
Abstract
Background
Methylated genes detected in sputum are promise biomarkers for lung cancer. Yet the current PCR technologies for quantification of DNA methylation and diagnostic value of the sputum biomarkers are not sufficient to be used for lung cancer early detection. The emerging droplet digital PCR (ddPCR) is a straightforward means for precise, direct, and absolute quantification of nucleic acids. Here, we investigate whether ddPCR can sensitively and robustly quantify DNA methylation in sputum for more precise diagnosis of lung cancer.
Results
First, the analytic performance of methylation-specific ddPCR (ddMSP) and quantitative methylation-specific PCR (qMSP) is determined in methylated and unmethylated DNA samples. Second, 29 genes, previously proposed as potential sputum biomarkers for lung cancer, are analyzed by using ddMSP in a training set of 127 lung cancer patients and 159 controls. ddMSP has higher sensitivity, precision, and reproducibility for quantification of methylation compared with qMSP (all p < 0.05). A classifier comprising four sputum methylation biomarkers for lung cancer is developed by using ddMSP, producing 86.6% sensitivity and 90.6% specificity, independent of stage and histology of lung cancer (all p > 0.05). The classifier has higher accuracy compared with sputum cytology (88.8 vs. 70.6%, p < 0.01). The diagnostic performance is confirmed in a testing set of 89 cases and 107 controls.
Conclusions
ddMSP is a robust tool for reliable quantification of DNA methylation in sputum, and the epigenetic classifier could help diagnose lung cancer at the early stage.
Similar content being viewed by others


Background
Lung cancer is the leading cause of cancer death among men and women [1]. More than 85% lung tumors are non-small cell lung cancers (NSCLCs), which consist of adenocarcinoma (AC), squamous cell carcinoma (SCC), and large cell carcinoma (LC). Cigarette smoking is the foremost cause of NSCLC [2]. People who smoke cigarettes are nearly 30 times more likely to get lung cancer or die from lung cancer than people who do not smoke. Even smoking a few cigarettes a day or smoking occasionally increases the risk of lung cancer. Individuals who quit smoking have a lower risk of lung cancer than if they had continued to smoke, but their risk is higher than the risk for people who never smoked. The National Lung Screening Trial (NLST) results show that using low-dose CT (LDCT) for the early detection of lung cancer in smokers can reduce the mortality by 20% as compared to chest X-rays [1]. Therefore, LDCT is recently recommended to be used for lung cancer early detection among smokers [3, 4]. However, LDCT is associated with over-diagnosis, excessive cost, and radiation exposure, limiting its clinical applications [3,4,5]. The development of noninvasive approaches that can accurately and cost-effectively diagnose early stage lung cancer among smokers remains clinically important [6].
Lung cancer develops from a field defect characterized by an accumulation of molecular abnormalities resulted from repeated exposure of the airway of the smokers to the tobacco-related carcinogens [7,8,9]. Regardless of the anatomic location relative to the tumors, the molecular alterations observed in the large bronchial airway might reflect the altered changes existed in lung tumors [9,10,11]. Sputum is defined as secretions from the airways and contains bronchial epithelial cells exfoliated from the airways or lungs [12]. Therefore, the analysis of exfoliated bronchial epitheliums in sputum for the molecular changes may provide a useful tool for noninvasively and cost-effectively diagnosing lung cancer.
DNA methylations of tumor suppressor genes (TSGs) are early molecular events in lung carcinogenesis and thus show great promise as biomarkers for early stage lung cancer [6, 11, 13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. Conventional qPCR-based platforms, particularly, methylation-specific PCR (qMSP), have been used for detecting DNA methylation of TSGs in sputum [13]. However, qMSP has some weaknesses, limiting its use in the clinical settings. For example, qMSP is an indirect approach, which requires internal controls for data normalization [35]. Furthermore, qMSP’s sensitivity for analyzing low copy number of genes is poor. This is particularly challenging for quantification of DNA methylation in bronchial epitheliums, as the large excess of non-epithelial cells in sputum could obscure detection of the relative scarcity of methylated DNA from the exfoliated bronchial epitheliums. A more sensitive, precise, and reproducibility method for quantification of methylated DNA in sputum would provide a useful means for noninvasive diagnosis of lung cancer.
Droplet digital PCR (ddPCR) is a direct method for quantitatively measuring nucleic acids [36,37,38,39,40,41,42,43,44,45], since it depends on limiting partition of the PCR volume, where a positive result of a large number of microreactions indicates the presence of a single molecule in a given reaction. The number of positive reactions, together with Poisson’s distribution, can be used to produce a straight and high-confidence measurement of the original target concentration [43]. Furthermore, ddPCR does not require the reliance on rate-based measurements, endogenous controls, and the use of calibration curves. In addition, previous studies including our own research have demonstrated that ddPCR can quantify low-abundance nucleic acids and has higher sensitivity and precision than does conventional PCR [36, 37, 46]. The objective of this study is to investigate whether methylation-specific ddPCR (ddMSP) could sensitively and robustly quantify DNA methylations in sputum and hence develop a biomarker-based classifier for early stage lung cancer.
Methods
Study population
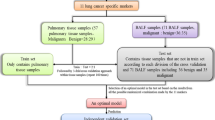
The study protocol was approved by the local Institutional Review Board. The participants in this study were recruited from the hospital at the point of their referral for suspected lung cancer between the ages of 55–80. Written informed consent was obtained from all enrolled subjects. Exclusion criteria included pregnancy, current pulmonary infection, surgery within 6 months, radiotherapy within 1 year, and life expectancy of < 1 year. Clinical diagnosis of lung cancer was made using histopathologic examinations of specimens obtained by CT-guided transthoracic needle biopsy, transbronchial biopsy, videotape-assisted thoracoscopic surgery, or surgical resection. The surgical pathologic staging was determined according to the TNM classification of the International Union Against Cancer with the 8th American Joint Committee on Cancer and the International Staging System for Lung Cancer. Histopathological classification was determined according to the World Health Organization classification. A total of 482 subjects including 216 lung cancer patients and 266 cancer-free smokers were recruited. The 216 lung cancer patients were diagnosed with NSCLC consisting of 55 stage I cases, 55 stage II cases, 50 stage III cases, and 56 stage IV cases. One hundred and twelve cases were AC, 91 were SCC, and 13 were LC. The 266 cancer-free patients who were smokers and served as control subjects had granulomatous inflammation (n = 117), nonspecific inflammatory changes (n = 105) or lung infections (n = 44). The cancer-free smokers had been followed for at least 2 years, and none had any evidence of cancer. No difference of age, gender, and smoking status was observed in the lung cancer cases vs. controls (All p > 0.05). To refine the biomarkers whose changes specific to NSCLC, the cases were matched to the controls on gender, age, race, and smoking status as a nested case-control study. The cases and controls were then randomly split into a training set and a testing set by using a random number generator. The training set consisted of 127 lung cancer patients and 159 cancer-free controls. The testing set comprised 89 lung cancer patients and 107 cancer-free controls. The demographic and clinical characteristics of the two cohorts are presented in Tables 1 and 2.
Sample collection and sputum cytology
Sputum was collected from the participants as described in previous reports [47,48,49,50,51,52,53,54]. Briefly, to reduce the percentage of oral epithelial cells in the sputum, subjects were asked to blow their nose, rinse their mouth, and swallow water to minimize contamination of squamous cells from postnasal drip and saliva. Sputum samples were then coughed in a sterile container and processed within 2 h. To further minimize oral squamous cell contamination, opaque or dense portions that looked different from saliva under the inverted microscope were selected using blunt forceps from expectorate. The samples were processed on ice in 4 volumes of 0.1% dithiothreitol (Sigma-Aldrich, St. Louis, Mo) followed by 4 volumes of phosphate-buffered saline (PBS) (Sigma-Aldrich). The cell suspension was filtered through 45-μm nylon gauzes (BNSH Thompson, Scarborough, ON, Canada). Absolute cell numbers and cell viability were quantitated by using a hemacytometer with trypan blue. Two cytocentrifuge slides were prepared from aliquots of cell suspension by using a cytospin machine (Shandon, Pittsburgh, PA) and were then stained with the Papanicolaou staining technique [12]. A sputum sample was considered adequate if lung macrophages or Curschmann spirals were present on the slides [11, 12]. Cytologic diagnosis was performed on the cytospin slides using the classification of Saccomanno et al. [12]. The remaining cells are stored at − 80 °C until used.
DNA isolation and bisulfite conversion
We extracted DNA from the specimens using DNeasy kit (Qiagen, Valencia, CA) as previously described [14]. We eluted DNA with 50 μL of elution buffer (10 mmol/L Tris-Cl, pH 8.5) (Sigma-Aldrich Corporation). DNA was quantified by using the Quantifiler Human DNA Quantification kit (Applied Biosystems, Foster City, CA). Bisulfite conversion was carried out on DNA by using the Zymo EZ DNA Methylation Kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol.
Serially diluted methylated/unmethylated DNA specimens
We purchased 100% methylated and 100% unmethylated control human DNA samples (Zymo Research). We isolated DNA from sputum of a healthy nonsmoker whose sputum DNA did not harbor DNA methylation of TSGs, including RASSF1A, 3OST2, and PRDM14 [14]. To determine limit of quantification (LOQ) of an assay, we diluted methylated DNA into the sputum DNA sample in the following concentrations: 100, 25, 6.25, 1.56, 0.39, 0.1, 0.04, and 0% methylated DNA. To determine limits of detection (LOD) of an assay, we prepared serially diluted samples containing 5000, 2500, 1250, 625, 313, 156, and 0 pg methylated DNA in H2O.
Quantification of DNA methylation in sputum by ddMSP
We added bisulfite-treated DNA (2 μL) to ddPCR mixture (18 μL) containing 2 × ddPCR Supermix for probes (no-dUTP), 750 nmol/L of each primer and 250 nmol/L of the corresponding probe in a final volume of 20 μL. Twenty-nine genes were selected for DNA methylation analysis, since the genes were previously reported as potential sputum methylation biomarkers for lung cancer [6, 13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. The 29 genes are 3OST2, APC, CDH1, CDO1, CXCL, CYGB, DAL-1, DAPK, DCR2, FAM19A4, FHIT, GATA, H-cadherin, HOXA9, JPH3, KIFLA, MAGE, p16, PAX5, PCDH20, PHACTR3, PRDM14, RARβ, RASSF1A, SOX17, SULF2, TAC1, TCF2L, and ZFP42 (Additional file 1: Table S1). Primers and probes of the targeted genes were designed in the studies [6, 13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. A thermocycling protocol (95 °C × 10 min; 40 cycles of [94 °C × 30s, 60 °C × 60s], 98 °C × 10 min) was undertaken in a Bio-Rad C1000 (Bio-Rad, Pleasanton, CA). The PCR plate was transferred to the QX100 Droplet Reader (Bio-Rad) for automatic reading of samples in all wells. We used QuantaSoft 1.7.4 analysis software (Bio-Rad) and Poisson statistics to compute droplet concentrations (copies/μL; PCR scale). Only tests that had at least 10,000 droplets were used for the ddMSP analysis [36, 37]. All assays were done in triplicates, and one no-template control and two interplate controls were carried along in each experiment.
Quantification of DNA methylation in sputum by qMSP
qMSP was done as previously described [13, 14]. The cycle threshold (Ct) values for each gene were determined. Ct values above 35 were censored according to previous recommendations [13, 14, 55,56,57,58]. To determine methylation level of target genes in a given sample, we normalized Ct values of the target genes in relation to that the of myoblast determination protein one (MYOD1) [13, 32]. The percentage of methylated reference (PMR) was defined as target gene/MYOD1 ratio of the sample divided by target gene/MYOD1 ratio of the calibrator DNA (methylated control DNA) and multiplying by 100 [14].
Comparison of tolerance of ddMSP and qMSP to PCR inhibitors
To determine tolerance of ddMSP and qMSP to inhibitory substances of PCR, we directly introduced inhibitors, sodium dodecyl sulfate (SDS), and heparin (Sigma-Aldrich Corporation), into the PCR reactions [59, 60]. Differences in the resulting inhibition curves and the half-maximal inhibitory concentrations (IC50) were assessed and compared as described previously [59, 60].
Statistical analysis
We used t test to determine significant differences of values of each gene between cases and controls. We used log transformation of the molecular results and applied Pearson’s correlation analysis to assess relationship between DNA methylation and demographic characteristics of subjects. We calculated coefficient of variations (CV) to determine the variation between different measurements. We performed the linear regression between different measurements of the assays and the amount of input DNA. We used the receiver-operator characteristic (ROC) curve and area under the curve (AUC) to determine accuracy, sensitivity, and specificity of each gene or the tests. We employed logistic regression models with constrained parameters as in least absolute shrinkage and selection operator (LASSO) based on ROC criterion to eliminate the irrelevant genes and optimize a composite biomarker panel (classifier). The optimal panel of biomarkers was blindly applied to the testing data set to confirm the diagnostic value by comparing the AUC with the goodness-of-fit statistics [61].
Results
ddMSP has higher sensitivity, precision, and reproducibility for quantification of DNA methylation compared with qMSP
In methylated DNA serially diluted in sputum DNA of a healthy nonsmoker, ddMSP generated at least 10,000 droplets passing through a fluorescence detector. The results suggested that the specimens were successfully “read” by ddPCR. ddMSP detected methylated genes (RASSF1A, 3OST2, and PRDM14) at a concentration of 0.04% (LOQ = 0.04%)(R2 = 0.966) (Fig. 1a), whereas qMSP detected the methylation at a concentration of 0.10% (LOQ = 0.10%)(R2 = 0.935) (p = 0.008) (Fig. 1b). There was excellent linearity between the methylated DNA input and values measured by both qMSP and ddMSP (all R2 ≥ 0.93). Furthermore, the dispersion of values of the four analyses of the specimen was lower with ddMSP than with qMSP. The repeated measurements by ddMSP had a lower CV value compared with those determined by qMSP (p = 0.03) (Additional file 1: Table S2). Therefore, ddMSP had a higher precision for quantification of methylation compared to qMSP (p = 0.03) (Additional file 1: Table S2).

The dynamic ranges and sensitivities of ddMSP and qMSP for quantification of DNA methylation. a In methylated DNA serially diluted in sputum DNA of a healthy nonsmoker, ddMSP can detect levels of methylated 3OST2 as low as 0.04% (LOQ = 0.04%). A negative template control (NTC) sample was also tested. R2 = 0.966 shows excellent linear correlation between measured concentration of methylated DNA and expected percentage of methylation. b qMSP can detect methylated 3OST2 at 0.10% (LOQ = 0.10%) in the same diluted samples with R2 of 0.935. c In 100% methylated DNA serially diluted into water, ddMSP can detect the smallest amount of methylated DNA at 156 pg/μL (156 pg/μL) with R2 of 0.959. d qMSP can detect the smallest amount of methylated DNA at 156 pg/μL (156 pg/μL) with R2 of 0.937
To determine the absolute LOD of the two platforms, 100% methylated DNA serially diluted into water and then tested by ddMSP and qMSP. The smallest amount of methylated DNA that can be reliably measured by ddMSP was 156 pg/μL (Fig. 1c), suggesting that ddMSP had a LOD of 156 pg/μL. qMSP produced more than 35 Ct values for the samples that had less than 313 pg methylated DNA per microliter, yielding a LOD of 313 pg/μL (Fig. 1d). Therefore, ddMSP had higher sensitivity as demonstrated by lower LOQ and LOD than did qMSP in the serial dilutions of DNA control samples (all p < 0.001).
To determine reproducibility of ddMSP and qMSP, the diluted samples were independently analyzed. The CVs of repeated measures by ddMSP on different days were more than twofold lower compared with those determined by qMSP (Additional file 1: Table S3). Furthermore, the CVs of repeated measures by different research staff using ddMSP were at least twofold lower than did those generated by qMSP (Additional file 1: Table S4). Therefore, ddMSP had a higher reproducibility than did qMSP for quantification of DNA methylation.
To evaluate analytic performance of ddMSP and qMSP in clinical sputum samples, sputum of 20 lung cancer patients and 20 cancer-free controls was tested for RASSF1A, whose aberrant methylation level was shown to be elevated in sputum of lung cancer patients [13, 14, 55,56,57,58]. Each well of the samples contained at least 10,000 droplets (Fig. 2a). Therefore, ddMSP analysis of DNA methylation could successfully be performed in clinical sputum specimens. RASSF1A analyzed by both the techniques displayed a high methylation level in lung cancer patients vs. controls (all p < 0.05). In the ddMSP assay, a specimen with ≥ one copy of DNA methylation of RASSF1Aper microliter was considered to be positive. When the criteria was used, of 20 sputum specimens of lung cancer patients, 11 (55%) had positive methylation of the gene detected by ddMSP. In the qMSP assay, a PMR ≥ 1% was classified as positive for RASSF1A in a given sample [62]. When the criteria was used, 9 (45%) were positive for RASSF1A by qMSP. The same 4 sputum specimens of control subjects had positive methylation of the gene detected by both ddMSP and qMSP. Therefore, ddMSP analysis of DNA methylation of RASSF1A in sputum had a higher sensitivity (55%) than did qMSP (45%) (p = 0.01) for distinguishing lung cancer patients from control subjects, while maintaining the same specificity (80%) (Additional file 1: Figure S1). Furthermore, the CVs of repeated measures by ddMSP on different days by different researchers were approximately twofold lower compared with those generated by qMSP. Altogether, in clinical sputum specimens, ddMSP also exhibited higher sensitivity, accuracy, and reproducibility than did qMSP for quantification of DNA methylation.

ddMSP and qMSP analyses of DNA methylation in clinical sputum samples. a ddMSP analysis of sputum samples of 20 cancer-free controls (normal subjects, N) and 20 patients diagnosed with lung tumor (T) for DNA methylation of RASSF1A. Each well of the sputum samples contained at least 10,000 droplets, suggesting that the clinical sputum specimens could be successfully “read” by ddPCR. b ddMSP analysis of 29 genes in a training set of 127 lung cancer patients and 159 controls developed an epigenetic classifier consisting of four DNA methylation biomarkers. The epigenetic classifier produced 0.92 AUC with 86.6% sensitivity and 90.6% specificity for diagnosis of lung cancer. c The epigenetic classifier had higher accuracy (88.8 vs. 70.63%, p = 0.004) and sensitivity (86.6 vs. 44.8%, p < 0.001) compared with sputum cytology, whereas keeping a similar specificity (90.6 vs. 91.2%, p = 0.34)
To compare the tolerance of ddMSP and qMSP to PCR inhibitors, we added SDS and heparin directly into the PCR reactions and then calculated log IC50 values from the resulting inhibition curves. We found greater than a half log increase in IC50 of ddMSP over qMSP for both SDS and heparin (all p < 0.05), implying that ddMSP tolerated the presence of the inhibitors better than qMSP.
Diagnostic performance of ddMSP quantified-sputum methylation biomarkers for lung cancer
We first evaluated DNA methylation of 29 genes in the training cohort of 127 NSCLC patients and 159 controls. All the 29 genes displayed a higher level of methylation in patients vs. controls (all p < 0.05). ROC curve and AUC analysis showed that the genes had 29–88% sensitivities and 26–92% specificities in differentiating lung cancer patients from healthy controls (Additional file 1: Table S1). Since methylation levels of genes did not follow a normal distribution, we used the log transformation of ddPCR results. We then applied multivariate logistic regression models with stepwise regression based on ROC curve to develop a prediction classifier. Four genes (HOXA9, RASSF1A, SOX17, and TAC1) were identified as the best biomarkers (all p < 0.001) and incorporated into a logistic classifier: Probability of lung cancer = ex/(1 + ex), where x = 1.69 + 1.48 × log (HOXA9) − 1.25 × log (RASSF1A) + 0.27 × log (SOX17) + 0.16 × log (TAC1). The logistic classifier produced 0.92 AUC for lung cancer detection (Fig. 2b). Furthermore, Pearson correlation among methylation levels of the four genes was low (p > 0.05), implying that their diagnostic values were complementary to each other. Using Youden’s index, we set up optimal cutoff at 1.28 for the prediction classifier. Subsequently, combined use of the four genes by simply calculating the equation produced 86.6% sensitivity and 90.6% specificity. In addition, including other genes in the prediction classifier did not improve the accuracy for lung cancer diagnosis. The prevalence of the DNA methylation of the four genes was related with pack-years of smoking (p = 0.03). Since the cases and controls were matched 1:1 by age, gender, and smoking status as a nested case-control study, we adjusted the parameters during model building. The logistic classifier did not show special association with stage and histological type of lung cancer, and patients’ age, gender, and smoking status (all p > 0.05). Moreover, the logistic classifier had higher accuracy (88.8 vs. 70.63%, p = 0.004) and sensitivity (86.6 vs. 44.8%, p < 0.001) than did sputum cytology, while maintaining a similar specificity (90.6 vs. 91.2%, p = 0.34) (Fig. 2c). The integrated use of the biomarkers and sputum cytology did not significantly increase the diagnostic value.
Validating the panel of ddMSP-quantified methylation biomarkers in a testing cohort
In the testing cohort, the panel of the four genes had 85.4% sensitivity and 91.6% specificity in differentiating lung cancer patients from controls (Table 3). In line with findings in the training set, the logistic classifier was not associated with patient’s age, gender, and smoking status, as well as histological type and stage of NSCLC (all p > 0.05). Moreover, the logistic classifier yielded higher accuracy (88.8 vs. 71.4%, p = 0.002) and sensitivity (85.4 vs. 46.1%, p < 0.001), while keeping a similar specificity (91.6 vs. 92.5%, p = 0.45), as compared with sputum cytology (Table 3). Taken together, the validation data confirmed the potential of the ddMSP-quantified sputum biomarkers as a sensitive classifier for the early detection of lung cancer.
Discussion
This current study presents the earliest assessment of ddPCR, an emerging technique, for quantitative detection of DNA methylation in sputum. We find that ddMSP can absolutely and robustly quantify DNA methylation in sputum without requiring external references. Therefore, determination of DNA methylation in sputum by ddMSP is highly efficient, and data handling is forthright. Furthermore, our head-to-head comparison of ddMSP and qMSP reveals that ddMSP displays higher precision and reproducibility in measuring copy number of DNA methylation in both control DNA samples and clinical sputum specimens. The sensitivity of conventional qMSP for analyzing low-abundance methylated DNA is poor. This is particularly challenging for the determination of DNA methylation in bronchial epitheliums, since the large excess of normal cells in sputum could obscure detection of the relative scarcity of methylated DNA. We find that ddMSP has a higher sensitivity to quantify cancer-specific methylation and thus could overcome the obstacle of qMSP. In addition, the total time required for ddMSP is about twofold shorter than did qMSP and might be further reduced when an automated system is used [63, 64]. Moreover, ddMSP is not expensive and tolerates the PCR inhibitors better compared with conventional qMSP. Altogether, ddMSP is a straightforward and robust approach for accurate quantification of DNA methylation in sputum.
Importantly, using ddMSP, we develop and validate a DNA methylation-based classifier that has higher accuracy and sensitivity compared with sputum cytology, the clinical gold standard. Furthermore, ddMSP analysis of the four genes by simply calculating the equation would be a convenient tool to be used in the clinics. In addition, the diagnostic performance of the logistic classifier is independent of stage and histological type of the NSCLC, as well as age, gender, and smoking status of subjects. Therefore, the classifier has an important characteristic if it is employed for more precisely and easily identifying early stage lung cancer among smokers.
However, some limitations do exist in this present study: (i) we evaluate ddMSP for quantification of DNA methylation using a retrospective cases and controls, which may produce selection bias and overfitting. Furthermore, the cases and controls are hospital-based patients, and not representative of the smokers in a screening setting for lung cancer early detection. We will perform a large trial to prospectively validate if the logistic classifier could help identify lung cancer at the early stage in a screening setting among smokers. (ii) the overall sensitivity and specificity of the DNA methylation-based classifier are 86.6 and 90.6%, which are not high enough for clinical diagnosis of NSCLC. The integration of the methylation biomarkers with other classes of biomarkers, such as microRNAs [36, 47,48,49,50,51,52, 65,66,67] or DNA mutations [33], is one path to improve the early detection of lung cancer [14]. (iii) since there is no sample left from the patients of the training and cohorts, we are not able to test the classification performance of qMSP in all the samples of the training and testing cohorts. We are consenting new cases and controls and collecting specimens and will then compare performance of qMSP and ddMSP in the same samples. (iv) like qMSP, ddMSP has the same limitations as do other PCR-based platforms. For instance, large experiments can become quite labor intensive to perform, when multiple genes are targeted. In this present study, we use 96-well PCR plates. Given the four genes to be analyzed with duplication, we only test 12 clinical samples at one time. In the future, we will use 484-well PCR plates, in which, we could simultaneously test 60 samples.
Conclusion
ddMSP could be a sensitive and robust tool for reliable quantitation of sputum methylation biomarkers. The ddMSP-quantified methylation classifier may provide a potential diagnostic test for the early detection of lung cancer and thus reduce the deaths and costs associated with the disease. Nevertheless, the continued development of this new technology, and further exploring its value for routine use for the early detection of lung cancer would be required.
References
Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, Fagerstrom RM. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365:395–409.
Blumer G. Cigarette smoking and cancer of the lung. Ill Med J. 1951;100:98–9.
Patz EF Jr, Pinsky P, Gatsonis C, Sicks JD, Kramer BS, Tammemagi MC. Overdiagnosis in low-dose computed tomography screening for lung cancer. JAMA Intern Med. 2014;174:269–74.
Aberle DR, Berg CD, Black WC, Church TR, Fagerstrom RM, Galen B. The national lung screening trial: overview and study design. Radiology. 2011;258:243–53.
Sant M, Allemani C, Santaquilani M, Knijn A, Marchesi F, Capocaccia R. EUROCARE-4. Survival of cancer patients diagnosed in 1995-1999. Results and commentary. Eur J Cancer. 2009;45:931–91.
Hubers AJ, Prinsen CF, Sozzi G, Witte BI, Thunnissen E. Molecular sputum analysis for the diagnosis of lung cancer. Br J Cancer. 2013;109:530–7.
Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4:707–17.
Grossman DC, Curry SJ, Owens DK, Barry MJ, Davidson KW, Doubeni CA. Screening for adolescent idiopathic scoliosis: US preventive services task force recommendation statement. JAMA. 2018;319:165–72.
Brody JS, Spira A. State of the art. Chronic obstructive pulmonary disease, inflammation, and lung cancer. Proc Am Thorac Soc. 2006;3:535–7.
Kadara H, Wistuba II. Field cancerization in non-small cell lung cancer: implications in disease pathogenesis. Proc Am Thorac Soc. 2012;9:38–42.
Belinsky SA, Liechty KC, Gentry FD, Wolf HJ, Rogers J, Vu K. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006;66:3338–44.
Saccomanno G, Saunders RP, Archer VE, Auerbach O, Kuschner M, Beckler PA. Cancer of the lung: the cytology of sputum prior to the development of carcinoma. Acta Cytol. 1965;9:413–23.
Hubers AJ, Heideman DA, Burgers SA, Herder GJ, Sterk PJ, Rhodius RJ. DNA hypermethylation analysis in sputum for the diagnosis of lung cancer: training validation set approach. Br J Cancer. 2015;112:1105–13.
Su Y, Fang H, Jiang F. Integrating DNA methylation and microRNA biomarkers in sputum for lung cancer detection. Clin Epigenetics. 2016;8:109.
Liu D, Peng H, Sun Q, Zhao Z, Yu X, Ge S. The indirect efficacy comparison of DNA methylation in sputum for early screening and auxiliary detection of lung cancer: a meta-analysis. Int J Environ Res Public Health. 2017; 23:14–9.
Hsu HS, Chen TP, Wen CK, Hung CH, Chen CY, Chen JT. Multiple genetic and epigenetic biomarkers for lung cancer detection in cytologically negative sputum and a nested case-control study for risk assessment. J Pathol. 2007;213:412–9.
Guzman L, Depix MS, Salinas AM, Roldan R, Aguayo F, Silva A. Analysis of aberrant methylation on promoter sequences of tumor suppressor genes and total DNA in sputum samples: a promising tool for early detection of COPD and lung cancer in smokers. Diagn Pathol. 2012;7:87.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6.
Wang X, Cao A, Peng M, Hu C, Liu D, Gu T. The value of chest CT scan and tumor markers detection in sputum for early diagnosis of peripheral lung cancer. Zhongguo Fei Ai Za Zhi. 2004;7:58–63.
Leng S, Wu G, Klinge DM, Thomas CL, Casas E, Picchi MA. Gene methylation biomarkers in sputum as a classifier for lung cancer risk. Oncotarget. 2017;8:63978–85.
Belinsky SA, Leng S, Wu G, Thomas CL, Picchi MA, Lee SJ. Gene methylation biomarkers in sputum and plasma as predictors for lung cancer recurrence. Cancer Prev Res (Phila). 2017;10:635–40.
Miyake M, Gomes Giacoia E, Aguilar Palacios D, Rosser CJ. Lung cancer risk assessment for smokers: gene promoter methylation signature in sputum. Biomark Med. 2012;6:512.
Leng S, Do K, Yingling CM, Picchi MA, Wolf HJ, Kennedy TC. Defining a gene promoter methylation signature in sputum for lung cancer risk assessment. Clin Cancer Res. 2012;18:3387–95.
Hwang SH, Kim KU, Kim JE, Kim HH, Lee MK, Lee CH. Detection of HOXA9 gene methylation in tumor tissues and induced sputum samples from primary lung cancer patients. Clin Chem Lab Med. 2011;49:699–704.
Liu Y, Lan Q, Shen M, Jin J, Mumford J, Ren D. Aberrant gene promoter methylation in sputum from individuals exposed to smoky coal emissions. Anticancer Res. 2008;28:2061–6.
Belinsky SA, Grimes MJ, Casas E, Stidley CA, Franklin WA, Bocklage TJ. Predicting gene promoter methylation in non-small-cell lung cancer by evaluating sputum and serum. Br J Cancer. 2007;96:1278–83.
Su SB, Yang LJ, Zhang W, Jin YL, Nie JH, Tong J. p16 and MGMT gene methylation in sputum cells of uranium workers. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2006;24:92–5.
Belinsky SA, Klinge DM, Dekker JD, Smith MW, Bocklage TJ, Gilliland FD. Gene promoter methylation in plasma and sputum increases with lung cancer risk. Clin Cancer Res. 2005;11:6505–11.
Olaussen KA, Soria JC, Park YW, Kim HJ, Kim SH, Ro JY. Assessing abnormal gene promoter methylation in paraffin-embedded sputum from patients with NSCLC. Eur J Cancer. 2005;41:2112–9.
Hubers AJ, Heideman DA, Duin S, Witte BI, de Koning HJ, Groen HJ. DNA hypermethylation analysis in sputum of asymptomatic subjects at risk for lung cancer participating in the NELSON trial: argument for maximum screening interval of 2 years. J Clin Pathol. 2017;7:250–4.
Hubers AJ, Brinkman P, Boksem RJ, Rhodius RJ, Witte BI, Zwinderman AH. Combined sputum hypermethylation and eNose analysis for lung cancer diagnosis. J Clin Pathol. 2014;67:707–11.
Hubers AJ, van der Drift MA, Prinsen CF, Witte BI, Wang Y, Shivapurkar N. Methylation analysis in spontaneous sputum for lung cancer diagnosis. Lung Cancer. 2014;84:127–33.
Hubers AJ, Heideman DA, Yatabe Y, Wood MD, Tull J, Taron M, et al. EGFR mutation analysis in sputum of lung cancer patients: a multitechnique study. Lung Cancer. 2013;82(1):38–43.
Hubers AJ, Heideman DA, Herder GJ, Burgers SA, Sterk PJ, Kunst PW. Prolonged sampling of spontaneous sputum improves sensitivity of hypermethylation analysis for lung cancer. J Clin Pathol. 2012;65:541–5.
Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 2013;10:1003–5.
Li N, Ma J, Guarnera MA, Fang H, Cai L, Jiang F. Digital PCR quantification of miRNAs in sputum for diagnosis of lung cancer. J Cancer Res Clin Oncol. 2014;140:145–50.
Ma J, Li N, Guarnera M, Jiang F. Quantification of plasma miRNAs by digital PCR for cancer diagnosis. Biomark Insights. 2013;8:127–36.
Bhat S, Herrmann J, Armishaw P, Corbisier P, Emslie KR. Single molecule detection in nanofluidic digital array enables accurate measurement of DNA copy number. Anal Bioanal Chem. 2009;394:457–67.
Kiss MM, Ortoleva-Donnelly L, Beer NR, Warner J, Bailey CG, Colston BW. High-throughput quantitative polymerase chain reaction in picoliter droplets. Anal Chem. 2008;80:8975–81.
Kreutz JE, Munson T, Huynh T, Shen F, Du W, Ismagilov RF. Theoretical design and analysis of multivolume digital assays with wide dynamic range validated experimentally with microfluidic digital PCR. Anal Chem. 2011;83:8158–68.
Pinheiro LB, Coleman VA, Hindson CM, Herrmann J, Hindson BJ, Bhat S. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem. 2012;84:1003–11.
Pohl G, Shih IM. Principle and applications of digital PCR. Expert Rev Mol Diagn. 2004;4:41–7.
Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96:9236–41.
Hayden RT, Gu Z, Ingersoll J, Abdul-Ali D, Shi L, Pounds S, et al. Comparison of droplet digital PCR to real-time PCR for quantitative detection of cytomegalovirus. J Clin Microbiol. 2013;51(2):540–6.
Day E, Dear PH, McCaughan F. Digital PCR strategies in the development and analysis of molecular biomarkers for personalized medicine. Methods. 2013;59:101–7.
Li H, Jiang Z, Leng Q, Bai F, Wang J, Ding X. A prediction model for distinguishing lung squamous cell carcinoma from adenocarcinoma. Oncotarget. 2017;8:50704–14.
Yu L, Todd NW, Xing L, Xie Y, Zhang H, Liu Z. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int J Cancer. 2010;127:2870–8.
Xing L, Todd NW, Yu L, Fang H, Jiang F. Early detection of squamous cell lung cancer in sputum by a panel of microRNA markers. Mod Pathol. 2010;23:1157–64.
Xie Y, Todd NW, Liu Z, Zhan M, Fang H, Peng H. Altered miRNA expression in sputum for diagnosis of non-small cell lung cancer. Lung Cancer. 2010;67:170–6.
Anjuman N, Li N, Guarnera M, Stass SA, Jiang F. Evaluation of lung flute in sputum samples for molecular analysis of lung cancer. Clin Transl Med. 2013;2:15.
Jiang F, Todd NW, Li R, Zhang H, Fang H, Stass SA. A panel of sputum-based genomic marker for early detection of lung cancer. Cancer Prev Res (Phila). 2010;3:1571–8.
Jiang F, Todd NW, Qiu Q, Liu Z, Katz RL, Stass SA. Combined genetic analysis of sputum and computed tomography for noninvasive diagnosis of non-small-cell lung cancer. Lung Cancer. 2009;66:58–63.
Qiu Q, Todd NW, Li R, Peng H, Liu Z, Yfantis HG. Magnetic enrichment of bronchial epithelial cells from sputum for lung cancer diagnosis. Cancer. 2008;114:275–83.
Li R, Todd NW, Qiu Q, Fan T, Zhao RY, Rodgers WH. Genetic deletions in sputum as diagnostic markers for early detection of stage I non-small cell lung cancer. Clin Cancer Res. 2007;13:482–7.
Li W, Deng J, Jiang P, Zeng X, Hu S, Tang J. Methylation of the RASSF1A and RARbeta genes as a candidate biomarker for lung cancer. Exp Ther Med. 2012;3:1067–71.
Lee SM, Lee WK, Kim DS, Park JY. Quantitative promoter hypermethylation analysis of RASSF1A in lung cancer: comparison with methylation-specific PCR technique and clinical significance. Mol Med Rep. 2012;5:239–44.
Fischer JR, Ohnmacht U, Rieger N, Zemaitis M, Stoffregen C, Manegold C. Prognostic significance of RASSF1A promoter methylation on survival of non-small cell lung cancer patients treated with gemcitabine. Lung Cancer. 2007;56:115–23.
Chen H, Suzuki M, Nakamura Y, Ohira M, Ando S, Iida T. Aberrant methylation of RASGRF2 and RASSF1A in human non-small cell lung cancer. Oncol Rep. 2006;15:1281–5.
Dingle TC, Sedlak RH, Cook L, Jerome KR. Tolerance of droplet-digital PCR vs real-time quantitative PCR to inhibitory substances. Clin Chem. 2013;59:1670–2.
Wilson IG. Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol. 1997;63:3741–51.
Lemeshow S, Hosmer DW Jr. A review of goodness of fit statistics for use in the development of logistic regression models. Am J Epidemiol. 1982;115:92–106.
Grote HJ, Schmiemann V, Geddert H, Bocking A, Kappes R, Gabbert HE. Methylation of RAS association domain family protein 1A as a biomarker of lung cancer. Cancer. 2006;108:129–34.
Kurimoto K, Hayashi M, Guerrero-Preston R, Koike M, Kanda M, Hirabayashi S. PAX5 gene as a novel methylation marker that predicts both clinical outcome and cisplatin sensitivity in esophageal squamous cell carcinoma. Epigenetics. 2017;12: 865–74.
Campomenosi P, Gini E, Noonan DM, Poli A, D'Antona P, Rotolo N. A comparison between quantitative PCR and droplet digital PCR technologies for circulating microRNA quantification in human lung cancer. BMC Biotechnol. 2016;16:60.
Shen J, Liao J, Guarnera MA, Fang H, Cai L, Stass SA. Analysis of MicroRNAs in sputum to improve computed tomography for lung cancer diagnosis. J Thorac Oncol. 2014;9:33–40.
Su J, Liao J, Gao L, Shen J, Guarnera MA, Zhan M. Analysis of small nucleolar RNAs in sputum for lung cancer diagnosis. Oncotarget. 2016;7:5131–42.
Xing L, Su J, Guarnera MA, Zhang H, Cai L, Zhou R. Sputum microRNA biomarkers for identifying lung cancer in indeterminate solitary pulmonary nodules. Clin Cancer Res. 2015;21:484–9.
Acknowledgements
This study was supported in part by a grant for cancer research from Jiangsu Province Hospital of Nanjing University of Chinese Medicine (Y. S.)
Author information
Authors and Affiliations
Contributions
YS, HF, and FJ conducted the experiments and participated in study design, coordination, and data interpretation, and preparing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was supported in part by a grant for cancer research from the Jiangsu Province Hospital of Nanjing University of Chinese Medicine.
The study was approved by the Nanjing University of Chinese Medicine Institutional Review Board and adhered to the Declaration of Helsinki, and all patients provided written informed consent.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Twenty-nine genes displayed a higher level of methylation in sputum of lung cancer patients vs. controls. Table S2. CVs of the four analyses by ddMSP and qMSP on the same specimens. Table S3. CVs of repeated measures by ddMSP and qMSP on different times. Table S4. CVs of repeated measures by different researches using ddMSP and qMSP. Figure S1. Sensitivities and specificities of measuring DNA methylation of RASSF1A by ddMSP and qMSP for diagnosis of lung cancer in sputum samples of 20 cancer-free controls (normal subjects, N) and 20 patients diagnosed with lung tumor (T). ddMSP analysis of DNA methylation of RASSF1A had a higher sensitivity (55%) than did qMSP (45%) (p = 0.01) for distinguishing lung cancer cases from controls, while maintaining the same specificity (80%). *, p = 0.01. (PDF 235 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Su, Y., Fang, H.B. & Jiang, F. An epigenetic classifier for early stage lung cancer. Clin Epigenet 10, 68 (2018). https://doi.org/10.1186/s13148-018-0502-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-018-0502-3