
Abstract
Background
5,10-Methylenetetrahydrofolate reductase (MTHFR) is a key enzyme in one-carbon metabolism that ensures the availability of methyl groups for methylation reactions. Two single-nucleotide polymorphisms (SNPs) in the MTHFR gene, 677C>T and 1298A>C, result in a thermolabile enzyme with reduced function. These variants, in both the maternal and/or fetal genes, have been associated with pregnancy complications including miscarriage, neural tube defects (NTDs), and preeclampsia (PE), perhaps due to altered capacity for DNA methylation (DNAm). In this study, we assessed the association between MTHFR 677TT and 1298CC genotypes and risk of NTDs, PE, or normotensive intrauterine growth restriction (nIUGR). Additionally, we assessed whether these high-risk genotypes are associated with altered DNAm in the placenta.
Results
In 303 placentas screened for this study, we observed no significant association between the occurrence of NTDs (N = 55), PE (early-onset: N = 28, late-onset: N = 20), or nIUGR (N = 21) and placental (fetal) MTHFR 677TT or 1298CC genotypes compared to healthy pregnancies (N = 179), though a trend of increased 677TT genotype in PE/IUGR together was observed (OR 2.53, p = 0.048). DNAm was profiled in 10 high-risk 677 (677TT + 1298AA), 10 high-risk 1298 (677CC + 1298CC), and 10 reference (677CC + 1298AA) genotype placentas. Linear modeling identified no significantly differentially methylated sites between high-risk 677 or 1298 and reference placentas at a false discovery rate < 0.05 and Δβ ≥ 0.05 using the Illumina Infinium HumanMethylation450 BeadChip. Using a differentially methylated region analysis or separating cytosine-guanine dinucleotides (CpGs) by CpG density to reduce multiple comparisons also did not identify differential methylation. Additionally, there was no consistent evidence for altered methylation of repetitive DNA between high-risk and reference placentas.
Conclusions
We conclude that large-scale, genome-wide disruption in DNAm does not occur in placentas with the high-risk MTHFR 677TT or 1298CC genotypes. Furthermore, there was no evidence for an association of the 1298CC genotype and only a tendency to higher 677TT in pregnancy complications of PE/IUGR. This may be due to small sample sizes or folate repletion in our Canadian population attenuating effects of the high-risk MTHFR variants. However, given our results and the conflicting results in the literature, investigations into alternative mechanisms that may explain the link between MTHFR variants and pregnancy complications, or in populations at risk of folate deficiencies, are warranted.
Similar content being viewed by others


Background
One-carbon metabolism (OCM) is a fundamental biochemical pathway that activates and transfers methyl (CH3) groups for purine synthesis and methylation of DNA, proteins, and lipids, making it important for processes such as DNA synthesis, cellular division, and proliferation. Both functional and dietary deficiencies are thought to contribute to altered OCM cycling. Several B vitamins act as substrates or cofactors for OCM, most notably vitamin B9 or folate, the transporter of methyl groups in OCM. Genetic variants in a central OCM enzyme, 5,10-methylenetetrahydrofolate reductase (MTHFR), have been heavily researched in association with human diseases, such as cardiovascular disease, pregnancy complications, and cancers [1,2,3,4]. MTHFR catalyzes the irreversible reduction of 5,10-methylenetetrahydrofolate (5,10-CH3-THF) to 5-methyltetrahydrofolate (5-CH3-THF). 5-CH3-THF is subsequently used as the substrate for the conversion of homocysteine to methionine, catalyzed by the enzyme methionine reductase. Methionine is then used to synthesize S-adenosylmethionine (SAM), the universal methyl donor for methylation reactions, including DNA methylation (DNAm), catalyzed by DNA methyltransferases (DNMTs). As such, MTHFR is key to directing one-carbon units toward DNAm reactions, which has motivated the investigation of alterations in DNAm as the mechanism underlying the association of genetic variants in MTHFR with various pathologies.
Two single-nucleotide polymorphisms (SNPs) in the MTHFR gene, 677C>T (rs1801133) and 1298A>C (rs1801131), result in reduced MTHFR function in vitro, particularly in the homozygous recessive state [5,6,7,8]. These variants are common in the population; globally, the variant allele frequencies are approximately 0.25–0.3 (dbSNP [9]), though frequencies vary between different populations. These variants have been associated with markers of altered OCM, such as increased levels of homocysteine and altered levels of blood folates [10,11,12,13,14,15,16], most consistently for the 677 variant. High-risk MTHFR genotypes (677TT and 1298CC) or variant alleles (677T and 1298C) have been found in association with a number of reproductive and developmental pathologies. MTHFR 677 and 1298 variants in affected pregnancies or parents have been associated with miscarriage [17,18,19] and neural tube defects [20,21,22,23,24,25]. The 677T allele and 677TT genotype in mothers have been associated with preeclampsia (PE), a maternal hypertensive disorder in pregnancy [26,27,28]. Associations between fetal-placental MTHFR 677TT genotypes have been identified [29], though these are not as well studied as the maternal variants.
Researchers have hypothesized that increased risk of pathology might be attributed to aberrant patterns in DNAm, resulting from altered OCM flux caused by these MTHFR variants [25, 30, 31]. While several studies have investigated the association of the MTHFR 677C>T and 1298A>C variants with altered DNAm, results are inconsistent; some have reported associations between the high-risk homozygous MTHFR genotypes and/or folate levels and altered DNAm [32,33,34,35,36,37], whereas others find no association [38,39,40,41]. As gene expression, DNAm patterns, and metabolic requirements are highly variable between tissues, even these conflicting results ascertained in adult, non-pregnant blood, may not generalize to pregnancy complications.
The placenta is a directly relevant tissue in which to study the interaction between MTHFR variants, altered DNAm, and pregnancy complications. Due to the demand for DNA synthesis, cellular division, and proliferation by the growing fetus and placenta, the requirement for folate during pregnancy increases by approximately 5–10 times the level of non-pregnant women [42]. High-affinity folate receptors on maternal-facing trophoblast cells allow the placenta to transport and concentrate folate from the maternal blood up to three times within the placenta [43, 44], ensuring the availability of this crucial nutrient during development. Consistent with studies in other tissues, the MTHFR 677T allele is associated with reduced MTHFR enzyme function in the placenta [45]. If OCM flux is impaired and DNAm patterns are altered in the placenta due to reduced variant MTHFR function, this could have implications for placental function and thus increase risk of pregnancy complications. Aberrant DNAm in the form of imprinting is known to have significant impact on placental development (reviewed in [46, 47]). Genome-wide or imprinted gene-specific alterations in DNAm have been noted in placental insufficiency complications of intrauterine growth restriction (IUGR) [48] and in early-onset PE [49,50,51]. Additionally, the placenta is a tissue that may be more likely to exhibit altered DNAm in response to reduced MTHFR enzyme function. The placenta exhibits a high degree of within- and between-individual variability in DNAm [52, 53], suggesting that it may be tolerant to changes in DNAm, allowing this organ to adapt to environmental conditions [52,53,54].
To date, no studies have investigated the association between DNAm in the placenta and the high-risk MTHFR 677TT and 1298CC genotypes. This may provide insight in to how the MTHFR variants have been previously associated with pregnancy complications, and potentially help to resolve the currently conflicting literature investigating the association between these variants and DNAm in other tissues. In this study, we evaluated whether fetal high-risk MTHFR genotypes were more prevalent in pregnancy complications of PE, IUGR, and neural tube defects (NTDs) using 303 placental DNA samples. The DNAm patterns of 30 placentas were heavily profiled using both site-specific and genome-wide techniques, including the Illumina Infinium HumanMethylation450 BeadChip array and repetitive DNA methylation, to understand the relationship between high-risk MTHFR 677TT and 1298CC genotypes and DNAm in the placenta.
Methods
Ethics and sample collection
Ethics approval for this study was obtained from the University of British Columbia/Children’s Hospital and Women’s Health Centre of British Columbia Research Ethics Board (H04-70488, H10-01028). Placentas were collected from term deliveries at BC Women’s Hospital and Health Centre and from second trimester stillbirths, elective terminations, and spontaneous abortions through the embryo-fetal pathology laboratory. Cases with a prenatally identified chromosomal abnormality were excluded.
A minimum of two distinct sites were sampled from the fetal side of each placenta after fetal membranes (amnion and chorion) were removed. Samples were washed thoroughly with PBS to remove maternal blood. DNA was extracted by a standard salting-out procedure modified from Miller et al. [55] and quality evaluated using a NanoDrop ND-1000 (Thermo Scientific). One site from each placenta was selected at random for genotyping. As DNAm varies significantly within the placenta [52, 53], DNA was combined in equal amounts from at least two sites to generate a more representative sample in which to evaluate placental DNAm.
Case characteristics
A total of 303 placentas were screened for MTHFR 677 and 1298 polymorphisms. These included 179 placentas from uncomplicated pregnancies, 48 from pregnancies associated with PE (28 early-onset PE, 20 late-onset PE), 21 from pregnancies associated with intrauterine growth restriction in the absence of maternal hypertension (normotensive IUGR, nIUGR), and 55 from pregnancies with a fetal NTD (Table 1). PE was defined according to the Society of Obstetricians and Gynaecologists of Canada (SOGC) criteria as pregnancies with (i) gestational hypertension (BP > 140/90 mmHg) and proteinuria (> 300 g/day) arising after 20 weeks of gestation; (ii) pre-existing hypertension with superimposed gestational hypertension, proteinuria, and/or one or more adverse maternal or fetal conditions; or (iii) gestational hypertension without proteinuria, with one or more adverse maternal or fetal conditions [56]. PE was subdivided into early-onset preeclampsia (EOPE), defined as a diagnosis of PE before 34 weeks of gestation, and late-onset preeclampsia (LOPE), a diagnosis of PE after 34 weeks of gestation [57]. IUGR commonly co-occurs with PE and was also defined following the SOGC criteria as birth weight < 3rd percentile, accounting for both fetal sex and gestational age (GA), or birth weight < 10th percentile with additional clinical findings indicative of poor growth such as uterine artery notching, absent or reversed end-diastolic velocity on Doppler ultrasound, or oligohydramnios [58]. nIUGR was defined as unexplained IUGR without the presence of maternal hypertension. NTDs were defined as a fetus diagnosed with spina bifida, anencephaly, or encephalocele on ultrasound and/or fetal autopsy.
MTHFR genotyping
Placental DNA was genotyped for the MTHFR 677 and 1298 polymorphisms using pyrosequencing. Primer sequences and reaction conditions can be found in Additional file 1: Table S1. Five microliters of PCR product was sequenced on a PyroMark Q96 MD Pyrosequencer (Qiagen) using standard protocols [59]. A subset of the genotyping results from the NTD group (N = 36) has been published elsewhere [60].
Population stratification
Minor allele frequencies for the MTHFR 677 and 1298 SNPs vary significantly between different populations [61,62,63,64], as do the prevalence of NTDs and PE/IUGR pathologies [65]. Both high-risk MTHFR genotypes vary by ethnicity and geography, indicating that selective pressures have influenced their frequencies [62, 66]. Therefore, prior to performing a genetic association analysis, we aimed to assess whether our pregnancy complication groups were matched for ancestry. Maternal self-reported ethnicity was available for only 67% of cases, and no information about the father’s ethnicity was available. We thus used a panel of 57 ancestry-informative marker (AIM) SNPs [67,68,69] that were developed to distinguish between African, European, East Asian, and South Asian ancestries to infer the ancestry of study samples and assess population stratification along three major axes of variation.
Two hundred seventy-seven placental villus DNA samples were successfully genotyped at 53 AIMs using the Sequenom iPlex Gold platform by the Génome Québec Innovation Centre at McGill University, Montréal, Canada, with a call rate > 0.9 for both SNPs and samples. Multidimensional scaling (MDS) with k = 3 dimensions was performed in our study samples (N = 277) in addition to individuals (N = 2157) from African, East Asian, European, and South Asian populations from the 1000 Genomes Project (1kGP) [64] using 50 of the AIM genotypes that were available in both cohorts. This method allows 1kGP samples to be used as ancestry reference populations for our admixed population and has been used to identify ancestry outliers [70, 71]. The first three MDS coordinates were extracted for each sample and used to describe ancestry along a continuum rather than in discrete groups. We believe this better reflects ancestry in admixed populations such as that in Vancouver, as well as potentially better representing variation within an ancestry group. Further description of this method is included in Additional file 2: Methods.
MTHFR genotype and DNAm
To assess the association of MTHFR genotype with DNAm, a subset of 30 control/mild pregnancy complication placentas were selected for in-depth DNAm profiling, hereafter referred to as the placental DNAm samples. None of these placentas had chromosomal abnormalities, as confirmed by multiplex ligation-dependent probe amplification in one or more sites per placenta. The effect of each MTHFR SNP was assessed independently by comparing 10 placentas with reference genotype at each MTHFR SNP (677CC + 1298AA), 10 placentas with the high-risk 677TT genotype in combination with the reference 1298AA (termed “high-risk 677”), and 10 placentas with high-risk 1298CC genotype in combination with the reference 677CC genotype (termed “high-risk 1298”). The reference, high-risk 677, and high-risk 1298 groups were matched by sex, gestational age, birth weight, and maternal reported ethnicity (Table 2). To obtain sufficient numbers in each genotype group, as the high-risk genotypes are relatively rare in our population and we additionally excluded heterozygotes at either locus, some mild pregnancy complication cases (LOPE without IUGR and nIUGR) were included. We previously found no evidence for altered placental DNAm associated with these phenotypes compared to controls in a separate study [51].
Illumina Infinium HumanMethylation450 BeadChip
Combined placental DNA from the 30 placental DNAm samples described in Table 2 was purified using the Qiagen DNeasy Blood and Tissue kit, and 750 ng of this DNA was bisulfite converted using the EZ DNA Methylation kit (Zymo Research) following the manufacturer’s protocols. Samples were processed following the Illumina Infinium HumanMethylation450 BeadChip (450k array) protocol [72] and scanned using the Illumina HiScan 2000. Raw intensity was read into Illumina Genome Studio software 2011.1, and background normalization was applied. Data processing was performed as described in Price et al. [73], including sample quality checks, probe filtering, data normalization, and batch correction. This processing pipeline resulted in a final N = 442,355 cytosine-guanine dinucleotide (CpG) sites from the 450k array for analysis.
Repetitive DNA methylation
In addition to the 450k array, genome-wide DNAm was also assessed using DNAm at repetitive Alu, LINE-1, and ribosomal DNA (rDNA) sequences [74] by pyrosequencing in the 30 placental DNAm samples. These sequences are dispersed throughout the genome, allowing DNAm to be measured at many sites using a single assay per repetitive sequence. DNAm at these three repetitive DNA sequences has been shown to be altered due to different environmental or biological factors [75,76,77,78,79]. Three hundred nanograms of purified combined placental villi DNA was bisulfite converted using the EZ DNA Methylation-Gold Kit (Zymo Research) following the manufacturer’s protocol. Alu and LINE-1 elements were amplified using primer sets designed to complement the L1H and AluSx consensus sequences [80], and rDNA repeats were amplified using primers designed to target the rDNA promoter [81] (Additional file 1: Table S1). PCR products were sequenced on a PyroMark Q96 MD Pyrosequencer (Qiagen) using standard protocols [59]. The DNAm status of each CpG dinucleotide (Alu: N = 3; LINE-1: N = 4; rDNA: N = 26) was evaluated using the PyroQ CpG software (Biotage). For each assay, the correlation of DNAm between CpGs was confirmed, and then an average DNAm was calculated across the CpGs within each assay in each sample.
Statistical analyses
Statistical analyses were conducted in R statistical software [82]. Deviation from Hardy-Weinberg equilibrium (HWE) at the two MTHFR SNPs in controls was assessed using an exact test for HWE. Differences in the distribution of ancestry MDS coordinate values between control and pregnancy complication groups (EOPE, LOPE, nIUGR, NTD) were assessed using the Kolmogorov-Smirnov test. The association between the frequency of MTHFR 677TT and/or 1298CC genotypes and pregnancy complications was assessed using a one-tailed Fisher’s exact test to test the hypothesis that the high-risk genotypes will be increased in pregnancy complications compared to controls. For the placental DNAm samples, 450k array-wide average DNAm and percent outlier probes per sample were calculated as in Price et al. [60]. Altered measures of genome-wide methylation, including 450k array-wide average, percent outlier probes, Alu, LINE-1, and rDNA methylation, were assessed using the Mann-Whitney test. All p values from statistical tests involving multiple comparisons (ancestry MDS coordinate values, altered genome-wide DNAm measures) were corrected for multiple testing using the Bonferroni method.
450k array site-specific differential methylation was also assessed as in Price et al. [60]. Briefly, a linear model with the MTHFR group as the main effect and fetal sex and gestational age included as covariates was fit to every CpG on the array (N = 442,355). Differential methylation results were then extracted for the comparison of high-risk 677 to reference and for that of high-risk 1298 to reference. These comparisons were used to calculate group differences in DNAm (delta-beta, Δβ). Significantly differentially methylated CpG sites were considered as those with a false discovery rate (FDR) < 0.05 and Δβ ≥ 0.05. Two dimension-reduction techniques were additionally used in the 450k array data: a differentially methylated region (DMR) analysis, as in Price et al. [60], and an assessment of differential DNAm depending on the CpG density of the surrounding region. 450k probes were separated into four groups based on the CpG density: high-density islands, island shores, intermediate-density islands, and non-islands, defined as per Price et al. [83], and the unadjusted p value distributions from the linear model were assessed in each CpG density group separately.
Results
Analysis of ancestry-informative markers identifies no significant population stratification
Prior to testing for the association of fetal MTHFR genotypes with NTDs or PE/IUGR groups, we sought to confirm that these pregnancy complication groups were not confounded with ancestry as the frequency of the MTHFR 677 and 1298 variants varies between different ancestry groups [61,62,63]. Self-reported ethnicity was available from mothers for only 67% (203/303) of samples, which were predominately of European (Caucasian) and East Asian ancestries. We thus described ancestry using coordinates 1, 2, and 3 obtained through MDS of genotypes at 50 AIMs (Additional file 3: Table S2) in 277 of our placental samples along with 2157 samples from the 1kGP [64] (Additional file 4: Figure S1). These three MDS coordinates were significantly different between the four major continental populations from 1kGP (Additional file 4: Figure S1). Furthermore, for those samples for which we had both maternal reported ethnicities in addition to AIMs (N = 181), the three ancestry MDS coordinates were highly concordant with maternal reported ethnicity (Additional file 5: Figure S2). These findings suggest that this method is adequate to describe major patterns of genetic ancestry. There was no significant difference in the distribution of ancestry MDS coordinate values 1, 2, and 3 between the NTD, PE, and nIUGR pathology groups in comparison to controls (Fig. 1). We thus concluded that our pathology groups do not show evidence of confounding by ancestry.

Distribution of ancestry derived from multidimensional scaling of AIM genotypes in control and pregnancy complication placentas. In N = 28 EOPE, N = 20 LOPE, N = 21 nIUGR, or N = 55 NTD placentas, there were no significant differences in the distribution of ancestry MDS coordinate values compared to N = 179 controls at either of the three ancestry MDS coordinates (Kolmogorov-Smirnov tests, Bonferroni-corrected p > 0.05). This suggests that there is no population stratification by ancestry in the groups selected for this study. EOPE early-onset preeclampsia, LOPE late-onset preeclampsia, nIUGR normotensive intrauterine growth restriction, NTD neural tube defect
MTHFR 677TT and 1298CC genotypes are not significantly associated with PE or NTDs
To investigate whether the MTHFR 677TT and 1298CC genotypes were associated with PE, nIUGR, or NTD pathologies, we genotyped placentas at these two loci from 179 control, 28 EOPE, 20 LOPE, 21 nIUGR, and 55 NTD pregnancies. Neither SNP deviated from HWE in controls (Additional file 6: Table S3). In our population of 303 placentas collected in Vancouver, Canada, the frequencies of the variant MTHFR 677T and 1298C alleles were 0.295 and 0.290, respectively. There was no significant increase in the frequency of the high-risk MTHFR 677TT or 1298CC genotypes in EOPE, LOPE, nIUGR, or NTD cases compared to controls (Table 3). There was, however, a tendency for increased MTHFR 677TT genotype in placentas from pregnancies complicated by placental pathologies (PE or nIUGR). When considered together (PE or nIUGR; N = 69), the increase in MTHFR 677TT frequency compared to controls was nominally significant (OR = 2.53, p = 0.048).
High-risk MTHFR 677 and 1298 genotypes are not associated with altered genome-wide DNAm in the placenta
Due to the central role that MTHFR plays in OCM, the high-risk MTHFR genotypes are often hypothesized to affect the cell’s ability to methylate DNA. We anticipated that such effects could potentially be more pronounced in the placenta due to its high demand for folate in pregnancy. We selected a subset of 30 placental samples with no, or mild, pathology in which to profile DNAm using both genome-wide and site-specific approaches. The selected samples were of three MTHFR genotype groups: (1) reference (N = 10; MTHFR 677CC + 1298AA), (2) high-risk 677 (N = 10; MTHFR 677TT + 1298AA), and (3) high-risk 1298 (N = 10; MTHFR 677CC + 1298CC). No cases with high-risk genotypes at both loci were available in our population to test.
First, these 30 placental DNAm samples were run on the 450k array, from which several measures of DNAm were obtained. Array-wide DNAm was calculated by averaging of 442,355 CpG sites in each sample. This array-wide measure of DNAm did not differ significantly between either of the high-risk MTHFR groups and the reference group (Table 4). Altered genome-wide DNAm might not be a characteristic of all carriers of the MTHFR variants; thus, we also calculated the percentage of outlier CpG sites from the 450k array for each sample to identify individuals exhibiting outlying patterns of DNAm [84]. Though there was no significant difference in outlier CpGs between the high-risk 677 and reference group (Table 4), there was a trend for more outlying CpG sites in the high-risk 1298 group than in the reference (Table 4, Bonferroni-corrected p = 0.058).
Next, the methylation of repetitive DNA sequences was assessed in the 30 placental DNAm samples. Repetitive DNAm assays target numerous sites in the genome that are not well covered by the 450k array, and thus give an additional measure of genome-wide DNAm. No significant alterations in the DNAm of Alu, LINE-1, or rDNA sequences were identified between either of the high-risk MTHFR genotype groups and the reference genotype group (Table 4). Slightly higher methylation was seen for the high-risk 677 group in all comparisons, though the range of values was similar. There was, however, a trend for decreased LINE-1 DNAm in the high-risk 1298 group compared to the reference group (nominal p = 0.052), but this is not meaningful after correction for multiple comparisons. Overall, we find no conclusive evidence for altered genome-wide DNAm in association with the high-risk 677 or high-risk 1298 genotypes in the placenta using these DNAm measures.
High-risk MTHFR 677 and 1298 genotypes are not associated with altered site-specific DNAm in the placenta
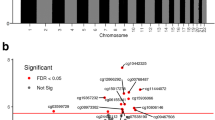
The DNAm status of individual CpG sites targeted by the 450k array in association with the high-risk MTHFR genotype groups was next assessed. A linear model was fit to each CpG site to test for differential methylation by the genotype group, including sex and gestational age at birth as covariates. None of the 442,355 CpG sites was differentially methylated at a FDR < 0.05 and methylation difference (Δβ) > 0.05 in either of the high-risk MTHFR genotype groups compared to the reference group (Fig. 2).

450k array-wide differential DNAm volcano plots in high-risk MTHFR 677 and high-risk 1298 placentas. Differential methylation was determined using a linear model with the MTHFR group as the main effect and fetal sex and gestational age included as covariates. The magnitude of difference (Δβ) between risk groups and reference group is depicted on the x-axis, and the significance of the comparison (−log10(adjusted p value)) is on the y-axis, for every CpG tested on the 450k array (N = 442,355). a Differential methylation results between high-risk 677 and reference placentas. b Differential methylation results between high-risk 1298 and reference placentas. Neither comparison identified any CpG sites differentially methylated between the high-risk MTHFR placentas and reference placentas. 450k array Illumina Infinium HumanMethylation450 BeadChip, FDR false discovery rate
Following this finding, two dimension-reduction techniques were utilized to explore whether identification of differences between high-risk MTHFR groups and controls in the 450k array data was limited due to a small sample size or large number of test sites. Due to structural or functional differences, some genomic regions may be more vulnerable to the effects of a reduced ability to methylate DNA potentially caused by the presence of variant MTHFR enzymes. As such, 450k probes were separated into four groups based on the CpG density of the surrounding region: high-density islands, island shores, intermediate-density islands, and non-islands. Additionally, a DMR finding tool was utilized to identify whether any DMRs existed between high-risk MTHFR genotype placentas and controls. Unadjusted p value distributions did not show differential methylation at any of the four CpG density groups between high-risk MTHFR and reference placentas (Additional file 7: Figure S3), nor were any significant DMRs identified. Given these results, we conclude that large-scale alterations in DNAm at CpG sites measured by the 450k array in the placenta are not commonly associated with high-risk 677 or 1298 MTHFR genotypes in our population.
Discussion
Altered DNAm has been proposed as a mechanism through which MTHFR 677TT and 1298CC genotypes have been associated with pregnancy complications and other pathologies [25, 30, 31]. In this study, we sought to investigate alterations in DNAm in association with high-risk MTHFR 677TT and 1298CC genotypes in the placenta, a crucial tissue for the development of the fetus and a healthy pregnancy. Despite deeply profiling DNAm in N = 10 high-risk 677, N = 10 high-risk 1298, and N = 10 reference placentas using a variety of measures, we identified no evidence for altered placental genome-wide or site-specific DNAm in association with high-risk MTHFR genetic variants.
Given the fundamental involvement of OCM in activating and transporting methyl units, if the variant MTHFR alleles influence DNAm, this effect is predicted to be widespread and not gene-specific [85]. By using the 450k array, we interrogated DNAm at over 440,000 sites in the placental genome, assessing specific CpG sites and also genome-wide trends. This array covers 99% of RefSeq genes and is widely dispersed across genomic features and, therefore, can provide an accurate reflection of genome-wide changes associated with specific genomic features or gene regulation. No significant differences in the numerous measures of altered 450k array genome-wide or site-specific DNAm were identified, despite additionally utilizing dimension-reduction techniques to account for a small sample size and large number of test sites. DNAm at repetitive DNA sequences, including Alu and LINE-1 repetitive elements and rDNA repeats, was also assessed, as they are not well covered by the array, and they allow us to interrogate numerous locations in the genome in one pyrosequencing assay. The Alu and LINE-1 repetitive elements have previously been used as surrogate measures for genome-wide DNAm [74, 86], and all three repetitive sequences have exhibited alterations in DNAm in certain pathologies and in response to environmental exposures [75,76,77,78,79]. Though small sample size limited our power to detect significant differences in DNAm in this study, we aimed to mitigate this by deeply profiling the 30 placental DNAm samples using a variety of measures of DNAm to assess whether any differential methylation exists in association with the high-risk MTHFR genotypes. Our study cannot fully exclude that subtle differences in placental DNAm exist in association with high-risk MTHFR genotypes or that a subset of at-risk placentas might show changes in DNAm while the groups as a whole did not. Despite this, the results from these numerous genome-wide assays reveal that at the very least, large magnitude and/or array-wide differential methylation does not commonly occur in association with high-risk MTHFR genotype in the placenta.
Our study is the first that has investigated the associations between MTHFR 677 and 1298 variants and altered DNAm in the placenta, and only the second that has done so using a genome-wide DNAm microarray platform. Numerous studies have investigated altered DNAm in association with MTHFR 677 and/or 1298 variants using different measures of genome-wide DNAm and/or targeted gene DNAm, summarized in Table 5. Results from these various studies, mainly in the blood, are conflicting. Certain studies have found associations between MTHFR 677 or 1298 polymorphisms and altered DNAm; however, many do not find significant associations with altered DNAm or only find altered DNAm in the presence of low levels of OCM nutrients (Table 5). Some of these inconsistencies may be explained by the use of different measures of altered DNAm (i.e., genome-wide, candidate site-specific, repetitive element DNAm) between studies, lack of multiple-test correction, use of different tissues, or inconsistencies in accounting for confounding variables. Nonetheless, the effect of the MTHFR 677 and 1298 variants on DNAm is clearly complex.
Several studies reviewed in Table 5 suggest that altered DNAm in association with MTHFR 677 and 1298 variants might only be present under limited folate conditions [33, 35]. The presence of folate stabilizes the variant MTHFR 677 enzyme [87], and adequate folate attenuates the effects of high-risk MTHFR 677TT genotype on increased homocysteine [88, 89]. Due to the retrospective nature of the study, we were unable to assess folate concentrations in the placenta or maternal blood and did not have complete information on maternal folic acid supplementation. Though folate status was unknown for the cases in this study, we assume that most of the women in our Canadian cohort were folate-replete due to folic acid fortification in cereal and grain, increased literacy around healthy pregnancies, and high uptake of gestational monitoring. Additionally, in a study of 368 pregnant women in Toronto, Canada, with similar demographics as our population in Vancouver, Plumptre et al. found that none of these women were folate deficient during pregnancy, even considering that 7% of women did not take folic acid supplements [90]. It is possible that in the presence of adequate folate levels, the activity of the variant MTHFR 677 or 1298 enzymes in the placentas of our study was not diminished enough to result in a compromised OCM and altered DNAm. Despite this potential limitation, investigating alterations in placental DNAm in association with MTHFR variants in a folate-replete population under the hypothesis that this may increase risk of pregnancy complications is still warranted. Fortification of grain products with folic acid has not entirely reduced the incidence of NTDs in folate-replete populations [91, 92]; in Canada, NTDs are the most common congenital abnormality [93]. Additionally, pathologies such as PE and IUGR are also present at a high frequency in folate-replete populations, and associations between PE and MTHFR have been observed in such populations [27], indicating a mechanism for association with pathology beyond low-folate/folic acid status.
In our study population, we found no significant association of NTDs, EOPE, LOPE, or nIUGR with high-risk 677TT or 1298CC placental genotypes, although there was a tendency to increased MTHFR 677TT in pregnancies affected by PE or IUGR as a whole (OR 2.53, p = 0.048). This trend is consistent with the literature noting an increased risk of PE in association with the 677T allele in both the maternal blood and the placenta [27, 29]. In a recent meta-analysis of 52 different studies, with a combined total of 7398 PE cases and 11,230 controls, Wu et al. identified a significantly increased risk of PE in association with the MTHFR 677T allele [28]. However, in 1103 cases and 988 controls, no association between the MTHFR 1298C allele and PE [28] was found. As for NTDs, our data was not suggestive of any association with fetal MTHFR 677TT or 1298CC genotype. Sample size limited our power to detect significant differences between study groups; however, few studies have investigated associations between placental/fetal MTHFR variants and PE/IUGR and NTD pathologies in the Canadian population post-folic acid fortification, and the main focus of the current research was to assess altered placental DNAm in association with high-risk MTHFR genotypes. Larger studies in NTDs have identified increased risk in association with the 677 variant [25], but there is inconsistent evidence for an association with the 1298CC genotype [24, 94].
Population stratification, the presence of systematic differences in allele frequencies between cases and controls, typically due to differences in ancestry, can be a limitation of genetic or epigenetic association studies. Specifically, false positive or negative results can be a consequence of failing to match study groups on this variable. To address this, we utilized a novel approach to assess population stratification in our study groups using three continuous variables of ancestry based on a MDS analysis of a panel of AIMs. This is similar to studies using MDS of genome-wide genotypes (i.e., from a SNP array or DNA sequencing) in study samples combined with ancestry reference populations to identify ancestry outliers, select homogeneous groups, or infer ancestry [70, 71, 95], and to studies that include principal components or MDS coordinates highly associated with ancestry in statistical models to correct for ancestry [96, 97]. Other potential confounding factors for our study, such as maternal smoking status, diet, and medications taken during pregnancy, were not well documented in all cases included in this study and thus could have resulted in heterogeneity between study groups that we were unable to account for in statistical modeling.
Currently, the evidence supporting the relationship between MTHFR 677 or 1298 variant and pathology or altered DNAm is not conclusive enough for physicians to support implementing MTHFR genetic testing as a clinical practice [98]. Despite this, MTHFR genotyping is available from 50 certified labs in the USA [98], and testing is widely promoted in the naturopathic field, where patients are told that a “faulty genotype” may explain a list of symptoms and diseases including “anxiousness, adrenal fatigue, brain fog, cervical dysplasia, increased risk of many cancers, low thyroid, leaky gut, high blood pressure, heart attacks, stroke, Alzheimer’s disease, diabetes, and miscarriages” (https://sciencebasedmedicine.org/dubious-mthfr-genetic-mutation-testing/). These patients are advised to take supplements containing “methyl folate” and “methyl B12” to increase methylation and decrease their risk of disease development (https://www.jillcarnahan.com/2013/05/12/mthfr-gene-mutation-whats-the-big-deal-about-methylation/). Our findings, coupled with variable results from other studies, suggest that these variants may not be of such strong concern in terms of DNAm, particularly in healthy individuals meeting folate requirements; however, studies with larger sample sizes are required to validate this. At the very least, the negative results from our study suggest that if these variants have an effect on placental and thereby newborn health in Canada, it may not be through altered DNA methylation.
Conclusions
DNA methylation (DNAm) alterations have been proposed to be the link between MTHFR 677C>T and 1298A>C variants and increased risk of pregnancy complications. In this novel study of DNAm in human placentas of high-risk MTHFR 677TT and 1298CC individuals, we did not find evidence of altered DNAm associated with these genotypes in numerous measures of genome-wide and CpG site-specific methylation. We conclude that widespread changes in DNAm do not occur in the placentas of MTHFR 677 and 1298 variant carriers in our folate-replete population. Further studies with larger sample sizes and/or in populations that are folate deficient may support or refute our results. The results from this study suggest that factors other than alterations in DNAm may contribute to the previously reported association between high-risk MTHFR genotypes and pathology.
Abbreviations
- 1kGP:
-
1000 Genomes Project
- 450k array:
-
Illumina Infinium HumanMethylation450 BeadChip
- AIMs:
-
Ancestry-informative markers
- CpG:
-
Cytosine-guanine dinucleotide
- DMR:
-
Differentially methylated region
- DNAm:
-
DNA methylation
- EOPE:
-
Early-onset preeclampsia
- FDR:
-
False discovery rate
- HWE:
-
Hardy-Weinberg equilibrium
- IUGR:
-
Intrauterine growth restriction
- LOPE:
-
Late-onset preeclampsia
- MDS:
-
Multidimensional scaling
- MTHFR:
-
5,10-Methylenetetrahydrofolate reductase
- nIUGR:
-
Normotensive intrauterine growth restriction
- NTD:
-
Neural tube defect
- OCM:
-
One-carbon metabolism
- PE:
-
Preeclampsia
- rDNA:
-
Ribosomal DNA
- SNP:
-
Single-nucleotide polymorphism
- Δβ:
-
Delta-beta
References
Botto LD, Yang Q. 5,10-methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am J Epidemiol. 2000;151(9):862–77.
Kim YI. Methylenetetrahydrofolate reductase polymorphisms, folate, and cancer risk: a paradigm of gene-nutrient interactions in carcinogenesis. Nutr Rev. 2000;58(7):205–9.
Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Schouten EG, MTHFR Studies Collaboration Group. MTHFR 677C-->T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA. 2002;288(16):2023–31.
Kumar A, Kumar P, Prasad M, Sagar R, Yadav AK, Pandit AK, Jali VP, Pathak A. Association of C677T polymorphism in the methylenetetrahydrofolate reductase gene (MTHFR gene) with ischemic stroke: a meta-analysis. Neurol Res. 2015;37(7):568–77.
Kang SS, Wong PW, Susmano A, Sora J, Norusis M, Ruggie N. Thermolabile methylenetetrahydrofolate reductase: an inherited risk factor for coronary artery disease. Am J Hum Genet. 1991;48(3):536–45.
van der Put NM, Gabreels F, Stevens EM, Smeitink JA, Trijbels FJ, Eskes TK, van den Heuvel LP, Blom HJ. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am J Hum Genet. 1998;62(5):1044–51.
Weisberg I, Tran P, Christensen B, Sibani S, Rozen R. A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol Genet Metab. 1998;64(3):169–72.
Weisberg IS, Jacques PF, Selhub J, Bostom AG, Chen Z, Curtis Ellison R, Eckfeldt JH, Rozen R. The 1298A-->C polymorphism in methylenetetrahydrofolate reductase (MTHFR): in vitro expression and association with homocysteine. Atherosclerosis. 2001;156(2):409–15.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Engbersen AM, Franken DG, Boers GH, Stevens EM, Trijbels FJ, Blom HJ. Thermolabile 5,10-methylenetetrahydrofolate reductase as a cause of mild hyperhomocysteinemia. Am J Hum Genet. 1995;56(1):142–50.
Bagley PJ, Selhub J. A common mutation in the methylenetetrahydrofolate reductase gene is associated with an accumulation of formylated tetrahydrofolates in red blood cells. Proc Natl Acad Sci U S A. 1998;95(22):13217–20.
Friedman G, Goldschmidt N, Friedlander Y, Ben-Yehuda A, Selhub J, Babaey S, Mendel M, Kidron M, Bar-On H. A common mutation A1298C in human methylenetetrahydrofolate reductase gene: association with plasma total homocysteine and folate concentrations. J Nutr. 1999;129(9):1656–61.
Dekou V, Whincup P, Papacosta O, Ebrahim S, Lennon L, Ueland PM, Refsum H, Humphries SE, Gudnason V. The effect of the C677T and A1298C polymorphisms in the methylenetetrahydrofolate reductase gene on homocysteine levels in elderly men and women from the British Regional Heart Study. Atherosclerosis. 2001;154(3):659–66.
Shelnutt KP, Kauwell GP, Chapman CM, Gregory JF 3rd, Maneval DR, Browdy AA, Theriaque DW, Bailey LB. Folate status response to controlled folate intake is affected by the methylenetetrahydrofolate reductase 677C-->T polymorphism in young women. J Nutr. 2003;133(12):4107–11.
Tanaka T, Scheet P, Giusti B, Bandinelli S, Piras MG, Usala G, Lai S, Mulas A, Corsi AM, Vestrini A, Sofi F, Gori AM, Abbate R, Guralnik J, Singleton A, Abecasis GR, Schlessinger D, Uda M, Ferrucci L. Genome-wide association study of vitamin B6, vitamin B12, folate, and homocysteine blood concentrations. Am J Hum Genet. 2009;84(4):477–82.
Clarke R, Bennett DA, Parish S, Verhoef P, Dotsch-Klerk M, Lathrop M, Xu P, Nordestgaard BG, Holm H, Hopewell JC, Saleheen D, Tanaka T, Anand SS, Chambers JC, Kleber ME, Ouwehand WH, Yamada Y, Elbers C, Peters B, Stewart AF, Reilly MM, Thorand B, Yusuf S, Engert JC, Assimes TL, Kooner J, Danesh J, Watkins H, Samani NJ, Collins R, Peto R, MTHFR Studies Collaborative Group. Homocysteine and coronary heart disease: meta-analysis of MTHFR case-control studies, avoiding publication bias. PLoS Med. 2012;9(2):e1001177.
Kim SY, Park SY, Choi JW, Kim DJ, Lee SY, Lim JH, Han JY, Ryu HM, Kim MH. Association between MTHFR 1298A>C polymorphism and spontaneous abortion with fetal chromosomal aneuploidy. Am J Reprod Immunol. 2011;66(4):252–8.
Behjati R, Modarressi MH, Jeddi-Tehrani M, Dokoohaki P, Ghasemi J, Zarnani AH, Aarabi M, Memariani T, Ghaffari M, Akhondi MA. Thrombophilic mutations in Iranian patients with infertility and recurrent spontaneous abortion. Ann Hematol. 2006;85(4):268–71.
Zetterberg H, Regland B, Palmer M, Ricksten A, Palmqvist L, Rymo L, Arvanitis DA, Spandidos DA, Blennow K. Increased frequency of combined methylenetetrahydrofolate reductase C677T and A1298C mutated alleles in spontaneously aborted embryos. Eur J Hum Genet. 2002;10(2):113–8.
De Marco P, Calevo MG, Moroni A, Arata L, Merello E, Finnell RH, Zhu H, Andreussi L, Cama A, Capra V. Study of MTHFR and MS polymorphisms as risk factors for NTD in the Italian population. J Hum Genet. 2002;47(6):319–24.
Parle-McDermott A, Mills JL, Kirke PN, O’Leary VB, Swanson DA, Pangilinan F, Conley M, Molloy AM, Cox C, Scott JM, Brody LC. Analysis of the MTHFR 1298A-->C and 677C-->T polymorphisms as risk factors for neural tube defects. J Hum Genet. 2003;48(4):190–3.
Rampersaud E, Melvin EC, Siegel D, Mehltretter L, Dickerson ME, George TM, Enterline D, Nye JS, Speer MC, NTD Collaborative Group. Updated investigations of the role of methylenetetrahydrofolate reductase in human neural tube defects. Clin Genet. 2003;63(3):210–4.
Kirke PN, Mills JL, Molloy AM, Brody LC, O’Leary VB, Daly L, Murray S, Conley M, Mayne PD, Smith O, Scott JM. Impact of the MTHFR C677T polymorphism on risk of neural tube defects: case-control study. BMJ. 2004;328(7455):1535–6.
Yadav U, Kumar P, Yadav SK, Mishra OP, Rai V. Polymorphisms in folate metabolism genes as maternal risk factor for neural tube defects: an updated meta-analysis. Metab Brain Dis. 2015;30(1):7–24.
Yang Y, Chen J, Wang B, Ding C, Liu H. Association between MTHFR C677T polymorphism and neural tube defect risks: a comprehensive evaluation in three groups of NTD patients, mothers, and fathers. Birth Defects Res A Clin Mol Teratol. 2015;103(6):488–500.
Sohda S, Arinami T, Hamada H, Yamada N, Hamaguchi H, Kubo T. Methylenetetrahydrofolate reductase polymorphism and pre-eclampsia. J Med Genet. 1997;34(6):525–6.
Chedraui P, Salazar-Pousada D, Villao A, Escobar GS, Ramirez C, Hidalgo L, Perez-Lopez FR, Genazzani A, Simoncini T. Polymorphisms of the methylenetetrahydrofolate reductase gene (C677T and A1298C) in nulliparous women complicated with preeclampsia. Gynecol Endocrinol. 2014;30(5):392–6.
Wu X, Yang K, Tang X, Sa Y, Zhou R, Liu J, Luo Y, Tang W. Folate metabolism gene polymorphisms MTHFR C677T and A1298C and risk for preeclampsia: a meta-analysis. J Assist Reprod Genet. 2015;32(5):797–805.
Chedraui P, Andrade ME, Salazar-Pousada D, Escobar GS, Hidalgo L, Ramirez C, Spaanderman ME, Kramer BW, Gavilanes AW. Polymorphisms of the methylenetetrahydrofolate reductase gene (C677T and A1298C) in the placenta of pregnancies complicated with preeclampsia. Gynecol Endocrinol. 2015;31(7):569–72.
Blom HJ, Shaw GM, den Heijer M, Finnell RH. Neural tube defects and folate: case far from closed. Nat Rev Neurosci. 2006;7(9):724–31.
McKay JA, Groom A, Potter C, Coneyworth LJ, Ford D, Mathers JC, Relton CL. Genetic and non-genetic influences during pregnancy on infant global and site specific DNA methylation: role for folate gene variants and vitamin B(12). PLoS One. 2012;7(3):e33290. https://doi.org/10.1371/journal.pone.0033290.
Stern LL, Mason JB, Selhub J, Choi SW. Genomic DNA hypomethylation, a characteristic of most cancers, is present in peripheral leukocytes of individuals who are homozygous for the C677T polymorphism in the methylenetetrahydrofolate reductase gene. Cancer Epidemiol Biomark Prev. 2000;9(8):849–53.
Friso S, Choi SW, Girelli D, Mason JB, Dolnikowski GG, Bagley PJ, Olivieri O, Jacques PF, Rosenberg IH, Corrocher R, Selhub J. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci U S A. 2002;99(8):5606–11.
Castro R, Rivera I, Ravasco P, Camilo M, Jakobs C, Blom H, de Almeida IT. 5,10-methylenetetrahydrofolate reductase (MTHFR) 677C-->T and 1298A-->C mutations are associated with DNA hypomethylation. J Med Genet. 2004;41(6):454–8.
Friso S, Girelli D, Trabetti E, Olivieri O, Guarini P, Pignatti PF, Corrocher R, Choi SW. The MTHFR 1298A>C polymorphism and genomic DNA methylation in human lymphocytes. Cancer Epidemiol Biomark Prev. 2005;14(4):938–43.
Weiner AS, Boyarskikh UA, Voronina EN, Mishukova OV, Filipenko ML. Methylenetetrahydrofolate reductase C677T and methionine synthase A2756G polymorphisms influence on leukocyte genomic DNA methylation level. Gene. 2014;533(1):168–72.
Llanos AA, Marian C, Brasky TM, Dumitrescu RG, Liu Z, Mason JB, Makambi KH, Spear SL, Kallakury BV, Freudenheim JL, Shields PG. Associations between genetic variation in one-carbon metabolism and LINE-1 DNA methylation in histologically normal breast tissues. Epigenetics. 2015;10(8):727–35.
Narayanan S, McConnell J, Little J, Sharp L, Piyathilake CJ, Powers H, Basten G, Duthie SJ. Associations between two common variants C677T and A1298C in the methylenetetrahydrofolate reductase gene and measures of folate metabolism and DNA stability (strand breaks, misincorporated uracil, and DNA methylation status) in human lymphocytes in vivo. Cancer Epidemiol Biomark Prev. 2004;13(9):1436–43.
Gomes MVM, Toffoli LV, Arruda DW, Soldera LM, Pelosi GG, Neves-Souza RD, Freitas ER, Castro DT, Marquez AS. Age-related changes in the global DNA methylation profile of leukocytes are linked to nutrition but are not associated with the MTHFR C677T genotype or to functional capacities. PLoS One. 2012;7(12):e52570. https://doi.org/10.1371/journal.pone.0052570.
de Arruda ITS, Persuhn DC, de Oliveira NFP. The MTHFR C677T polymorphism and global DNA methylation in oral epithelial cells. Genet Mol Biol. 2013;36(4):490–3.
Wang L, Shangguan S, Chang S, Yu X, Wang Z, Lu X, Wu L, Zhang T. Determining the association between methylenetetrahydrofolate reductase (MTHFR) gene polymorphisms and genomic DNA methylation level: a meta-analysis. Birth Defects Res A Clin Mol Teratol. 2016;106(8):667–74.
Antony AC. In utero physiology: role of folic acid in nutrient delivery and fetal development. Am J Clin Nutr. 2007;85(2):598S–603S.
Solanky N, Requena Jimenez A, D’Souza SW, Sibley CP, Glazier JD. Expression of folate transporters in human placenta and implications for homocysteine metabolism. Placenta. 2010;31(2):134–43.
Laanpere M, Altmae S, Stavreus-Evers A, Nilsson TK, Yngve A, Salumets A. Folate-mediated one-carbon metabolism and its effect on female fertility and pregnancy viability. Nutr Rev. 2010;68(2):99–113.
Daly SF, Molloy AM, Mills JL, Lee YJ, Conley M, Kirke PN, Weir DG, Scott JM. The influence of 5,10 methylenetetrahydrofolate reductase genotypes on enzyme activity in placental tissue. Br J Obstet Gynaecol. 1999;106(11):1214–8.
Frost JM, Moore GE. The importance of imprinting in the human placenta. PLoS Genet. 2010;6(7):e1001015. https://doi.org/10.1371/journal.pgen.1001015.
Varmuza S, Miri K. What does genetics tell us about imprinting and the placenta connection? Cell Mol Life Sci. 2015;72(1):51–72.
Bourque DK, Avila L, Penaherrera M, von Dadelszen P, Robinson WP. Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta. 2010;31(3):197–202.
Yuen RK, Penaherrera MS, von Dadelszen P, McFadden DE, Robinson WP. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet. 2010;18(9):1006–12.
Gao WL, Li D, Xiao ZX, Liao QP, Yang HX, Li YX, Ji L, Wang YL. Detection of global DNA methylation and paternally imprinted H19 gene methylation in preeclamptic placentas. Hypertens Res. 2011;34(5):655–61.
Wilson SL, Leavey K, Cox B, Robinson WP. Mining DNA methylation alterations towards a classification of placental pathologies. Hum Mol Genet. 2018;27(1):135–46.
Avila L, Yuen RK, Diego-Alvarez D, Penaherrera MS, Jiang R, Robinson WP. Evaluating DNA methylation and gene expression variability in the human term placenta. Placenta. 2010;31(12):1070–7.
Penaherrera MS, Jiang R, Avila L, Yuen RK, Brown CJ, Robinson WP. Patterns of placental development evaluated by X chromosome inactivation profiling provide a basis to evaluate the origin of epigenetic variation. Hum Reprod. 2012;27(6):1745–53.
Novakovic B, Yuen RK, Gordon L, Penaherrera MS, Sharkey A, Moffett A, Craig JM, Robinson WP, Saffery R. Evidence for widespread changes in promoter methylation profile in human placenta in response to increasing gestational age and environmental/stochastic factors. BMC Genomics. 2011;12:529. https://doi.org/10.1186/1471-2164-12-529.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
Magee LA, Helewa M, Moutquin JM, von Dadelszen P, Hypertension Guideline Committee, Strategic Training Initiative in Research in the Reproductive Health Sciences (STIRRHS) Scholars. Diagnosis, evaluation, and management of the hypertensive disorders of pregnancy. J Obstet Gynaecol Can. 2008;30(3 Suppl):S1–48.
von Dadelszen P, Magee LA, Roberts JM. Subclassification of preeclampsia. Hypertens Pregnancy. 2003;22(2):143–8.
Kramer MS, Platt RW, Wen SW, Joseph KS, Allen A, Abrahamowicz M, Blondel B, Breart G, Fetal/Infant Health Study Group of the Canadian Perinatal Surveillance System. A new and improved population-based Canadian reference for birth weight for gestational age. Pediatrics. 2001;108(2):E35.
Royo JL, Hidalgo M, Ruiz A. Pyrosequencing protocol using a universal biotinylated primer for mutation detection and SNP genotyping. Nat Protoc. 2007;2(7):1734–9.
Price EM, Penaherrera MS, Portales-Casamar E, Pavlidis P, Van Allen MI, McFadden DE, Robinson WP. Profiling placental and fetal DNA methylation in human neural tube defects. Epigenetics Chromatin. 2016;9:6. https://doi.org/10.1186/s13072-016-0054-8. eCollection 2016
Pepe G, Camacho Vanegas O, Giusti B, Brunelli T, Marcucci R, Attanasio M, Rickards O, De Stefano GF, Prisco D, Gensini GF, Abbate R. Heterogeneity in world distribution of the thermolabile C677T mutation in 5,10-methylenetetrahydrofolate reductase. Am J Hum Genet. 1998;63(3):917–20.
Wilcken B, Bamforth F, Li Z, Zhu H, Ritvanen A, Redlund M, Stoll C, Alembik Y, Dott B, Czeizel A, Gelman-Kohan Z, Scarano G, Bianca S, Ettore G, Tenconi R, Bellato S, Scala I, Mutchinick O, Lopez M, de Walle H, Hofstra R, Joutchenko L, Kavteladze L, Bermejo E, Martinez-Frias M, Gallagher M, Erickson J, Vollset S, Mastroiacovo P, Andria G, Botto L. Geographical and ethnic variation of the 677C>T allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): findings from over 7000 newborns from 16 areas world wide. J Med Genet. 2003;40(8):619–25.
Esfahani ST, Cogger EA, Caudill MA. Heterogeneity in the prevalence of methylenetetrahydrofolate reductase gene polymorphisms in women of different ethnic groups. J Am Diet Assoc. 2003;103(2):200–7.
1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
Bryant AS, Worjoloh A, Caughey AB, Washington AE. Racial/ethnic disparities in obstetric outcomes and care: prevalence and determinants. Am J Obstet Gynecol. 2010;202(4):335–43.
Wang X, Fu J, Li Q, Zeng D. Geographical and ethnic distributions of the MTHFR C677T, A1298C and MTRR A66G gene polymorphisms in Chinese populations: a meta-analysis. PLoS One. 2016;11(4):e0152414. https://doi.org/10.1371/journal.pone.0152414.
Phillips C, Salas A, Sanchez JJ, Fondevila M, Gomez-Tato A, Alvarez-Dios J, Calaza M, de Cal MC, Ballard D, Lareu MV, Carracedo A, SNPforID Consortium. Inferring ancestral origin using a single multiplex assay of ancestry-informative marker SNPs. Forensic Sci Int Genet. 2007;1(3–4):273–80.
Fondevila M, Phillips C, Santos C, Freire Aradas A, Vallone PM, Butler JM, Lareu MV, Carracedo A. Revision of the SNPforID 34-plex forensic ancestry test: assay enhancements, standard reference sample genotypes and extended population studies. Forensic Sci Int Genet. 2013;7(1):63–74.
Phillips C, Freire Aradas A, Kriegel AK, Fondevila M, Bulbul O, Santos C, Serrulla Rech F, Perez Carceles MD, Carracedo A, Schneider PM, Lareu MV. Eurasiaplex: a forensic SNP assay for differentiating European and South Asian ancestries. Forensic Sci Int Genet. 2013;7(3):359–66.
Tardif JC, Rheaume E, Lemieux Perreault LP, Gregoire JC, Feroz Zada Y, Asselin G, Provost S, Barhdadi A, Rhainds D, L’Allier PL, Ibrahim R, Upmanyu R, Niesor EJ, Benghozi R, Suchankova G, Laghrissi-Thode F, Guertin MC, Olsson AG, Mongrain I, Schwartz GG, Dube MP. Pharmacogenomic determinants of the cardiovascular effects of dalcetrapib. Circ Cardiovasc Genet. 2015;8(2):372–82.
Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, Schmidt MK, Chang-Claude J, Bojesen SE, Bolla MK, Wang Q, Dicks E, Lee A, Turnbull C, Rahman N, Breast and Ovarian Cancer Susceptibility Collaboration, Fletcher O, Peto J, Gibson L, Dos Santos Silva I, Nevanlinna H, Muranen TA, Aittomaki K, Blomqvist C, Czene K, Irwanto A, Liu J, Waisfisz Q, Meijers-Heijboer H, Adank M, Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON), van der Luijt RB, Hein R, Dahmen N, Beckman L, Meindl A, Schmutzler RK, Muller-Myhsok B, Lichtner P, Hopper JL, Southey MC, Makalic E, Schmidt DF, Uitterlinden AG, Hofman A, Hunter DJ, Chanock SJ, Vincent D, Bacot F, Tessier DC, Canisius S, Wessels LF, Haiman CA, Shah M, Luben R, Brown J, Luccarini C, Schoof N, Humphreys K, Li J, Nordestgaard BG, Nielsen SF, Flyger H, Couch FJ, Wang X, Vachon C, Stevens KN, Lambrechts D, Moisse M, Paridaens R, Christiaens MR, Rudolph A, Nickels S, Flesch-Janys D, Johnson N, Aitken Z, Aaltonen K, Heikkinen T, Broeks A, Veer LJ, van der Schoot CE, Guenel P, Truong T, Laurent-Puig P, Menegaux F, Marme F, Schneeweiss A, Sohn C, Burwinkel B, Zamora MP, Perez JI, Pita G, Alonso MR, Cox A, Brock IW, Cross SS, Reed MW, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N, Henderson BE, Schumacher F, Le Marchand L, Andrulis IL, Knight JA, Glendon G, Mulligan AM, kConFab Investigators, Australian Ovarian Cancer Study Group, Lindblom A, Margolin S, Hooning MJ, Hollestelle A, van den Ouweland AM, Jager A, Bui QM, Stone J, Dite GS, Apicella C, Tsimiklis H, Giles GG, Severi G, Baglietto L, Fasching PA, Haeberle L, Ekici AB, Beckmann MW, Brenner H, Muller H, Arndt V, Stegmaier C, Swerdlow A, Ashworth A, Orr N, Jones M, Figueroa J, Lissowska J, Brinton L, Goldberg MS, Labreche F, Dumont M, Winqvist R, Pylkas K, Jukkola-Vuorinen A, Grip M, Brauch H, Hamann U, Bruning T, GENICA (Gene Environment Interaction and Breast Cancer in Germany) Network, Radice P, Peterlongo P, Manoukian S, Bonanni B, Devilee P, Tollenaar RA, Seynaeve C, van Asperen CJ, Jakubowska A, Lubinski J, Jaworska K, Durda K, Mannermaa A, Kataja V, Kosma VM, Hartikainen JM, Bogdanova NV, Antonenkova NN, Dork T, Kristensen VN, Anton-Culver H, Slager S, Toland AE, Edge S, Fostira F, Kang D, Yoo KY, Noh DY, Matsuo K, Ito H, Iwata H, Sueta A, Wu AH, Tseng CC, Van Den Berg D, Stram DO, Shu XO, Lu W, Gao YT, Cai H, Teo SH, Yip CH, Phuah SY, Cornes BK, Hartman M, Miao H, Lim WY, Sng JH, Muir K, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Shen CY, Hsiung CN, Wu PE, Ding SL, Sangrajrang S, Gaborieau V, Brennan P, McKay J, Blot WJ, Signorello LB, Cai Q, Zheng W, Deming-Halverson S, Shrubsole M, Long J, Simard J, Garcia-Closas M, Pharoah PD, Chenevix-Trench G, Dunning AM, Benitez J, Easton DF. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45(4):353-61, 361e1-2.
Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6(6):692–702.
Price EM, Robinson WP. Adjusting for batch effects in DNA methylation microarray data, a lesson learned. Front Genet. 2018;9:83.
Price EM, Cotton AM, Penaherrera MS, McFadden DE, Kobor MS, Robinson W. Different measures of “genome-wide” DNA methylation exhibit unique properties in placental and somatic tissues. Epigenetics. 2012;7(6):652–63.
Powell MA, Mutch DG, Rader JS, Herzog TJ, Huang TH, Goodfellow PJ. Ribosomal DNA methylation in patients with endometrial carcinoma: an independent prognostic marker. Cancer. 2002;94(11):2941–52.
Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit. Environ Health Perspect. 2008;116(11):1547–52.
Baccarelli A, Wright R, Bollati V, Litonjua A, Zanobetti A, Tarantini L, Sparrow D, Vokonas P, Schwartz J. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology. 2010;21(6):819–28.
Zhu ZZ, Hou L, Bollati V, Tarantini L, Marinelli B, Cantone L, Yang AS, Vokonas P, Lissowska J, Fustinoni S, Pesatori AC, Bonzini M, Apostoli P, Costa G, Bertazzi PA, Chow WH, Schwartz J, Baccarelli A. Predictors of global methylation levels in blood DNA of healthy subjects: a combined analysis. Int J Epidemiol. 2012;41(1):126–39.
Teschler S, Gotthardt J, Dammann G, Dammann RH. Aberrant DNA methylation of rDNA and PRIMA1 in borderline personality disorder. Int J Mol Sci. 2016;17(1) https://doi.org/10.3390/ijms17010067.
Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, Bertazzi PA, Yang AS. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67(3):876–80.
Qiao Y, Mondal K, Trapani V, Wen J, Carpenter G, Wildin R, Price EM, Gibbons RJ, Eichmeyer J, Jiang R, DuPont B, Martell S, Lewis SM, Robinson WP, O’Driscoll M, Wolf FI, Zwick ME, Rajcan-Separovic E. Variant ATRX syndrome with dysfunction of ATRX and MAGT1 genes. Hum Mutat. 2014;35(1):58–62.
R Core Team: R: a language and environment for statistical computing. 2016
Price ME, Cotton AM, Lam LL, Farre P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6(1):4. https://doi.org/10.1186/1756-8935-6-4.
Hanna CW, McFadden DE, Robinson WP. DNA methylation profiling of placental villi from karyotypically normal miscarriage and recurrent miscarriage. Am J Pathol. 2013;182(6):2276–84.
O’Neill RJ, Vrana PB, Rosenfeld CS. Maternal methyl supplemented diets and effects on offspring health. Front Genet. 2014;5:289.
Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32(3):e38.
Guenther BD, Sheppard CA, Tran P, Rozen R, Matthews RG, Ludwig ML. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol. 1999;6(4):359–65.
de Bree A, Verschuren WM, Bjorke-Monsen AL, van der Put NM, Heil SG, Trijbels FJ, Blom HJ. Effect of the methylenetetrahydrofolate reductase 677C-->T mutation on the relations among folate intake and plasma folate and homocysteine concentrations in a general population sample. Am J Clin Nutr. 2003;77(3):687–93.
Hustad S, Midttun Ã, Schneede J, Vollset SE, Grotmol T, Ueland PM. The methylenetetrahydrofolate reductase 677C-->T polymorphism as a modulator of a B vitamin network with major effects on homocysteine metabolism. Am J Hum Genet. 2007;80(5):846–55.
Plumptre L, Masih SP, Ly A, Aufreiter S, Sohn KJ, Croxford R, Lausman AY, Berger H, O’Connor DL, Kim YI. High concentrations of folate and unmetabolized folic acid in a cohort of pregnant Canadian women and umbilical cord blood. Am J Clin Nutr. 2015;102(4):848–57.
De Wals P, Tairou F, Van Allen MI, Uh SH, Lowry RB, Sibbald B, Evans JA, Van den Hof MC, Zimmer P, Crowley M, Fernandez B, Lee NS, Niyonsenga T. Reduction in neural-tube defects after folic acid fortification in Canada. N Engl J Med. 2007;357(2):135–42.
Williams J, Mai CT, Mulinare J, Isenburg J, Flood TJ, Ethen M, Frohnert B, Kirby RS, Centers for Disease Control and Prevention. Updated estimates of neural tube defects prevented by mandatory folic acid fortification—United States, 1995-2011. MMWR Morb Mortal Wkly Rep. 2015;64(1):1–5.
Irvine B, Luo W, Leon JA. Congenital anomalies in Canada 2013: a perinatal health surveillance report by the Public Health Agency of Canada’s Canadian Perinatal Surveillance System. Health Promot Chronic Dis Prev Can. 2015;35(1):21–2.
Wang XW, Luo YL, Wang W, Zhang Y, Chen Q, Cheng YL. Association between MTHFR A1298C polymorphism and neural tube defect susceptibility: a metaanalysis. Am J Obstet Gynecol. 2012;206(3):251.e1–7.
Oskoui M, Gazzellone MJ, Thiruvahindrapuram B, Zarrei M, Andersen J, Wei J, Wang Z, Wintle RF, Marshall CR, Cohn RD, Weksberg R, Stavropoulos DJ, Fehlings D, Shevell MI, Scherer SW. Clinically relevant copy number variations detected in cerebral palsy. Nat Commun. 2015;6 https://doi.org/10.1038/ncomms8949.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–9.
Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N, Toro R, Wittfeld K, Abramovic L, Andersson M, Aribisala BS, Armstrong NJ, Bernard M, Bohlken MM, Boks MP, Bralten J, Brown AA, Chakravarty MM, Chen Q, Ching CR, Cuellar-Partida G, den Braber A, Giddaluru S, Goldman AL, Grimm O, Guadalupe T, Hass J, Woldehawariat G, Holmes AJ, Hoogman M, Janowitz D, Jia T, Kim S, Klein M, Kraemer B, Lee PH, Olde Loohuis LM, Luciano M, Macare C, Mather KA, Mattheisen M, Milaneschi Y, Nho K, Papmeyer M, Ramasamy A, Risacher SL, Roiz-Santianez R, Rose EJ, Salami A, Samann PG, Schmaal L, Schork AJ, Shin J, Strike LT, Teumer A, van Donkelaar MM, van Eijk KR, Walters RK, Westlye LT, Whelan CD, Winkler AM, Zwiers MP, Alhusaini S, Athanasiu L, Ehrlich S, Hakobjan MM, Hartberg CB, Haukvik UK, Heister AJ, Hoehn D, Kasperaviciute D, Liewald DC, Lopez LM, Makkinje RR, Matarin M, Naber MA, McKay DR, Needham M, Nugent AC, Putz B, Royle NA, Shen L, Sprooten E, Trabzuni D, van der Marel SS, van Hulzen KJ, Walton E, Wolf C, Almasy L, Ames D, Arepalli S, Assareh AA, Bastin ME, Brodaty H, Bulayeva KB, Carless MA, Cichon S, Corvin A, Curran JE, Czisch M, de Zubicaray GI, Dillman A, Duggirala R, Dyer TD, Erk S, Fedko IO, Ferrucci L, Foroud TM, Fox PT, Fukunaga M, Gibbs JR, Goring HH, Green RC, Guelfi S, Hansell NK, Hartman CA, Hegenscheid K, Heinz A, Hernandez DG, Heslenfeld DJ, Hoekstra PJ, Holsboer F, Homuth G, Hottenga JJ, Ikeda M, Jack CR Jr, Jenkinson M, Johnson R, Kanai R, Keil M, Kent JW, Jr KP, Kwok JB, Lawrie SM, Liu X, Longo DL, McMahon KL, Meisenzahl E, Melle I, Mohnke S, Montgomery GW, Mostert JC, Muhleisen TW, Nalls MA, Nichols TE, Nilsson LG, Nothen MM, Ohi K, Olvera RL, Perez-Iglesias R, Pike GB, Potkin SG, Reinvang I, Reppermund S, Rietschel M, Romanczuk-Seiferth N, Rosen GD, Rujescu D, Schnell K, Schofield PR, Smith C, Steen VM, Sussmann JE, Thalamuthu A, Toga AW, Traynor BJ, Troncoso J, Turner JA, Valdes Hernandez MC, van’t Ent D, van der Brug M, van der Wee NJ, van Tol MJ, Veltman DJ, Wassink TH, Westman E, Zielke RH, Zonderman AB, Ashbrook DG, Hager R, Lu L, FJ MM, Morris DW, Williams RW, Brunner HG, Buckner RL, Buitelaar JK, Cahn W, Calhoun VD, Cavalleri GL, Crespo-Facorro B, Dale AM, Davies GE, Delanty N, Depondt C, Djurovic S, Drevets WC, Espeseth T, Gollub RL, Ho BC, Hoffmann W, Hosten N, Kahn RS, Le Hellard S, Meyer-Lindenberg A, Muller-Myhsok B, Nauck M, Nyberg L, Pandolfo M, Penninx BW, Roffman JL, Sisodiya SM, Smoller JW, van Bokhoven H, van Haren NE, Volzke H, Walter H, Weiner MW, Wen W, White T, Agartz I, Andreassen OA, Blangero J, Boomsma DI, Brouwer RM, Cannon DM, Cookson MR, de Geus EJ, Deary IJ, Donohoe G, Fernandez G, Fisher SE, Francks C, Glahn DC, Grabe HJ, Gruber O, Hardy J, Hashimoto R, Hulshoff Pol HE, Jonsson EG, Kloszewska I, Lovestone S, Mattay VS, Mecocci P, McDonald C, McIntosh AM, Ophoff RA, Paus T, Pausova Z, Ryten M, Sachdev PS, Saykin AJ, Simmons A, Singleton A, Soininen H, Wardlaw JM, Weale ME, Weinberger DR, Adams HH, Launer LJ, Seiler S, Schmidt R, Chauhan G, Satizabal CL, Becker JT, Yanek L, van der Lee SJ, Ebling M, Fischl B, Longstreth WT, Jr GD, Schmidt H, Nyquist P, Vinke LN, van Duijn CM, Xue L, Mazoyer B, Bis JC, Gudnason V, Seshadri S, Ikram MA, Alzheimer’s Disease Neuroimaging Initiative, CHARGE Consortium, EPIGEN, IMAGEN, SYS, Martin NG, Wright MJ, Schumann G, Franke B, Thompson PM, Medland SE. Common genetic variants influence human subcortical brain structures. Nature. 2015;520(7546):224–9.
Hickey SE, Curry CJ, Toriello HV. ACMG practice guideline: lack of evidence for MTHFR polymorphism testing. Genet Med. 2013;15(2):153–6.
Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10.
van Mil NH, Bouwland-Both MI, Stolk L, Verbiest MM, Hofman A, Jaddoe VW, Verhulst FC, Eilers PH, Uitterlinden AG, Steegers EA, Tiemeier H, Steegers-Theunissen RP. Determinants of maternal pregnancy one-carbon metabolism and newborn human DNA methylation profiles. Reproduction. 2014;148(6):581–92.
Song MA, Brasky TM, Marian C, Weng DY, Taslim C, Llanos AA, Dumitrescu RG, Liu Z, Mason JB, Spear SL, Kallakury BV, Freudenheim JL, Shields PG. Genetic variation in one-carbon metabolism in relation to genome-wide DNA methylation in breast tissue from heathy women. Carcinogenesis. 2016;37(5):471–80.
Jung AY, Smulders Y, Verhoef P, Kok FJ, Blom H, Kok RM, Kampman E, Durga J. No effect of folic acid supplementation on global DNA methylation in men and women with moderately elevated homocysteine. PLoS One. 2011;6(9):e24976.
Ono H, Iwasaki M, Kuchiba A, Kasuga Y, Yokoyama S, Onuma H, Nishimura H, Kusama R, Ohnami S, Sakamoto H, Yoshida T, Tsugane S. Association of dietary and genetic factors related to one-carbon metabolism with global methylation level of leukocyte DNA. Cancer Sci. 2012;103(12):2159–64.
Hanks J, Ayed I, Kukreja N, Rogers C, Harris J, Gheorghiu A, Liu CL, Emery P, Pufulete M. The association between MTHFR 677C>T genotype and folate status and genomic and gene-specific DNA methylation in the colon of individuals without colorectal neoplasia. Am J Clin Nutr. 2013;98(6):1564–74.
Deroo LA, Bolick SC, Xu Z, Umbach DM, Shore D, Weinberg CR, Sandler DP, Taylor JA. Global DNA methylation and one-carbon metabolism gene polymorphisms and the risk of breast cancer in the Sister Study. Carcinogenesis. 2014;35(2):333–8.
Louie K, Minor A, Ng R, Poon K, Chow V, Ma S. Evaluation of DNA methylation at imprinted DMRs in the spermatozoa of oligozoospermic men in association with MTHFR C677T genotype. Andrology. 2016;4(5):825–31.
Shelnutt KP, Kauwell GP, Gregory JF 3rd, Maneval DR, Quinlivan EP, Theriaque DW, Henderson GN, Bailey LB. Methylenetetrahydrofolate reductase 677C-->T polymorphism affects DNA methylation in response to controlled folate intake in young women. J Nutr Biochem. 2004;15(9):554–60.
Axume J, Smith SS, Pogribny IP, Moriarty DJ, Caudill MA. The MTHFR 677TT genotype and folate intake interact to lower global leukocyte DNA methylation in young Mexican American women. Nutr Res. 2007;27(1):1365–17.
La Merrill M, Torres-Sanchez L, Ruiz-Ramos R, Lopez-Carrillo L, Cebrian ME, Chen J. The association between first trimester micronutrient intake, MTHFR genotypes, and global DNA methylation in pregnant women. J Matern Fetal Neonatal Med. 2012;25(2):133–7.
Aarabi M, San Gabriel MC, Chan D, Behan NA, Caron M, Pastinen T, Bourque G, MacFarlane AJ, Zini A, Trasler J. High-dose folic acid supplementation alters the human sperm methylome and is influenced by the MTHFR C677T polymorphism. Hum Mol Genet. 2015;24(22):6301–13.
Acknowledgements
We thank all the participants of this study for kindly donating the samples. We also thank Dr. Deborah McFadden, Dr. Peter von Dadelszen, Dr. Margot Van Allen, Dr. Hayley Bos, Kristal Louie, Luana Avila, and Ruby Jiang for the sample acquisition and processing; Dr. Elodie Portales-Casamar for her advice in the ancestry analysis; Dr. Maria Peñaherrera for the array processing and advice in the manuscript preparation; Samantha Wilson and Chaini Konwar for their advice in the data analysis and manuscript preparation; and the Kobor Lab for the generous use of their facilities.
Funding
This work was supported by Canadian Institutes of Health Research grants FRN106430 and FRN49520 (to WPR). WPR receives salary support through an investigatorship award from the BC Children’s Hospital Research Institute (BCCHRI).
Availability of data and materials
The 450k array dataset generated and analyzed in this current study has been deposited in NCBI’s Gene Expression Omnibus [99] and is accessible through GEO Series accession number GSE108567 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108567). The remaining data are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
GFDG contributed to the study design, ran the pyrosequencing assays, performed the statistical analysis, interpreted the results, and wrote the draft of the manuscript. EMP contributed to the study design, ran the assays, performed the statistical analysis, interpreted the results, and contributed to the draft of the manuscript. CWH contributed to the study design and ran the assays and 450k arrays. WPR conceived of and supported the study and contributed to the statistical analyses and interpretation. All authors read, critically revised, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval for this study was obtained from the University of British Columbia/Children’s Hospital and Women’s Health Centre of British Columbia Research Ethics Board (H04-70488, H10-01028). For all control, PE, nIUGR, and some NTD cases (N = 261), mothers provided informed written consent to participate prior to delivery or termination of pregnancy. For certain NTD cases obtained retrospectively from pathological autopsy specimens (N = 42), biospecimens were de-identified and unlinked to clinical data. For all cases, only non-identifiable information is presented in this publication.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. PCR and pyrosequencing conditions. (DOCX 22 kb)
Additional file 2:
Methods. (DOCX 20 kb)
Additional file 3:
Table S2. Global minor allele frequencies of 50 AIM SNPs used to assess ancestry. (DOCX 25 kb)
Additional file 4:
Figure S1. Distribution of ancestry coordinates derived from MDS of 50 AIM SNP genotypes. (DOCX 166 kb)
Additional file 5:
Figure S2. Distribution of N = 277 study samples along 3 MDS ancestry coordinates. (DOCX 138 kb)
Additional file 6:
Table S3. MTHFR 677 and 1298 genotype counts and Hardy-Weinberg equilibrium. (DOCX 20 kb)
Additional file 7:
Figure S3. Distribution of unadjusted p-values by CpG density between high-risk 677 or high-risk 1298 placentas compared to reference placentas. (DOCX 230 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Del Gobbo, G.F., Price, E., Hanna, C.W. et al. No evidence for association of MTHFR 677C>T and 1298A>C variants with placental DNA methylation. Clin Epigenet 10, 34 (2018). https://doi.org/10.1186/s13148-018-0468-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-018-0468-1