adulthood and residential traffic exposure. BMC Med. 2017;15(1):205.
Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol. 2012;13(6):397–404.
Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470(7334):359–65.
Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464(7288):520–8.
Blasco MA. The epigenetic regulation of mammalian telomeres. Nat Rev Genet. 2007;8(4):299–309.
Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, Blasco MA. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol. 2006;8(4):416–24.
Benetti R, Garcia-Cao M, Blasco MA. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat Genet. 2007;39(2):243–50.
Wong JY, De Vivo I, Lin X, Grashow R, Cavallari J, Christiani DC. The association between global DNA methylation and telomere length in a longitudinal study of boilermakers. Genet Epidemiol. 2014;38(3):254–64.
Dong Y, Huang Y, Gutin B, Raed A, Dong Y, Zhu H. Associations between Global DNA methylation and telomere length in healthy adolescents. Sci Rep. 2017;7(1):4210.
Jena NR. DNA damage by reactive species: Mechanisms, mutation and repair. J Biosci. 2012;37(3):503–17.
Wachsman JT. DNA methylation and the association between genetic and epigenetic changes: relation to carcinogenesis. Mutat Res. 1997;375(1):1–8.
Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, Messina S, Iuliano R, Fusco A, Santillo MR, Muller MT, Chiariotti L, Gottesman ME, Avvedimento EV. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 2007;3(7):e110.
O'Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, Casero RA, Sears CL, Baylin SB. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20(5):606–19.
Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions. J Biol Chem. 1992;267(1):166–72.
Weitzman SA, Turk PW, Milkowski DH, Kozlowski K. Free radical adducts induce alterations in DNA cytosine methylation. Proc Natl Acad Sci U S A. 1994;91(4):1261–4.
Donkena KV, Young CY, Tindall DJ. Oxidative stress and DNA methylation in prostate cancer. Obstet Gynecol Int. 2010;2010:302051.
Martin EM, Fry RC. Environmental influences on the epigenome: exposure- associated DNA methylation in human populations. Annu Rev Public Health. 2018;39(1):309–33.
Hou L, Zhang X, Wang D, Baccarelli A. Environmental chemical exposures and human epigenetics. Int J Epidemiol. 2012;41(1):79–105.
Reichard JF, Schnekenburger M, Puga A. Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem Biophys Res Commun. 2007;352(1):188–92.
Jiang CL, He SW, Zhang YD, Duan HX, Huang T, Huang YC, Li GF, Wang P, Ma LJ, Zhou GB, Cao Y. Air pollution and DNA methylation alterations in lung cancer: a systematic and comparative study. Oncotarget. 2017;8(1):1369–91.
Martin EM, Fry RC. A cross-study analysis of prenatal exposures to environmental contaminants and the epigenome: support for stress-responsive transcription factor occupancy as a mediator of gene-specific CpG methylation patterning. Environ Epigenet. 2016;2(1):dvv011. https://doi.org/10.1093/eep/dvv011.
Benetti R, Gonzalo S, Jaco I, Munoz P, Gonzalez S, Schoeftner S, Murchison E, Andl T, Chen T, Klatt P, Li E, Serrano M, Millar S, Hannon G, Blasco MA. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008;15(9):998.
Tsamou M, Martens DS, Cox B, Madhloum N, Vrijens K, Nawrot TS. Sex-specific associations between telomere length and candidate miRNA expression in placenta. J Transl Med. 2018;16(1):254.
He J, Jiang BH. Interplay between reactive oxygen species and microRNAs in cancer. Curr Pharmacol Rep. 2016;2(2):82–90.
Wiesen JL, Tomasi TB. Dicer is regulated by cellular stresses and interferons. Mol Immunol. 2009;46(6):1222–8.
Ungvari Z, Tucsek Z, Sosnowska D, Toth P, Gautam T, Podlutsky A, Csiszar A, Losonczy G, Valcarcel-Ares MN, Sonntag WE, Csiszar A. Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J Gerontol A Biol Sci Med Sci. 2013;68(8):877–91.
Liu J, Zhang C, Zhao Y, Feng Z. MicroRNA Control of p53. J Cell Biochem. 2017;118(1):7–14.
Dai L, Mehta A, Mordukhovich I, Just AC, Shen J, Hou L, Koutrakis P, Sparrow D, Vokonas PS, Baccarelli AA, Schwartz JD. Differential DNA methylation and PM2.5 species in a 450 K epigenome-wide association study. Epigenetics. 2017;12(2):139–48.
Rosa MJ, Just AC, Guerra MS, Kloog I, Hsu HL, Brennan KJ, Garcia AM, Coull B, Wright RJ, Tellez Rojo MM, Baccarelli AA, Wright RO. Identifying sensitive windows for prenatal particulate air pollution exposure and mitochondrial DNA content in cord blood. Environ Int. 2017;98:198–203.
Plusquin M, Saenen ND, Nawrot TS. Epigenetics and the exposome. In: Dagnino S, Macherone A, editors. Unraveling the Exposome. Cham: Springer; 2019.
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, Turecki G, Delaney A, Varhol R, Thiessen N, Shchors K, Heine VM, Rowitch DH, Xing X, Fiore C, Schillebeeckx M, Jones SJ, Haussler D, Marra MA, Hirst M, Wang T, Costello JF. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–7.
Miousse IR, Chalbot MC, Aykin-Burns N, Wang X, Basnakian A, Kavouras IG, Koturbash I. Epigenetic alterations induced by ambient particulate matter in mouse macrophages. Environ Mol Mutagen. 2014;55(5):428–35.
Breton CV, Marsit CJ, Faustman E, Nadeau K, Goodrich JM, Dolinoy DC, Herbstman J, Holland N, LaSalle JM, Schmidt R, Yousefi P, Perera F, Joubert BR, Wiemels J, Taylor M, Yang IV, Chen R, Hew KM, Freeland DM, Miller R, Murphy SK. Small-magnitude effect sizes in epigenetic end points are important in children’s environmental health studies: the Children’s Environmental Health and Disease Prevention Research Center’s Epigenetics Working Group. Environ Health Perspect. 2017;125(4):511–26.
Alfano R, Herceg Z, Nawrot TS, Chadeau-Hyam M, Ghantous A, Plusquin M. The impact of air pollution on our epigenome: how far is the evidence? (a systematic review). Curr Environ Health Rep. 2018;5(4):544-578.
Felix JF, Joubert BR, Baccarelli AA, Sharp GC, Almqvist C, Annesi-Maesano I, Arshad H, Baiz N, Bakermans-Kranenburg MJ, Bakulski KM, Binder EB, Bouchard L, Breton CV, Brunekreef B, Brunst KJ, Burchard EG, Bustamante M, Chatzi L, Cheng Munthe-Kaas M, Corpeleijn E, Czamara D, Dabelea D, Davey Smith G, De Boever P, Duijts L, Dwyer T, Eng C, Eskenazi B, Everson TM, Falahi F, Fallin MD, Farchi S, Fernandez MF, Gao L, Gaunt TR, Ghantous A, Gillman MW, Gonseth S, Grote V, Gruzieva O, Haberg SE, Herceg Z, Hivert MF, Holland N, Holloway JW, Hoyo C, Hu D, Huang RC, Huen K, Jarvelin MR, Jima DD, Just AC, Karagas MR, Karlsson R, Karmaus W, Kechris KJ, Kere J, Kogevinas M, Koletzko B, Koppelman GH, Kupers LK, Ladd-Acosta C, Lahti J, Lambrechts N, Langie SAS, Lie RT, Liu AH, Magnus MC, Magnus P, Maguire RL, Marsit CJ, McArdle W, Melen E, Melton P, Murphy SK, Nawrot TS, Nistico L, Nohr EA, Nordlund B, Nystad W, Oh SS, Oken E, Page CM, Perron P, Pershagen G, Pizzi C, Plusquin M, Raikkonen K, Reese SE, Reischl E, Richiardi L, Ring S, Roy RP, Rzehak P, Schoeters G, Schwartz DA, Sebert S, Snieder H, Sorensen TIA, Starling AP, Sunyer J, Taylor JA, Tiemeier H, Ullemar V, Vafeiadi M, Van Ijzendoorn MH, Vonk JM, Vriens A, Vrijheid M, Wang P, Wiemels JL, Wilcox AJ, Wright RJ, Xu CJ, Xu Z, Yang IV, Yousefi P, Zhang H, Zhang W, Zhao S, Agha G, Relton CL, Jaddoe VWV, London SJ. Cohort profile: Pregnancy And Childhood Epigenetics (PACE) Consortium. Int J Epidemiol. 2018;47(1):22–3u.
Mostafavi N, Vermeulen R, Ghantous A, Hoek G, Probst-Hensch N, Herceg Z, Tarallo S, Naccarati A, Kleinjans JCS, Imboden M, Jeong A, Morley D, Amaral AFS, van Nunen E, Gulliver J, Chadeau-Hyam M, Vineis P, Vlaanderen J. Acute changes in DNA methylation in relation to 24 h personal air pollution exposure measurements: a panel study in four European countries. Environ Int. 2018;120:11–21.
Fiorito G, Vlaanderen J, Polidoro S, Gulliver J, Galassi C, Ranzi A, Krogh V, Grioni S, Agnoli C, Sacerdote C, Panico S, Tsai MY, Probst-Hensch N, Hoek G, Herceg Z, Vermeulen R, Ghantous A, Vineis P, Naccarati A. Oxidative stress and inflammation mediate the effect of air pollution on cardio- and cerebrovascular disease: a prospective study in nonsmokers. Environ Mol Mutagen. 2018;59(3):234–46.
Breton CV, Gao L, Yao J, Siegmund KD, Lurmann F, Gilliland F. Particulate matter, the newborn methylome, and cardio-respiratory health outcomes in childhood. Environmental epigenetics. 2016;2(2):dvw005.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinforma. 2012;13:86.
Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics (Oxford, England). 2014;30(10):1431–9.
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105(44):17046–9.
Yehuda R, Lehrner A, Bierer LM. The public reception of putative epigenetic mechanisms in the transgenerational effects of trauma. Environ Epigenet. 2018;4(2):dvy018.
Acknowledgements
Not applicable
Funding
This review is supported by grants from the European Research Council (ERC-2012-StG 310898/POC INCALO) and FWO (G082317 N).
Author information
Authors and Affiliations
Contributions
All authors were responsible in contributing to a specific part of the review: RA and HB wrote the introduction; NDS, KYN, and BGJ contributed to the part of DNA methylation; KV wrote the part of miRNA; NDS, KYN, and DSM wrote the part of the interplay between biological processes; DSM wrote the part on the aging phenotype and the proposed air pollution hypothesis; and NDS, BGJ, MP, and TSN wrote the challenges and opportunities. All authors critically reviewed the whole manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Saenen, N.D., Martens, D.S., Neven, K.Y. et al. Air pollution-induced placental alterations: an interplay of oxidative stress, epigenetics, and the aging phenotype?. Clin Epigenet 11, 124 (2019). https://doi.org/10.1186/s13148-019-0688-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-019-0688-z