
Abstract
The correlation between epigenetics and human reproduction represents a very interesting field of study, mainly due to the possible transgenerational effects related to epigenetic modifications of male and female gametes. In the present review, we focused our attention to the role played by epigenetics on male reproduction, evidencing at least four different levels at which sperm epigenetic modifications could affect reproduction: (1) spermatogenesis failure; (2) embryo development; (3) outcome of assisted reproduction technique (ART) protocols, mainly as concerning genomic imprinting; and (4) long-term effects during the offspring lifetime. The environmental agents responsible for epigenetic modifications are also examined, suggesting that the control of paternal lifestyle prior to conception could represent in the next future a novel hot topic in the management of human reproduction.
Similar content being viewed by others


Background
You shall not bow down to them or worship them; for I, the Lord your God, am a jealous God, punishing the children for the sin of the parents to the third and fourth generation of those who hate me
(Esodus, 20.5)
Infertility represents a growing emergency in western countries, affecting about one out of seven couples who attempt to generate a child. In 2010, an estimated 48.5 million couples worldwide were infertile, against 42 million in 1990 [1]. In about 50 % of the cases, this condition is ascribable to the male partner, mainly due to a failure in the spermatogenesis process causing azoospermia or oligozoospermia at the sperm count [2]. Despite the large number of tools available for the identification of the pathogenesis of male infertility, in many cases, no specific cause is detected and no personalized therapeutic protocol can be established. A large number of studies have investigated in the last decades the presence of genetic alterations responsible for the failure of spermatogenesis, which are nevertheless identified only in 15–30 % of infertile males, even when stringent selection criteria are used [3, 4]. Despite the identification of several rare genetic variants associated to disruption of spermatogenesis, so far, the only two categories of genetic alterations responsible for a significant portion of cases of male infertility, and thus commonly tested in the clinical practice, are represented by chromosomal alterations and Yq microdeletions [5–10]. The presence of other genetic mechanisms, such as partial Yq microdeletions [11–14], specific Y-chromosome haplogroups [15–18], and polymorphism in genes related to mitochondrial function [19–21] as risk factors for infertility have been suggested, but with inconclusive results. More recently, the presence of X-linked copy number variants (CNVs) in infertile males has been reported by several studies [22, 23], but the overall incidence of these variants accounts only for a limited portion of all cases.
In recent years, great interest has been raised by the novel acquisitions on the epigenetic mechanisms of regulation of gene expression. Epigenetics can be defined as the study of mitotically or meiotically heritable modifications in the function of specific genes not related to modification in the DNA sequence [24]. This novel field of study has obtained large relevance also in the world of mass media, usually transmitting the “take-home message” that human destiny is not written inside genes, since environmental agents or experiences can influence human heredity [25]. As a matter of fact, the interaction between genes and environment in the determination of human phenotypes is very well known from many years, but the real novelties provided by the studies on epigenetics are that (1) environmental agents can modify the expression of specific genes without changing their sequence or copy number, and (2) these modifications can be transmitted to the offspring, so that either rare congenital diseases or the susceptibility to common diseases appearing during the lifetime can be the result of a gene-environment interaction that occurred in one parent of a subject, not in the subject himself. In this view, epigenetic studies represent a breakthrough in the field of human reproduction. In fact, since epigenetic modifications can be transmitted to the offspring, they obviously involve germ cells, and in some cases, they could affect gametogenesis as well as the embryo development, thus representing a potential cause of infertility of the couple. Moreover, since epigenetic alterations do not induce modification in the gene sequence or copy number, they could account for at least a portion of cases of male infertility in which no genetic abnormalities are detected using the conventional techniques of genetic analysis.
Several studies have investigated in the last years the role played by epigenetic modification in male gametogenesis and in male infertility. The aim of this review is to analyze the state-of-art of this field of research in order to give an answer to the following questions. (1) Can epigenetic mechanisms be related to the quality of the spermatogenesis process? (2) Can sperm epigenetic alterations affect embryo development? (3) Is there a relationship between sperm epigenetic modifications and outcome of assisted reproduction technique (ART) procedure? (4) Which environmental agents can be responsible for epigenetic modifications of sperm DNA?
Molecular basis of epigenetics
The main epigenetic mechanisms of gene expression regulation are represented by DNA methylation, histone modifications, and small, non-coding RNAs.
DNA methylation
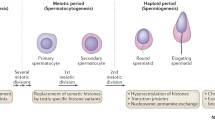
In mammalians, DNA methylation occurs at the 5′- position of cytosine residues, mainly within CpG dinucleotides, 60–80 % of which are methylated within the promoter regions of genes [24]. Methylation of CpG dinucleotides within the promoter regions leads to the silencing of transcription process, mediated by modifications in the condensation status of the chromatin. The process of DNA methylation is catalyzed by enzymes known as DNA methyltransferases (DNMTs), which can be classified in “de novo” DNMTs (which methylate specific chromosomal sequences during early embryogenesis), and maintenance methyltransferases (DNMT1), faithfully restoring the methylation patterns after each DNA replication cycle [26]. The process of DNA methylation is closely related with gametogenesis, since primordial germ cells (PGCs), when entering the developing gonad, undergo a process of deep decrease of DNA methylation, which will be subsequently restored in the prenatal life in males and during post-natal follicle development in females (Fig. 1) [27].

Epigenetic modifications during spermatogenesis. During the different steps of spermatogenesis, several epigenetic modifications involving DNA methylations and histone modifications occur. (1) PGCs undergo a process of demethylation involving DNA (with erasure of genomic imprinting) and histones (namely, K4 and K9 residues of H3). Also, a process of H4 deacetylation is present. DNMT3A, DNMT3B, and DNMT3L are expressed at this time. (2) In spermatogonia, a progressive DNA methylation occurs, with establishment of paternal methylation. (3) In spermatocytes, H3K9 and H3K4 methylation is observed. (4) In round spermatids, H4 becomes hyperacetylated, DNMT1 is expressed, and the transition from histones to TPs occurs. (5) Elongated spermatids show a maintenance of DNA methylation, together with H3K9 demethylation. The transition from TPs to protamines occurs at this step. (6) In spermatozoa, the genomic imprinting is maintained
Cytosine methylation can occur also in non-CpG sites (CpA > CpT > CpC), although the significance of these variants is still unknown. Methylation at non-CpG sites was previously considered to be largely present in the brain, embryonic stem cells (ESCs), induced pluripotent stem cells, and in oocytes [28–32]. However, Ichiyanagi et al. observed methylation at non-CpG sites in male germ cells as well, demonstrating that the level of the non-CpG methylation is higher in prospermatogonia and decreases along with mitotic division. In addition, this study also suggested the absence of a template-dependent mechanism for copying non-CpG methylation in prospermatogonia [33].
Another kind of epigenetic DNA modification is represented by 5-hydroxymethylcytosine (5hmC), an intermediate of DNA demethylation with important regulatory functions in various biological and pathological processes in the mammalian genome such as transcriptional regulation, DNA methylation regulation, and tumorigenesis [34]. Gan et al. showed that the highly ordered 5hmC alterations are critical for the differentiation of spermatogenic cells in the mouse [35]. Recently, Wang et al. for the first time compared 5-hydroxymethylcytosine profiles in normal, abnormal, and globozoospermia sperms, identifying 6664, 9029, and 6318 genes containing 5hmC, respectively [36]. In addition, since some 5hmC-containing genes are significantly involved in spermatogenesis, sperm motility, and morphology, the authors suggested that the 5hmC distribution differences may contribute to the sperm phenotype [36].
DNA methylation was originally investigated by Southern blot and methylation-sensitive restriction endonuclease digestion followed by PCR amplification [37]. Nowadays, gene-specific techniques are available including bisulfite sequencing, combined bisulfite restriction analysis (COBRA) [38], MethyLight [39], and bisulfite pyrosequencing [40], all based on bisulfite conversion of cytosine residue to uracil, leaving 5-methylcytosine residue unaffected [41], detectable by DNA sequencing. All the abovementioned techniques are sensitive, specific, and relatively inexpensive but not suitable for analysis of the whole genome, which includes about 28 million CpGs.
The next-generation sequencing (NGS) approach represents a new powerful tool for the study of human DNA methylome [42]. The NGS DNA methylation procedures are based on three main steps: digestion of genomic DNA with methyl-sensitive restriction enzymes, affinity-based enrichment of methylated DNA fragments, and chemical conversion methods [43, 44].
Another large-scale approach is represented by whole-genome bisulfite sequencing (WGBS), which is able to analyze DNA methylation profiles of whole genomes at single-base resolution [45]. Recently, several studies have reported WGBS accurate data on methylome both of human embryonic stem cells [46] and induced pluripotent stem cells [47].
Histone modifications
Modifications of histone tails represent other epigenetic chromatin marks critical for transcriptional regulation. Post-translational modifications of histone tails include methylation, acetylation, phosphorylation, ubiquitination, ribosylation, and sumoylation. Histone marks are dynamic process, since histone modifications can be easily induced and removed by a wide range of enzymes [48]. The most relevant histone change is represented by acetylation at lysine residues on the amino-terminal tail domains, whose correct levels are maintained by the combined action of two enzymes known as histone acetyl transferase (HAT) and histone deacetylase (HDAC). Generally, acetylation reduces the affinity of histones for DNA, making genes functionally active; on the other hand, histone deacetylation leads to chromatin condensation, making genes transcriptionally inactive. Conversely, histone methylation is a key regulator for both activation and inactivation of transcription. For example, lysine 4 of histone H3 (H3-K4) methylation is linked to gene expression, while H3K9 and H3K27 di- and tri-methylation is associated with gene silencing [49, 50].
Histone H3T is the testis-specific H3 variant in mammals. Tachiwana et al. showed that H3T nucleosomes can be assembled by Nap2 chaperone and that this structure is significantly unstable as compared to the conventional H3.1-containing nucleosomes [51, 52]. Furthermore, these authors suggested that the unstable H3T-containing nucleosome structure can influence the chromatin re-organization essential for spermatogenesis [52].
Histone modifications were originally investigated by Western blot with specific antibodies to modified histones, but nowadays, they are mainly studied by mass-spectrometry-based proteomic technologies [53]. Recently, NGS technologies have been developed able to analyze whole-genome histone modifications. These techniques are based on chromatin immunoprecipitation followed by sequencing maps of the genome-wide binding pattern of chromatin-associated proteins, which includes modified histones (ChIP-seq) [44, 54].
Small non-coding RNAs
A third mechanism of epigenetic regulation of gene activity is represented by small RNAs and large intergenic non-coding RNAs (lincRNAs). Small RNAs, not encoding proteins, are located in the nucleus of spermatozoa and play an essential role as epigenetic modifiers both in recognizing and preserving the DNA that remains bound to histones during the transition to protamine in spermiogenesis and in early embryonic development [55]. On the other hand, lincRNAs act at the level of chromatin allowing to select the histone modification enzymes, such as in the case of lincRNAs HOTAIR which acts as a scaffold for PRC2 and LSD1 enzymes to regulate lysine 27 methylation and lysine 4 demethylation in H3 histone [56].
Small non-coding RNAs are generally identified by RT-PCR analysis, in situ hybridization, or small RNA sequencing studies. Furthermore, the recent development of microarray technologies has permitted the global analysis of spermatozoal microRNAs (miRNAs) evidencing different expression profiles between fertile and infertile men [57, 58].
Genomic imprinting
A specific feature of epigenetic control of gene function is represented by genomic imprinting, a process leading to the expression of a specific set of genes (about 70–80, the majority of which clustered in 16 specific chromosomal regions) based on their maternal or paternal origin [59]. Genomic imprinting plays a key role in the regulation of resource acquisition by the offspring from the mother during prenatal and early postnatal life. Paternally and maternally imprinted genes play different roles in this mechanism, being many paternally expressed alleles able to increase resource transfer to the child, which is on the other side reduced by maternally expressed genes (“parental conflict hypothesis”) [60]. The correct balance between the activity of maternally and paternally imprinted genes can be disrupted by different mechanisms, such as chromosome deletions, uniparental disomy (UPD), or alterations in the imprinting center. In human, alterations of the process of genomic imprinting cause several congenital diseases mainly involving fetal growth (e.g., Beckwith–Wiedemann syndrome, Russell–Silver syndrome), hormone systems after birth (e.g., Albright hereditary osteodystrophy, pseudohypoparathyroidism 1A, transient neonatal diabetes mellitus), or behavior (e.g., Prader–Willi syndrome, Angelman syndrome) [60]. Moreover, imprinting alterations have been suggested as responsible for intrauterine growth restriction, in turn associated with an increased risk of cardiovascular disease, diabetes, and mental defects later in life [60, 61].
The discovery of the role played by epigenetic modifications on the function of paternal genome has prompted novel attention on the function of sperm DNA during embryo development. In fact, sperms have been historically viewed as specialized cells with the unique function of delivering the 23 paternal chromosomes to the oocyte, considered as the only gamete playing an active role in driving embryo development, thanks to its availability of cellular organelles, RNAs, and cellular machinery. Actually, the role played by the male gamete in embryo development appears to be more relevant than previously hypothesized. In this view, a full knowledge of the epigenetics of sperm could provide novel information about germ cell biology, paternal effects on embryogenesis, and the pluripotency of embryonic stem cells [62].
Epigenetics and spermatogenesis
The process of spermatogenesis
The formation of a mature sperm requires different processes, namely (1) mitotic proliferation of spermatogonia; (2) meiotic divisions; and (3) morphological differentiation of sperm precursors (spermiogenesis), leading to the generation of highly specialized cells characterized by the presence of a head, an intermediate portion, and a flagellum. Such a specific organization of the male germ cells is necessary to allow sperms to traverse a potentially hostile female reproductive tract, penetrate the cumulus oophorus and the zona pellucida, penetrate the oocyte, and finally complete multiple post-penetration events [62, 63]. During fetal life, spermatogenesis begins in the wall of the seminiferous tubules from undifferentiated diploid cells known as spermatogonia, which undergo several mitotic divisions in order to increase the pool of available precursors of germ cells. At puberty, some spermatogonia are transformed in type I spermatocytes, which undergo the first meiotic division producing haploid type II spermatocytes. A second meiotic division occurs in these cells, originating haploid spermatids. The last phase of spermatogenesis is represented by spermiogenesis, characterized by a morphological and structural transformation complex process of the round spermatid. This step, occurring without further cell division, leads to the production of mature sperm, characterized by the differentiation of the flagellum and the acrosome, essential prerequisites for sperm motility and fertilization capacity.
Histone–protamine replacement as the main epigenetic change in sperms
In addition to the typical morphology and motility, sperms are characterized also by a highly organized chromatin structure. In fact, sperm chromatin during spermiogenesis undergoes further condensation, due to the replacement of 90–95 % of the histones with one or more sperm-specific basic proteins, known as protamines [64]. This modification induces the formation of disulfide bonds (SS) that confer extreme stability to the core of the sperm nucleus, producing a number of relevant effects, such as improvement of sperm motility, protection from oxidative stress and toxic agents present within female reproductive tract, and block of the transcriptional activity of the sperm DNA [65]. The complex mechanism of histone–protamine transition is a finely regulated multi-step process. In the first step, the histones in round spermatids are replaced by a heterogeneous group of nuclear proteins (transition proteins (TP)), as the result of histone hyperacetylation [66] (Fig. 1). The second step takes place in elongating spermatids, determining the replacement of TP1 and TP2 with protamines [62, 67] (Fig. 1). Protamines have different functions: they allow the compaction of the nucleus ensuring the genetic integrity of the sperm and play an important role in epigenetic imprinting [62]. Mature spermatid nuclei present two types of protamines: the P1 protamine and the P2 family of protamines, constituted by P2 (the most abundant), P3, and P4 members. P1/P2 ratio appears to be critical for male fertility [68, 69]. In fact, the P1/P2 ratio, which in fertile males is close to 1 (range 0.8–1.2), is altered in infertile patients [69, 70]. Patients with a P1/P2 ratio <0.8 present inadequate DNA condensation and important alterations in sperm parameters, such as motility, counts, and structure [68–71]. Moreover, Aoki et al. demonstrated that low P1/P2 ratios are also associated with an increased DNA fragmentation, which is also inversely correlated with global sperm P1 and P2 concentrations, suggesting a protective role of the protamines against sperm DNA damage [72]. There is also evidence that subfertility can be correlated with an excess of protamine P2 precursors (pre-P2), determined by an alteration of the process leading to the mature protamine P2 formation [68, 71, 73, 74].
DNA methylation and histone modifications during spermatogenesis
Various and specific epigenetic marks are required during male gametogenesis for proper maturation of gametes. In fact, before meiosis, the first epigenetic events take place in the form of progressive demethylation–remethylation of DNA. During meiosis, DNMT3A, DNMT3B, and cofactor DNMT3L activity regulates the levels of de novo DNA methylation, completing this process after birth at the stage of pachytene spermatocyte [75]. Subsequently, the methylation profile is maintained by DNMT1 activity. In addition to the above-described processes, also histone modifications (methylation and acetylation) occur, which modify DNA accessibility to transcription factors (Fig. 1). In fact, specific enzymes such as histone methyltransferase (HMT) and histone demethylase (HDM) regulate lysine 9 of histone H3 (H3-K9) and lysine 4 of histone H3 (H3-K4) methylation patterns. Generally, histone H3-K9 methylation is high in meiosis but is removed at the end of this process, promoting gene activation, whereas histone H3-K4 methylation, which decreases during meiosis, is associated to DNA silencing [50] (Fig. 1). In addition, during spermatogenesis, several enzymes, such as HAT and HDAC, regulate the processes of acetylation and deacetylation of H3 and H4 lysine residues. During spermiogenesis, hyperacetylation of H4 plays a crucial role for correct histone to protamine transition and allows nucleosome disassembly in elongating spermatids [66, 76] (Fig. 1).
Epigenetic alterations and spermatogenesis disruption
The above-described epigenetic marks in germ line genes play a key role in the proper spermatogenesis processes, and several studies have demonstrated that aberrant epigenetic modification of genes expressed in the testes are associated with male infertility. Navarro-Costa et al. for the first time hypothesized that one of the factors of male gametogenic defects could be represented by epigenetic alterations of specific genes, evidencing increased methylation defects of the germ line regulator DAZL gene in different quality-fractioned sperm populations of oligoasthenoteratozoospermic (OAT) patients as compared to normozoospermic (NZ) men [77]. On the contrary, no variation in the methylation state of the DAZL gene promoter between NZ e OAT men was observed. This study also highlighted the existence of homogeneous DAZL methylation levels when comparing the normal sperm-enriched fractions of NZ men. This evidence has been subsequently confirmed on a larger scale by Krausz et al. who detected no differences in the DNA methylation status of several genes in the different sperm subpopulations of normozoospermic individuals [78]. Subsequently, other studies have identified more genes whose epigenetic modifications are related to alteration of both semen parameters and fertility maintenance. Hammoud et al., by analyzing seven imprinted loci (LIT1, MEST, SNRPN, PLAGL1, PEG3, H19, and IGF2), correlated alterations in the DNA methylation pattern of oligozoospermic patients with abnormal protamine levels [70]. Interestingly, alterations in the mRNA levels of the genes involved in the histone–protamine transition have been evidenced also by studies carried out by investigating the global testis transcriptome of normal and oligozoospermic patients by microarray analysis [79]. Other studies have demonstrated that DNA hypermethylation of the promoter of several genes (such as MTHFR, PAX8, NTF3, SFN, HRAS, RASGFR1, GTL2, PLAG1, D1RAS3, MEST, KCNQ1, LT1, SNRPN and others) plays a critical role in male infertility, being associated to alterations of sperm concentration, motility, and morphology, while hypomethylation of the IGF2/H19 imprinting control region 1 (ICR1) has been detected in patients with low concentration and sperm motility as compared to normozoospermic controls [70, 80–86].
Due to the increased knowledge about the epigenetic alterations occurring in sperm DNA of infertile patients, it has become clear that specific errors in the processes of epigenetic control may occur during each stage of spermatogenesis, adversely affecting male fertility and embryonic development [49]. In fact, epigenetic alterations occurring in mitosis can affect the expression of specific genes involved in the first steps of spermatogenesis, leading to a decreased efficiency of the process. When the meiotic stage is involved, these alterations can induce double-strand breaks or chromosomal non-disjunction. Finally, during spermiogenesis, epigenetic alterations can involve histone-to-protamine transition and histone removal and degradation, inducing protamine replacement errors [49]. Taken together, all these evidences suggest that different features of male infertility, such as alterations in sperm count or morphology, DNA fragmentation, chromosomal aneuploidies, and alterations in the chromatin package, could be all related to epigenetic mechanisms occurring at different stages of spermatogenesis.
Epigenetics and embryo development
DNA methylation and histone modifications play a crucial role in the process of genome reprogramming during early embryogenesis [87–89]. Preimplantation embryo development is a dynamic process characterized by deep gene expression profile change and modifications in the histone and chromatin organization [87, 90–92]. After fertilization, the genome of paternal and maternal origin is subjected to a process of reprogramming: at first, the male pronucleus is demethylated [93, 94], and after the formation of the zygote, the chromosomes of both parents are demethylated by a passive mechanism erasing most parts of the methylation marks except those involved in the process of genomic imprinting [95]. The methylation of imprinted genes is erased only in PGC, cells of epiblast that give rise to male and female gametes. Here, an extraordinary epigenetic regulation occurs in the early stages of embryonic development, when the methylation is erased and specific genes of pluripotency (OCT4 and NANOG) are expressed. De novo methylation starts in the inner cell mass of the blastocyst and the levels of methylated DNA increase in primitive ectoderm, while methylation is inhibited in the trophoblast and in the primitive endoderm [96, 97]. This alternation of demethylation and remethylation can be explained by the necessity during preimplantation to activate zygotic genes essential for early development, while de novo methylation could establish a state of global silencing in order to suppress retrotransposons [87]. In post-implantation embryos, the maintenance of DNA methylation is crucial for embryonic development. In female embryos, X inactivation, due to increased expression and accumulation of Xist RNA, occurs [98]. The X chromosome is inactivated to compensate the number of X-linked genes in males and females, and the process of inactivation takes place after the implantation of female embryos or during the process of differentiation of ESCs [99, 100]. A reactivation of the X chromosome occurs in the inner cell mass of the blastocyst and in the epiblast, followed by a random inactivation, a biological process in which both X chromosomes have the same probability of being inactivated [101, 102].
A first relevant role played by paternal genome in the above-described process is represented by genomic imprinting. In fact, at least three paternally imprinted genes, H19-IGF2, RASGRF, and DLK1-GTL2 are considered among the most relevant for embryonic development and placentation [103–105]. However, genomic imprinting does not appear to represent the only mechanism of paternal control on embryo development. As a matter of fact, it has been demonstrated that the presence of modified histones in the spermatozoa could represent a potential paternal contribution in epigenetic reprogramming of the zygote regardless of the imprinting process [106–108]. In fact, despite the exchange of histones with protamine is essential for the maturation of sperm, a residual percentage of genome (5–15 %) retains the nucleosomal organization [109]. These retained nucleosomes play a key role in the contribution of the paternal genome in embryonic development [110]. In fact, the paternal DNA packaging in spermatozoa seems to have a potential role in transmitting an epigenetic profile to the zygote in early embryogenesis [111], since the nucleosome retention takes place in hypomethylated regions corresponding to the promoters of developmental transcription and signaling factors which are the target of transcription factors such as OCT4, NANOG, SOX2, and KLF4 [110]. In this view, it is possible to hypothesize that an alteration of the correct distribution of nucleosome retention within sperm DNA could produce an impairment of embryo development. As a matter of fact, Hammoud et al. demonstrated the presence in infertile men of randomly distributed histone retention genome-wide, with alteration of the methylation status of candidate developmental promoters and imprinted loci [111]. Moreover, genes in histone-bound regions appear more susceptible to DNA damage induced by smoking, obesity, and aging as compared to protamine-bound regions, due to the incapacity of sperm to repair DNA damage [112]. Histone-bound regions play a crucial role in the activation of paternal genome transcription in the early embryo, since while paternal protamines are replaced by maternal histones in the first 4 to 6 h after fertilization, this does not occur for paternal histones, which are thus likely inherited by the embryo [113]. Thus, the reported data above suggest that, in some instances, epigenetic defects of the sperm could induce not only a poor sperm quality, but also a decreased ability of development of the generated embryo after the fusion of the gametes, providing a possible explanation for a number of early pregnancy loss after both in vivo and in vitro fertilization.
Epigenetics and ART
A large amount of literature data, including human and animal studies, has raised concerns about an increased risk of different diseases in the offspring generated by the use of ART [114, 115]. Several evidences have suggested that the majority of these abnormal conditions are related to epigenetic alterations.
Data from ART in animal models
Early studies carried out in mice had showed that alterations affecting the development and growth of the fetuses were linked to ovulation induction, manipulation of eggs, or embryo culture in vitro [116–119]. A confirmation came from Khosla et al. showed that preimplantation mouse embryos cultured in the presence of serum can change the expression and methylation of several imprinted genes (such as H19, IGF2, GRB10, and GRB7) and that these aberrant epigenetic modifications lead to abnormal fetal growth in ART animals [120].
Moving to different animal models, Young et al. evidenced the presence of the “large offspring syndrome” (LOS) (large size at birth, increased birth weight, breathing difficulties, reluctance to suckle, and sudden perinatal death) in sheep and cattle derived from cultured embryo [121]. Subsequently, the pathogenesis of LOS was suggested to be associated with epigenetic abnormalities, leading to loss of imprinting and over-expression of IGF2 receptor gene [122, 123]. Factors in the ART procedures, triggering these imprinting errors, were not clearly identified, but the onset of the syndrome appeared to be dependent by in vitro culture conditions [122, 123].
Data from ART in human
In human, a larger prevalence of syndromes related to imprinting alteration, particularly Beckwith-Wiedemann and Angelman syndromes, has been reported in children born after ART when compared to non-ART children [124–126]. Interestingly, the phenotype of Beckwith-Wiedemann syndrome, an overgrowth condition characterized by large size at birth, macroglossia, and visceromegaly, closely remembers the previously described LOS in cows and sheep generated by in vitro fertilization, suggesting a common mechanism of origin of these conditions. On the other hand, also a disproportionate number of low birth weight cases has been observed in ART children [127].
The association between ART and altered DNA epigenetic profiling has been demonstrated also by studies based on the analysis of the methylation status of CpG sites in the promoters of 700 genes of placenta and cord blood obtained from children conceived in vitro and in vivo [128]. This analysis showed hypomethylation of most CpG sites in the placenta and hypermethylation of most CpG sites in cord blood in the group of children conceived by in vitro fertilization as compared to naturally conceived children. Interestingly, the genes showing different expressions in the two groups appeared to be involved in chronic metabolic disorders including obesity, type II diabetes, and high blood pressure [128]. These data are in agreement with other studies reporting an increased risk of disturbs in body fat composition, changes in blood pressure, and increase in the late infancy growth velocity in children generated by ART procedures as compared to control children [129–131]. In addition, other studies showed a high risk of obesity or type II diabetes in adult life in children generated by ART [132]. These results strongly suggest that the effect of ART procedures could be manifested not only at birth, but also in late infancy or even in adult life.
As in animal models, the in vitro culture conditions have been suggested as a main cause of epigenetic defects in the offspring generated by ART also in human [133]. In addition, it has been proposed that the low birth weight observed after ART, as well as in small for gestational age and very premature children, is the result of an unfavorable embryonic, fetal, or neonatal environment, involving also epigenetic mechanisms, potentially related to metabolic alterations in late childhood [134]. However, several evidences have suggested that a crucial role could be also played by epigenetic defects in the sperms used in ART protocols. In fact, several authors suggested that DNA methylation changes at imprinted loci are inherited from the sperm of men with oligozoospermia [82, 135, 136]. This could suggest that the increase of DNA methylation variations in ART depends, at least in part, on the presence of epigenetic defects of male gamete. In this view, some authors have proposed the possible usefulness of analyzing the imprinting methylation status in the routine sperm examination for ART treatment [136]. Moreover, it has been hypothesized that the use of sperms with an abnormal P1/P2 ratio or defects of histone–protamine may be responsible of imprinting diseases in the offspring conceived with ART [50]. Unfortunately, this argument remains poorly understood, as demonstrated by other studies reporting that epigenetic abnormalities detected in sperms of oligozoospermic patients do not appear to be associated with ART outcome, suggesting that further studies are required in order to shed light on the relationship between sperm epimutation and alterations in children generated by ART [137].
Environmental agents inducing epigenetic modifications
Several environmental and lifestyle factors (stress, physical activity, alcohol intake, smoke, shift work) are known to affect male and female fertility [138], and in many cases, they have been shown to influence the occurrence of epigenetic modifications with implications for human diseases [139]. The presence of an environmental epigenetic inheritance through gametes has been evidenced by studies carried out on different animal models [140]. A few studies have suggested that food or physical activity can influence histone modifications and miRNA expression. Dashwood et al. demonstrated that a single intake of cruciferous vegetables inhibits HDAC activity in mononuclear cells of peripheral blood promoting H3 and H4 acetylation [141], while other studies demonstrated that exposure to cigarette smoke causes a down-regulation of mir-34b, mir-421, mir450-b, mir-466, and mir-469 [142]. Anyway, the largest body of evidence comes from studies investigating the environmental effects on DNA methylation. Alterations in this process have been demonstrated to be induced in specific genome regions by toxic chemicals, high intake of alcohol and mother’s diet, or smoking during intrauterine life [143, 144]. Further information in this field has been provided by studies investigating the role played by paternal exposures to various pollutants and lifestyle-related conditions on the health status of the offspring and of the future generations.
Paternal exposure to toxins or ionizing radiation
Great attention has been devoted to the effects of paternal exposures to environmental toxins or low-dose ionizing radiation, and of paternal lifestyle [145]. Several studies had previously demonstrated the presence of a strong association between paternal occupational exposures to chemicals and harmful health outcomes in the offspring. Feychting at al. demonstrated an increased risk of nervous system tumors related to paternal occupational exposure to pesticides and of leukemia related to woodwork by fathers [146]. Reid et al. evidenced the presence of high exposure to exhausts by paternal grandmothers of children with acute lymphoblastic leukemia [147]. However, many of these conditions are likely related to the presence of mutations in sperm DNA, thus representing a genetic, rather than epigenetic, mechanism. Is there any evidence supporting the presence of epigenetic mechanism driving the effects to the offspring of the paternal exposure to chemicals? Once again, the most relevant data in support of this hypothesis come from studies on animal models, showing that male exposure to pesticides or other harmful chemicals can be responsible for defects in the gametes and abnormal development of the offspring mainly via altered DNA methylation patterns in the germ line [148, 149]. Anway et al. evidenced that a transient embryonic exposure to the endocrine disruptor vinclozolin during gonadal sex determination in rats produced several diseases affecting the prostate, kidney, immune system, testis, as well as different cancers in the subsequent generations, suggesting a potential transgenerational effect [148]. Similar results were obtained by Guerrero-Bosagna et al., who showed that transient exposure of the F0 generation gestating female to vinclozolin during gonadal sex determination caused adult onset disease in the F3 generation male and female mice [149].
Also, ionizing radiations have been recently invoked as a risk factor for alterations of DNA methylation. These radiations trigger a series of processes on the cells as genotoxic alterations including DNA breaks, but the actual mechanism leading to a transgenerational effect is still poorly understood. Dubrova et al. suggested an epigenetic mechanism of transmission of the radiation-exposure signal through sperm, likely involving DNA methylation and affecting DNA repair processes [150]. These authors suggested that the persistence of instability into the germ line of unexposed offspring of irradiated mice could be responsible of mosaicism in germ cells, a well-known mechanism in the origin of human genetic disorders [150].
More recently, it has been suggested that a crucial role in transgenerational radiation effects, such as genomic and epigenomic instability, could be played by the Piwi-interacting RNAs (piRNA) pathway, involved in the maintenance of genomic stability by facilitating DNA methylation of transposable elements and also implicated in other epigenetic alterations affecting a variety of cellular regulation processes [151]. Another experiment on animal models supports transgenerational epigenetic changes as a result of parental exposure to genotoxic stressors, as irradiation, nutrition, and intake of anti-androgen compounds [152]. For example, it has been demonstrated that treatment with the anti-androgen compound vinclozolin on female mice induces epigenetic effects in the sperm of their offspring as compared to controls [153]. This study highlighted an increased methylation of the differentially methylated domains (DMDs) of maternal PEG1, PEG3, and SNRPN genes and decreased methylation of paternal H19 and GTL2 genes.
Paternal diet
One of the most intriguing topics in the field of epigenetic modifications of the germ line is represented by the influence played by the paternal diet on gametogenesis. The first evidences of this association came from animal models. Carone et al. demonstrated that male mice fed with low-protein diet generated an offspring showing an increased expression of genes involved in the synthesis of lipids and cholesterol, as compared to the offspring of control male mice fed with a normal diet. Based on these results, authors suggested that cholesterol and lipid metabolism in an offspring can be strongly affected by paternal diet [154]. This study was carried out by a whole-genome characterization of cytosine methylation patterns and RNA content in sperm obtained from mice submitted to low-protein or caloric restriction diets and controls. Authors detected similar cytosine methylation patterns in all three conditions, thus suggesting that the sperm epigenome is largely unaffected by these diets and that changes in relatively few loci can have profound effects in the developing animal [154]. However, in a more recent study, Radford et al. demonstrated that in utero undernourishment perturbs the adult sperm methylome, suggesting that alterations in gamete methylation could induce alterations in chromatin architecture, transcriptional networks differentiation, or tissue structure, and in turn is able to contribute to the intergenerational transmission of environmentally induced diseases [155].
In another animal study, Ng et al. showed the presence of pancreatic alterations, with early onset impaired insulin secretion and glucose tolerance worsening with time, in the female offspring of male mice fed with a high-fat diet [156]. This effect was mediated by the altered expression in adult female offspring of 642 pancreatic islet genes, belonging to 13 functional clusters, including cation and ATP binding, and cytoskeleton and intracellular transport. Fullston et al. demonstrated the presence of altered global methylation in mature sperm and abnormal testis transcription of male mice consuming a high-fat diet, with metabolic disturbances in the next generations [157]. In addition to experimental data on animal models, very interesting data about the role played by diet on the epigenetic modifications and on the consequent transgenerational effects are available in human as well. During the winter of 1944–45 of World War II, in the Netherlands, as a reprisal against the activity of the Dutch government-in-exile aimed to disrupt the transport of German reinforcements and troops, the Germans banned all food and fuel transports to Netherlands, inducing a severe famine, with the official daily rations for the general adult population decreasing gradually from about 1800 calories (December 1943) to below 800 calories (April 1945). The situation improved in a very short time after the liberation of the Netherlands on May 1945, with the rations raising up to over 2000 calories a day by June 1945 [158]. The famine caused a severe mortality in the population of Amsterdam, but nevertheless, several babies were conceived and birthed during that period. Several decades later, a number of studies investigated the health status of people born in Amsterdam during the famine period in order to shed light on the effects of malnutrition on the health of the offspring in adult life. In a first time, these studies evidenced an association with chronic diseases in adult life in the offspring (coronary heart disease, atherogenic lipid profile, obesity, raised levels of plasma fibrinogen, and decreased levels of factor VII), strongly related to the timing in gestation of exposure to famine [158, 159]. However, by analyzing the results of these epidemiological studies with the aid of molecular tools, it has become clear that the Dutch famine families study has provided the first direct evidence for epigenetic programming through prenatal famine exposure. In fact, it was clearly demonstrated that periconceptional exposure to famine produced an under-methylation (likely related to a deficiency in methyl donors) in the differentially methylated region (DMR) of the maternally imprinted IGF2 gene [160], suggesting that early undernutrition can cause epigenetic changes persisting throughout life. On the other hand, there was no variation in IGF2 methylation status in individuals exposed to famine in later gestation. Further studies evidenced that persistent changes in DNA methylation represent a common consequence of prenatal famine exposure and that they can be affected by the sex of the exposed individual and the gestational timing of the exposure [161]. More recently, it has been demonstrated that prenatal malnutrition-associated DMRs (P-DMRs) mostly occur in regulatory regions of genes showing differential expression during early development [162].
All the above-reported studies suggest that undernutrition plays a direct effect on the fetus during the early phases of development. However, why should we exclude that the target of the famine could be represented also by gametes other than by embryo? Several evidences strongly suggest a role played by paternal diet on the healthy status of children, as confirmed by the above-described results on animal models. In human, the link between grandparental nutrition and grandchild’s growth was at first examined by Bygren et al., who demonstrated that a surfeit of food in the environment when the paternal grandfather was a boy was related to a shortening of the proband survival [163]. The same group subsequently demonstrated that a limited availability of food during the father’s prepuberal age was related to a low cardiovascular disease mortality of the proband, while paternal grandfather exposure to a surfeit of food during the same period was related to increased diabetes mortality in the proband [164], suggesting epigenetic inheritance as a strong candidate for these phenomena [165]. Soubry et al. demonstrated the presence of alterations in the methylation status at multiple imprint regulatory regions in children with obese parents, suggesting a preconceptional influence of parental lifestyle and nutrition on the programming of imprint marks during gametogenesis [166]. In particular, the significant association between paternal obesity and altered methylation in the offspring suggests the susceptibility of the developing sperm to environmental insults. Very recently, the evidence of a role played by changes in paternal grandmothers’ early food supply on the risk of cardiovascular mortality of the female grandchildren have also suggested an X-linked epigenetic inheritance via spermatozoa [167]. However, it has also been stressed that a true transgenerational inheritance in response to diet should be examined on the third generation, which represents the actual first “unexposed” one, being the first filial generation directly exposed to the maternal diet, and deriving the second from gametes exposed in utero [168]. Despite these limitations, the possible presence of transgenerational effects related to the epigenetic effect of paternal diet remains a very interesting topic.
The four windows of epigenetic susceptibility
A crucial question concerning the role played by environmental agents in the epigenetic modifications of the male gamete is the following: when are the effects of such exposures transferred to the male gamete? Soubry et al. identified four potential windows of susceptibility during the development of the paternal germ line and zygote [145]. The first window is represented by paternal embryonic development, when PGC undergo genome-wide epigenetic erasure during migration to the genital ridge. Defects in this process, as well as in the maintenance of some protected regions, could be caused by internal or external factors during early development. The second window is represented by paternal prepuberty, since in this period, de novo methylation at imprinted gene loci occurs. The third window can be identified in the period in which spermatogenesis, and in particular the development from spermatogonium to spermatocytes, occurs, since methylation patterns are established during this time. This window appears to be a very important one, representing the reproductive period of the subject, when a careful evaluation of his lifestyle, with prevention of environmental stressors, could be used as a preventive strategy. Finally, the fourth window is represented by the periconception period and the zygote stage, when histone retention in certain genes could represent a potential mechanism for inheritance of environmentally induced epigenetic marks. While studies investigating the effect of environmental agents in the latter three windows are currently carried out, mostly on animal models but also in humans, it is more difficult to investigate epigenetic modifications in PGCs. However, the recently reported discovery that amniotic fluid stem cells (AFSCs) share a number of features with PGCs [169] provides a novel cellular model for the study of the effect played by environmental agents in altering the epigenetic processes occurring in these cells, opening new scenarios in this field of study.
Finally, it must be stressed that aging represents another possible risk factor for epigenetic modifications increasing the risk of neuropsychiatric disorders such as autism and schizophrenia. Jenkins et al. have recently identified 139 regions significantly and consistently hypomethylated and 8 regions significantly hypermethylated with aging, with a total of 117 genes involved [170]. These authors evidenced that a portion of the age-related changes in sperm DNA methylation involves genes previously associated with schizophrenia and bipolar disorder.
Conclusions
As evidenced by the large amount of studies carried out in this field, epigenetic mechanisms play a key role in the proper function of the male gamete, and alterations in these mechanisms can widely affect human reproduction. The effect of epigenetic modification of sperm gene function can affect the reproductive outcome in at least four different levels: (1) impairment of male fertility due to alterations in sperm number and morphology; (2) alterations of embryo development; (3) poor outcome of the ART protocols; and (4) risk of pathologies in the adultness for the offspring (Fig. 2). Due to the huge interest devoted to this topic by the scientific community, related to the possible implications in the field of human reproduction and health, our knowledge about the above-discussed mechanisms are increasing day by day. Recent studies have highlighted the molecular mechanisms underlying the epigenetic transgenerational inheritance of some disease. Guerrero-Bosagna et al. have demonstrated the presence of unique consensus DNA sequence motifs, zinc finger motifs, and G-quadruplex sequences in transgenerational DMR in sperm, which, by the interaction of molecular factors, could induce alterations of the chromatin structure and accessibility of proteins with DNA methyltransferases altering de novo DNA methylation patterns [171].

Epigenetic alterations induced by lifestyle and environmental factors (diet, smoking, radiation, alcohol consumption, etc.) can have substantial effects on the sperm function. As a first consequence, these modifications can induce sperm alterations leading to impairment of male fertility. When fertilization occurs, spontaneously or by ART, transgenerational epigenetic effects can be observed, in details leading to (1) alterations of embryo development, (2) congenital diseases at birth, and (3) late onset diseases (obesity, hypertension, diabetes, etc.) in the adult life
It can be suggested that in the next future, the study of epigenetics and epigenomics will likely represent a crucial step in the diagnostic workup of the infertile male, especially in cases submitted to ART, where it will be necessary to select adequately functional sperm to avoid the epigenetic alteration impact on the procedure. So far, the application of the analysis of epimutations in the male gamete in the clinical practice is hampered by the lack of complete information about the involved genes and by the use of expensive, low-throughput techniques. However, due to the large number of ongoing studies in this field, a clearer picture of the situation should be available in a short time, and the set-up of specific assays will likely reduce the costs and the time of these analyses. Further information will likely be provided by studies investigating the role played by sperm non-coding RNA in male fertility, which represents a very promising field of study [172]. Another very exciting field is represented by the potential role played by the non-sperm fraction of the seminal fluid, since postejaculatory effects on sperm survival and functional competence have been reported [173]. Surprisingly, seminal plasma may affect offspring independently of sperm, by stimulating the production of embryotrophic cytokines and growth factors by the female reproductive tract [173]. The alteration of this process induces abnormal fat deposition and metabolic phenotype in the offspring, particularly in the males [174]. Finally, great attention should be devoted to the role played by environmental agents both in determining and in repairing epigenetic alterations. In fact, the identification of the specific doses and times of action of agents able to induce epigenetic alteration of sperm DNA or to restore the functional conditions will be likely of great help in the treatment of spermatogenetic defects and/or poor outcome of both normal and in vitro fertilization. Most importantly, the possibility that paternal lifestyle could affect the health of the offspring during lifetime opens a novel and exacting scenario in the prevention of common, late onset diseases [175].
Abbreviations
- 5hmC:
-
5-hydroxymethylcytosine
- AFSCs:
-
amniotic fluid stem cells
- ART:
-
assisted reproduction techniques
- CNVs:
-
copy number variants
- COBRA:
-
combined bisulfite restriction analysis
- DMDs:
-
differentially methylated domains
- DMR:
-
differentially methylated region
- DNMTs:
-
DNA methyltransferases
- ESCs:
-
embryonic stem cells
- H3-K4:
-
histone H3 lysine 4
- H3-K9:
-
histone H3 lysine 9
- HAT:
-
histone acetyl transferase
- HDAC:
-
histone deacetylase
- HDM:
-
histone demethylase
- HMT:
-
histone methyltransferase
- ICR1:
-
imprinting control region 1
- lincRNAs:
-
large intergenic non-coding RNAs
- LOS:
-
large offspring syndrome
- NGS:
-
next-generation sequencing
- NZ:
-
normozoospermic
- OAT:
-
oligoasthenoteratozoospermic
- P-DMRs:
-
prenatal malnutrition-associated differentially methylated regions
- PGCs:
-
primordial germ cells
- piRNA:
-
Piwi-interacting RNAs
- TP:
-
transition proteins
- UPD:
-
uniparental disomy
- WGBS:
-
whole-genome bisulfite sequencing
References
Mascarenhas MN, Flaxman SR, Boerma T, Vanderpoel S, Stevens GA. National, regional, and global trends in infertility prevalence since 1990: a systematic analysis of 277 health surveys. PLoS Med. 2012;9:e1001356.
Krausz C. Male infertility: pathogenesis and clinical diagnosis. Best Pract Res Clin Endocrinol Metab. 2011;25:271–85.
Ferlin A, Raicu F, Gatta V, Zuccarello D, Palka G, Foresta C. Male infertility: role of genetic background. Reprod Biomed Online. 2007;14:734–45.
Esteves SC. A clinical appraisal of the genetic basis in unexplained male infertility. J Hum Reprod Sci. 2013;6:176–82.
Stuppia L, Gatta V, Calabrese G, Franchi PG, Morizio E, Bombieri C, et al. A quarter of men with idiopathic oligo-azoospermia display chromosomal abnormalities and microdeletions of different types in interval 6 of Yq11. Hum Genet. 1998;102:566–70.
Clementini E, Palka C, Iezzi I, Stuppia L, Guanciali-Franchi P, Tiboni GM. Prevalence of chromosomal abnormalities in 2078 infertile couples referred for assisted reproductive techniques. Hum Reprod. 2005;20:437–42.
Stuppia L, Gatta V, Antonucci I, Giuliani R, Scioletti AP, Palka G. Genetic testing in couples undergoing assisted reproduction technique protocols. Expert Opin Med Diagn. 2009;3:571–83.
McLachlan RI, O’Bryan MK. State of the art for genetic testing of infertile men. J Clin Endocrinol Metab. 2010;95:1013–24.
O’Flynn O’Brien KL, Varghese AC, Agarwal A. The genetic causes of male factor infertility: a review. Fertil Steril. 2010;93:1–12.
Kovac JR, Pastuszak AW, Lamb DJ. The use of genomics, proteomics, and metabolomics in identifying biomarkers of male infertility. Fertil Steril. 2013;99:998–1007.
Moro E, Ferlin A, Yen PH, Franchi PG, Palka G, Foresta C. Male infertility caused by a de novo partial deletion of the DAZ cluster on the Y chromosome. J Clin Endocrinol Metab. 2000;85:4069–73.
Machev N, Saut N, Longepied G, Terriou P, Navarro A, Levy N, et al. Sequence family variant loss from the AZFc interval of the human Y chromosome, but not gene copy loss, is strongly associated with male infertility. J Med Genet. 2004;41:814–25.
Hucklenbroich K, Gromoll J, Heinrich M, Hohoff C, Nieschlag E, Simoni M. Partial deletions in the AZFc region of the Y chromosome occur in men with impaired as well as normal spermatogenesis. Hum Reprod. 2005;20:191–97.
Rozen SG, Marszalek JD, Irenze K, Skaletsky H, Brown LG, Oates RD, et al. AZFc deletions and spermatogenic failure: a population-based survey of 20,000 Y chromosomes. Am J Hum Genet. 2012;91:890–96.
Previderé C, Stuppia L, Gatta V, Fattorini P, Palka G, Tyler-Smith C. Y-chromosomal DNA haplotype differences in control and infertile Italian subpopulations. Eur J Hum Genet. 1999;7:733–36.
Paracchini S, Stuppia L, Gatta V, Palka G, Moro E, Foresta C, et al. Y-chromosomal DNA haplotypes in infertile European males carrying Y-microdeletions. J Endocrinol Invest. 2000;23:671–76.
Paracchini S, Stuppia L, Gatta V, De Santo M, Palka G, Tyler-Smith C. Relationship between Y-chromosomal DNA haplotype and sperm count in Italy. J Endocrinol Invest. 2002;25:993–95.
Arredi B, Ferlin A, Speltra E, Bedin C, Zuccarello D, Ganz F, et al. Y-chromosome haplogroups and susceptibility to azoospermia factor c microdeletion in an Italian population. J Med Genet. 2007;44:205–08.
Rovio AT, Marchington DR, Donat S, Schuppe HC, Abel J, Fritsche E, et al. Mutations at the mitochondrial DNA polymerase (POLG) locus associated with male infertility. Nat Genet. 2001;29:261–62.
Brusco A, Michielotto C, Gatta V, Foresta C, Matullo G, Zeviani M, et al. The polymorphic polyglutamine repeat in the mitochondrial DNA polymerase gamma gene is not associated with oligozoospermia. J Endocrinol Invest. 2006;29:1–4.
Poongothai J. Mitochondrial DNA, polymerase gamma gene polymorphism is not associated with male infertility. J Assist Reprod Genet. 2013;30:1109–14.
Krausz C, Giachini C, Lo Giacco D, Daguin F, Chianese C, Ars E, et al. High resolution X chromosome-specific array-CGH detects new CNVs in infertile males. PLoS ONE. 2012;7:e44887.
Chianese C, Gunning AC, Giachini C, Daguin F, Balercia G, Ars E, et al. X chromosome-linked CNVs in male infertility: discovery of overall duplication load and recurrent, patient-specific gains with potential clinical relevance. PLoS ONE. 2014;9:e97746.
Riggs AD, Martienssen RA, Russo VEA. Introduction. In Epigenetic mechanisms of gene regulation. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1996.
Cloud J. Why Your DNA Isn’t Your Destiny, Time Magazine. 2010.
Liyanage VR, Jarmasz JS, Murugeshan N, Del Bigio MR, Rastegar M, Davie JR. DNA modifications: function and applications in normal and disease States. Biology (Basel). 2014;3:670–723.
van Montfoort AP, Hanssen LL, de Sutter P, Viville S, Geraedts JP, de Boer P. Assisted reproduction treatment and epigenetic inheritance. Hum Reprod Update. 2012;18:171–97.
Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97:5237–42.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22.
Ziller MJ, Muller F, Liao J, Zhang Y, Gu H, Bock C, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7:e1002389.
Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440.
Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–31.
Ichiyanagi T, Ichiyanagi K, Miyake M, Sasaki H. Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res. 2013;41:738–45.
Ye C, Li L. 5-hydroxymethylcytosine: a new insight into epigenetics in cancer. Cancer Biol Ther. 2014;15:10–5.
Gan H, Wen L, Liao S, Lin X, Ma T, Liu J, et al. Dynamics of 5-hydroxymethylcytosine during mouse spermatogenesis. Nat Commun. 2013;4:1995.
Wang XX, Sun BF, Jiao J, Chong ZC, Chen YS, Wang XL, et al. Genome-wide 5-hydroxymethylcytosine modification pattern is a novel epigenetic feature of globozoospermia. Oncotarget. 2015;6:6535–43.
Razin A, Riggs AD. DNA methylation and gene function. Science. 1980;210:604–10.
Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–34.
Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:E32.
Dupont JM, Tost J, Jammes H, Gut IG. De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal Biochem. 2004;333:119–27.
Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827–31.
Mardis ER. A decade’s perspective on DNA sequencing technology. Nature. 2011;470:198–203.
Laird PW. Principles and challenges of genome wide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203.
Mensaert K, Denil S, Trooskens G, Van Criekinge W, Thas O, De Meyer T. Next-generation technologies and data analytical approaches for epigenomics. Environ Mol Mutagen. 2014;55:155–70.
Miura F, Enomoto Y, Dairiki R, Ito T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012;40:e136.
Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–31.
Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705.
Dada R, Kumar M, Jesudasan R, Fernández JL, Gosálvez J, Agarwal A. Epigenetics and its role in male infertility. J Assist Reprod Genet. 2012;29:213–23.
Boissonnas CC, Jouannet P, Jammes H. Epigenetic disorders and male subfertility. Fertil Steril. 2013;99:624–31.
Tachiwana H, Osakabe A, Kimura H, Kurumizaka H. Nucleosome formation with the testis-specific histone H3 variant, H3t, by human nucleosome assembly proteins in vitro. Nucleic Acids Res. 2008;36:2208–18.
Tachiwana H, Kagawa W, Osakabe A, Kawaguchi K, Shiga T, Hayashi-Takanaka Y, et al. Structural basis of instability of the nucleosome containing a testis-specific histone variant, human H3T. Proc Natl Acad Sci U S A. 2010;107:10454–9.
Shah B, Kozlowski RL, Han J, Borchers CH. Emerging mass spectrometry-based technologies for analyses of chromatin changes: analysis of histones and histone modifications. Methods Mol Biol. 2011;773:259–303.
Rivera CM, Ren B. Mapping human epigenomes. Cell. 2013;155:39–55.
Hamatani T. Human spermatozoal RNAs. Fertil Steril. 2012;97:275–81.
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–93.
Hamatani T. Spermatozoal RNA, profiling towards a clinical evaluation of sperm quality. Reprod Biomed Online. 2011;22:103–5.
Kotaja N. MicroRNAs and spermatogenesis. Fertil Steril. 2014;101:1552–62.
Barlow DP. Genomic imprinting: a mammalian epigenetic discovery model. Annu Rev Genet. 2011;45:379–403.
Constância M, Kelsey G, Reik W. Resourceful imprinting. Nature. 2004;432:53–7.
Barker DJ, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235–39.
Carrell DT. Epigenetics of the male gamete. Fertil Steril. 2012;97:267–74.
Yanagimachi R. Male gamete contributions to the embryo. Ann N Y Acad Sci. 2005;1061:203–7.
Oliva R. Protamines and male infertility. Hum Reprod Update. 2006;12:417–35.
Balhorn R, Corzett M, Mazrimas JA. Formation of intraprotamine disulfides in vitro. Arch Biochem Biophys. 1992;296:384–93.
Sonnack V, Failing K, Bergmann M, Steger K. Expression of hyperacetylated histone H4 during normal and impaired human spermatogenesis. Andrologia. 2002;34:384–90.
Meistrich ML, Mohapatra B, Shirley CR, Zhao M. Roles of transition nuclear proteins in spermiogenesis. Chromosoma. 2003;111:483–88.
de Yebra L, Ballesca JL, Vanrell JA, Bassas L, Oliva R. Complete selective absence of protamine p2 in humans. J Biol Chem. 1993;268:10553–57.
Carrell DT, Liu L. Altered protamine 2 expression is uncommon in donors of known fertility, but common among men with poor fertilizing capacity, and may reflect other abnormalities of spermiogenesis. J Androl. 2001;22:604–10.
Hammoud SS, Purwar J, Pflueger C, Cairns BR, Carrell DT. Alterations in sperm DNA methylation patterns at imprinted loci in two classes of infertility. Fertil Steril. 2010;94:1728–33.
de Yebra L, Ballesca JL, Vanrell JA, Corzett M, Balhorn R, Oliva R. Detection of p2 precursors in the sperm cells of infertile patients who have reduced protamine p2 levels. Fertil Steril. 1998;69:755–59.
Aoki VW, Moskovtsev SI, Willis J, Liu L, Mullen JB, Carrell DT. DNA integrity is compromised in protamine-deficient human sperm. J Androl. 2005;26:741–48.
de Mateo S, Ramos L, de Boer P, Meistrich M, Oliva R. Protamine 2 precursors and processing. Protein Pept Lett. 2011;18:778–85.
Torregrosa N, Domínguez-Fandos D, Camejo MI, Shirley CR, Meistrich ML, Ballescà JL, et al. Protamine 2 precursors, protamine 1/protamine 2 ratio, DNA integrity and other sperm parameters in infertile patients. Hum Reprod. 2006;21:2084–89.
Oakes CC, la Salle S, Smiraglia DJ, Robaire B, Trasler JM. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev Biol. 2007;307:368–79.
Oliva R, Mezquita C. Marked differences in the ability of distinct protamines to disassemble nucleosomal core particles in vitro. Biochemistry. 1986;25:6508–11.
Navarro-Costa P, Nogueira P, Carvalho M, Leal F, Cordeiro I, Calhaz-Jorge C, et al. Incorrect DNA methylation of the DAZL promoter CpG island associates with defective human sperm. Hum Reprod. 2010;25:2647–54.
Krausz C, Sandoval J, Sayols S, Chianese C, Giachini C, Heyn H, et al. Novel insights into DNA methylation features in spermatozoa: stability and peculiarities. PLoS ONE. 2012;7:e44479.
Gatta V, Raicu F, Ferlin A, Antonucci I, Scioletti AP, Garolla A, et al. Testis transcriptome analysis in male infertility: new insight on the pathogenesis of oligo-azoospermia in cases with and without AZFc microdeletion. BMC Genomics. 2010;11:401.
Houshdaran S, Cortessis VK, Siegmund K, Yang A, Laird PW, Sokol RZ. Widespread epigenetic abnormalities suggest a broad DNA methylation erasure defect in abnormal human sperm. PLoS ONE. 2007;2:e1289.
Kobayashi H, Sato A, Otsu E, Hiura H, Tomatsu C, Utsunomiya T, et al. Aberrant DNA methylation of imprinted loci in sperm from oligospermic patients. Hum Mol Genet. 2007;16:2542–51.
Marques CJ, Costa P, Vaz B, Carvalho F, Fernandes S, Barros A, et al. Abnormal methylation of imprinted genes in human sperm is associated with oligozoospermia. Mol Hum Reprod. 2008;14:67–74.
Khazamipour N, Noruzinia M, Fatehmanesh P, Keyhanee M, Pujol P. MTHFR promoter hypermethylation in testicular biopsies of patients with non-obstructive azoospermia: the role of epigenetics in male infertility. Hum Reprod. 2009;24:2361–64.
Poplinski A, Tüttelmann F, Kanber D, Horsthemke B, Gromoll J. Idiopathic male infertility is strongly associated with aberrant methylation of MEST and IGF2/H19 ICR1. Int J Androl. 2010;33:642–49.
Wu W, Shen O, Qin Y, Niu X, Lu C, Xia Y, et al. Idiopathic male infertility is strongly associated with aberrant promoter methylation of methylenetetrahydrofolate reductase (MTHFR). PLoS ONE. 2010;5:e13884.
Rajender S, Avery K, Agarwal A. Epigenetics, spermatogenesis and male infertility. Mutat Res. 2011;727:62–71.
Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–73.
Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–38.
Rivera RM, Ross JW. Epigenetics in fertilization and preimplantation embryo development. Prog Biophys Mol Biol. 2013;113:423–32.
Monk M, Adams RL, Rinaldi A. Decrease in DNA methylase activity during preimplantation development in the mouse. Development. 1991;112:189–92.
Shi L, Wu J. Epigenetic regulation in mammalian preimplantation embryo development. Reprod Biol Endocrinol. 2009;7:59.
Huang JC, Lei ZL, Shi LH, Miao YL, Yang JW, Ouyang YC, et al. Comparison of histone modifications in in vivo and in vitro fertilization mouse embryos. Biochem Biophys Res Commun. 2007;354:77–83.
Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Embryogenesis: demethylation of the zygotic paternal genome. Nature. 2000;403:501–2.
Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, et al. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10:475–78.
Rougier N, Bourc’his D, Gomes DM, Niveleau A, Plachot M, Pàldi A, et al. Chromosome methylation patterns during mammalian preimplantation development. Genes Dev. 1998;12:2108–13.
Chapman V, Forrester L, Sanford J, Hastie N, Rossant J. Cell lineage-specific undermethylation of mouse repetitive DNA. Nature. 1984;307:284–86.
Rossant J, Sanford JP, Chapman VM, Andrews GK. Undermethylation of structural gene sequences in extraembryonic lineages of the mouse. Dev Biol. 1986;117:567–73.
Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996;379:131–37.
Mak W, Baxter J, Silva J, Newall AE, Otte AP, Brockdorff N. Mitotically stable association of polycomb group proteins and Enx1 with the inactive X chromosome in trophoblast stem cells. Curr Biol. 2004;12:1016–20.
Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science. 2004;303:644–49.
Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008;42:733–72.
Barakat TS, Jonkers I, Monkhorst K, Gribnau J. X-changing information on X inactivation. Exp Cell Res. 2010;316:679–87.
Davis TL, Trasler JM, Moss SB, Yang GJ, Bartolomei MS. Acquisition of the H19 methylation imprint occurs differentially on the parental alleles during spermatogenesis. Genomics. 1999;58:18–28.
Davis TL, Yang GJ, McCarrey JR, Bartolomei MS. The H19 methylation imprint is erased and re-established differentially on the parental alleles during male germ cell development. Hum Mol Genet. 2000;9:2885–94.
Li JY, Lees-Murdock DJ, Xu GL, Walsh CP. Timing of establishment of paternal methylation imprints in the mouse. Genomics. 2004;84:952–60.
Miller D, Ostermeier GC. Towards a better understanding of RNA carriage by ejaculate spermatozoa. Hum Reprod Update. 2006;12:757–67.
Miller D. Ensuring continuity of the paternal genome: potential roles for spermatozoal RNA in mammalian embryogenesis. Soc Reprod Fertil Suppl. 2007;65:373–89.
Gannon JR, Emery BR, Jenkins TG, Carrell DT. The sperm epigenome: implications for the embryo. Adv Exp Med Biol. 2014;791:53–66.
Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460:473–78.
Carrell DT, Hammoud SS. The human sperm epigenome and its potential role in embryonic development. Mol Hum Reprod. 2010;16:37–47.
Hammoud SS, Nix DA, Hammoud AO, Gibson M, Cairns BR, Carrell DT. Genome-wide analysis identifies changes in histone retention and epigenetic modifications at developmental and imprinted gene loci in the sperm of infertile men. Hum Reprod. 2011;26:2558–69.
Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–5.
Ward WS. Function of sperm chromatin structural elements in fertilization and development. Mol Hum Reprod. 2010;16:30–6.
Pinborg A, Henningsen AK, Malchau SS, Loft A. Congenital anomalies after assisted reproductive technology. Fertil Steril. 2013;99:327–32.
Shufaro Y, Laufer N. Epigenetic concerns in assisted reproduction: update and critical review of the current literature. Fertil Steril. 2013;99:605–6.
Bowman P, McLaren A. Viability and growth of mouse embryos after in vitro culture and fusion. J Embryol Exp Morphol. 1970;23:693–704.
Roemer I, Reik W, Dean W, Klose J. Epigenetic inheritance in the mouse. Curr Biol. 1997;7:277–80.
Dean W, Bowden L, Aitchison A, Klose J, Moore T, Meneses JJ, et al. Altered imprinted gene methylation and expression in completely ES cell-derived mouse fetuses: association with aberrant phenotypes. Development. 1998;125:2273–82.
Doherty AS, Mann MR, Tremblay KD, Bartolomei MS, Schultz RM. Differential effects of culture on imprinted H19 expression in the preimplantation mouse embryo. Biol Reprod. 2000;62:1526–35.
Khosla S, Dean W, Brown D, Reik W, Feil R. Culture of preimplantation mouse embryos affects fetal development and the expression of imprinted genes. Biol Reprod. 2001;64:918–26.
Young LE, Sinclair KD, Wilmut I. Large offspring syndrome in cattle and sheep. Rev Reprod. 1998;3:155–63.
Young LE, Fernandes K, McEvoy TG, Butterwith SC, Gutierrez CG, Carolan C, et al. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat Genet. 2001;27:153–54.
Young LE, Schnieke AE, McCreath KJ, Wieckowski S, Konfortova G, Fernandes K, et al. Conservation of IGF2-H19 and IGF2R imprinting in sheep: effects of somatic cell nuclear transfer. Mech Dev. 2003;120:1433–42.
Gosden R, Trasler J, Lucifero D, Faddy M. Rare congenital disorders, imprinted genes, and assisted reproductive technology. Lancet. 2003;361:1975–77.
DeBaun MR, Niemitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet. 2003;72:156–60.
Gicquel C, Gaston V, Mandelbaum J, Siffroi JP, Flahault A, Le Bouc Y. In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am J Hum Genet. 2003;72:1338–41.
Schieve LA, Meikle SF, Ferre C, Peterson HB, Jeng G, Wilcox LS. Low and very low birth weight in infants conceived with use of assisted reproductive technology. N Engl J Med. 2002;346:731–37.
Katari S, Turan N, Bibikova M, Erinle O, Chalian R, Foster M, et al. DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum Mol Genet. 2009;18:3769–78.
Ceelen M, van Weissenbruch MM, Roos JC, Vermeiden JP, van Leeuwen FE, Delemarre-van de Waal HA. Body composition in children and adolescents born after in vitro fertilization or spontaneous conception. J Clin Endocrinol Metab. 2007;92:3417–23.
Ceelen M, van Weissenbruch MM, Vermeiden JP, van Leeuwen FE, Delemarre-Van de Waal HA. Cardiometabolic differences in children born after in vitro fertilization: follow-up study. J Clin Endocrinol Metab. 2008;93:1682–88.
Ceelen M, van Weissenbruch MM, Prein J, Smit JJ, Vermeiden JP, Spreeuwenberg M, et al. Growth during infancy and early childhood in relation to blood pressure and body fat measures at age 8–18 years of IVF children and spontaneously conceived controls born to subfertile parents. Hum Reprod. 2009;24:2788–95.
Chen M, Norman RJ, Heilbronn LK. Does in vitro fertilisation increase type 2 diabetes and cardiovascular risk? Curr Diabetes Rev. 2011;7:426–32.
Niemitz EL, Feinberg AP. Epigenetics and assisted reproductive technology: a call for investigation. Am J Hum Genet. 2004;74:599–609.
Cutfield WS, Hofman PL, Mitchell M, Morison IM. Could epigenetics play a role in the developmental origins of health and disease? Pediatr Res. 2007;61:68R–75R.
Filipponi D, Feil R. Perturbation of genomic imprinting in oligozoospermia. Epigenetics. 2009;4:27–30.
Kobayashi H, Hiura H, John RM, Sato A, Otsu E, Kobayashi N, et al. DNA methylation errors at imprinted loci after assisted conception originate in the parental sperm. Eur J Hum Genet. 2009;17:1582–91.
Montjean D, Ravel C, Benkhalifa M, Cohen-Bacrie P, Berthaut I, Bashamboo A, et al. Methylation changes in mature sperm deoxyribonucleic acid from oligozoospermic men: assessment of genetic variants and assisted reproductive technology outcome. Fertil Steril. 2013;100:1241–47.
Sharma R, Biedenharn KR, Fedor JM, Agarwal A. Lifestyle factors and reproductive health: taking control of your fertility. Reprod Biol Endocrinol. 2013;11:66.
Alegría-Torres JA, Baccarelli A, Bollati V. Epigenetics and lifestyle. Epigenomics. 2011;3:267–77.
Wei Y, Schatten H, Sun QY. Environmental epigenetic inheritance through gametes and implications for human reproduction. Hum Reprod Update 2014
Dashwood R, Ho E. Dietary histone deacetylase inhibitors: from cells to mice to man. Semin Cancer Biol. 2007;17:363–69.
Izzotti A, Larghero P, Longobardi M, Cartiglia C, Camoirano A, Steele VE, et al. Dose-responsiveness and persistence of microRNA expression alterations induced by cigarette smoke in mouse lung. Mutat Res. 2011;717:9–16.
Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243–51.
Wilhelm-Benartzi CS, Houseman EA, Maccani MA, Poage GM, Koestler DC, Langevin SM, et al. In utero exposures, infant growth, and DNA methylation of repetitive elements and developmentally related genes in human placenta. Environ Health Perspect. 2012;120:296–302.
Soubry A, Hoyo C, Jirtle RL, Murphy SK. A paternal environmental legacy: evidence for epigenetic inheritance through the male germ line. Bioessays. 2014;36:359–71.
Feychting M, Plato N, Nise G, Ahlbom A. Paternal occupational exposures and childhood cancer. Environ Health Perspect. 2001;109:193–6.
Reid A, Glass DC, Bailey HD, Milne E, Armstrong BK, Alvaro F, et al. Parental occupational exposure to exhausts, solvents, glues and paints, and risk of childhood leukemia. Cancer Causes Control. 2011;22:1575–85.
Anway MD, Leathers C, Skinner MK. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology. 2006;147:5515–23.
Guerrero-Bosagna C, Covert TR, Haque MM, Settles M, Nilsson EE, Anway MD, et al. Epigenetic transgenerational inheritance of vinclozolin induced mouse adult onset disease and associated sperm epigenome biomarkers. Reprod Toxicol. 2012;34:694–707.
Dubrova YE, Plumb M, Gutierrez B, Boulton E, Jeffreys AJ. Transgenerational mutation by radiation. Nature. 2000;405:37.
Merrifield M, Kovalchuk O. Epigenetics in radiation biology: a new research frontier. Front Genet. 2013;4:40.
Jirtle RL, Skinner MK. Environtal epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–62.
Stouder C, Paoloni-Giacobino A. Transgenerational effects of the endocrine disruptor vinclozolin on the methylation pattern of imprinted genes in the mouse sperm. Reproduction. 2010;139:373–79.
Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–96.
Radford EJ, Ito M, Shi H, Corish JA, Yamazawa K, Isganaitis E, et al. In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science. 2014;345:1255903.
Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature. 2010;467:963–66.
Fullston T, Ohlsson Teague EM, Palmer NO, Deblasio MJ, Mitchell M, Corbett M, et al. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J. 2013;27:4226–43.
Painter RC, Roseboom TJ, Bleker OP. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod Toxicol. 2005;20:345–52.
Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol. 2001;185:93–8.
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–49.
Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–53.
Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592.
Bygren LO, Kaati G, Edvinsson S. Longevity determined by paternal ancestors’ nutrition during their slow growth period. Acta Biotheor. 2001;49:53–9.
Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur J Hum Genet. 2002;10:682–88.
Kaati G, Bygren LO, Pembrey M, Sjöström M. Transgenerational response to nutrition, early life circumstances and longevity. Eur J Hum Genet. 2007;15:784–90.
Soubry A, Murphy SK, Wang F, Huang Z, Vidal AC, Fuemmeler BF et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int J Obes (Lond) 2015; 39:650-657.
Bygren LO, Tinghög P, Carstensen J, Edvinsson S, Kaati G, Pembrey ME, et al. Change in paternal grandmothers early food supply influenced cardiovascular mortality of the female grandchildren. BMC Genet. 2014;15:12.
Vanhees K, Vonhögen IG, van Schooten FJ, Godschalk RW. You are what you eat, and so are your children: the impact of micronutrients on the epigenetic programming of offspring. Cell Mol Life Sci. 2014;71:271–85.
Antonucci I, Di Pietro R, Alfonsi M, Centurione MA, Centurione L, Sancilio S, et al. Human second-trimester amniotic fluid cells are able to create embryoid body-like structures “in vitro” and to show typical expression profiles of embryonic and primordial germ cells. Cell Transplant. 2014;23:1501–15.
Jenkins TG, Aston KI, Pflueger C, Cairns BR, Carrell DT. Age-associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility. PLoS Genet. 2014;10:e1004458.
Guerrero-Bosagna C, Weeks S, Skinner MK. Identification of genomic features in environmentally induced epigenetic transgenerational inherited sperm epimutations. PLoS ONE. 2014;9:e100194.
Lane M, Robker RL, Robertson SA. Parenting from before conception. Science. 2014;345:756–60.
Robertson SA. Seminal plasma and male factor signalling in the female reproductive tract. Cell Tissue Res. 2005;322:43–52.
Bromfield JJ, Schjenken JE, Chin PY, Care AS, Jasper MJ, Robertson SA. Maternal tract factors contribute to paternal seminal fluid impact on metabolic phenotype in offspring. Proc Natl Acad Sci U S A. 2014;111:2200–5.
Hughes V. Epigenetics: the sins of the father. Nature. 2014;507:22–4.
Acknowledgements
This work was supported by grants form the G. d’Annunzio University to Liborio Stuppia (Project “Amniotic Fluid Stem cells in regenerative medicine: differentiation into germ line cells and evaluation of transgenerational epigenetic alteration”) and Patrizia Ballerini (“Role of the purinergic system in stem cells”).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None of the authors has any competing interests in the manuscript.
Authors’ contributions
LS and MF carried out the search of literature about epigenetics and male infertility, epigenetics and ART, and environmental factors inducing epigenetic modifications. They also wrote the relative sections of the manuscript as well as the background and the conclusions. PB contributed to the editing of the section describing chemical agents inducing sperm DNA epigenetic alterations. VG performed the search of the literature about the tools for the epigenome analysis. IA performed the search of literature about epigenetics and embryo development and wrote the relative section as well as the conclusions. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Stuppia, L., Franzago, M., Ballerini, P. et al. Epigenetics and male reproduction: the consequences of paternal lifestyle on fertility, embryo development, and children lifetime health. Clin Epigenet 7, 120 (2015). https://doi.org/10.1186/s13148-015-0155-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-015-0155-4