
Abstract
Background
The prevalence of chronic diseases such as cancer, type 2 diabetes, metabolic syndrome (MetS), and cardiovascular disease increases with age in all populations. Epigenetic features are hypothesized to play important roles in the pathophysiology of age-associated diseases, but a map of these markers is lacking. We searched for genome-wide age-associated methylation signatures in peripheral blood of individuals at high risks for MetS by profiling 485,000 CpG sites in 192 individuals of Northern European ancestry using the Illumina HM450 array. Subjects (ages 6–85 years) were part of seven extended families, and 73% of adults and 32% of children were overweight or obese.
Results
We found 22,122 genome-wide significant age-associated CpG sites (P α=0.05 = 3.65 × 10−7 after correction for multiple testing) of which 14,155 are positively associated with age while 7,967 are negatively associated. By applying a positional density-based clustering algorithm, we generated a map of epigenetic ‘hot-spots’ of age-associated genomic segments, which include 290 age-associated differentially methylated CpG clusters (aDMCs), of which 207 are positively associated with age. Gene/pathway enrichment analyses were performed on these clusters using FatiGO. Genes localized to both the positively (n = 241) and negatively (n = 16) age-associated clusters are significantly enriched in specific KEGG pathways and GO terms. The most significantly enriched pathways are the hedgehog signaling pathway (adjusted P = 3.96 × 10−3) and maturity-onset diabetes of the young (MODY) (adjusted P = 6.26 × 10−3) in the positive aDMCs and type I diabetes mellitus (adjusted P = 3.69 × 10−7) in the negative aDMCs. We also identified several epigenetic loci whose age-associated change rates differ between subjects diagnosed with MetS and those without.
Conclusion
We conclude that in a family cohort at high risk for MetS, age-associated epigenetic features enrich in biological pathways important for determining the fate of fat cells and for insulin production. We also observe that several genes known to be related to MetS show differential epigenetic response to age in individuals with and without MetS.
Similar content being viewed by others

Background
Chronic diseases such as cancer, type 2 diabetes (T2D), metabolic syndrome (MetS), cardiovascular disease, and dementia constitute the most common health problems seen in developed societies (and increasingly, in developing societies), and their prevalence increases with age in all populations [1-4]. It is well established that environmental exposures, especially in early life, can alter the risk of various chronic diseases later in life [5,6], and while the mechanisms involved in this ‘programming’ of future risk are not yet understood in detail, epigenetic changes are believed to play an important role in this process [7,8].
Epigenetic mechanisms mediate the interaction between gene and environment throughout the lifespan; while the underlying genetic sequence does not change, environmental influences can alter epigenetic marks and thus alter gene expression and induce long-term changes in phenotype and disease susceptibility [9]. The gradual accumulation of epigenetic changes in critical genes may contribute to the observed age-related increase in the prevalence of various chronic disorders [10-13]. Epigenetic changes are known to be heritable across more than one generation of offspring in plants and mammals [14-19], and there is evidence that transgenerational epigenetic inheritance also occurs in humans [20-23]. Such transgenerational inheritance of epigenetic states may contribute to the observed inherited risk of various chronic disorders, including metabolic disorders [24].
DNA methylation is one of the most extensively studied epigenetic mechanisms and plays an important role in the process of development and differentiation [25]. There is evidence from both human and animal sources that prenatal nutritional deprivation can permanently alter DNA methylation at multiple loci, and these changes play a role in the observed alteration of future risk of chronic diseases like obesity, insulin resistance, and diabetes [26-32]. It is also known that DNA methylation patterns continue to change after birth, at least partly in response to environmental influences [33-35]. For example, studies show that identical twins have broadly similar epigenetic profiles in utero but these profiles gradually diverge as they get older [36-38]. Several studies have looked at the effect of aging on genome-wide DNA methylation in adults, and these studies show that age-dependent methylation changes are found in a variety of tissues and correlate well enough with age that the methylation status of selected loci can be used to predict the age of a subject [35,39-41].
These age-related methylation changes may play a role in the observed age-related risk of various chronic diseases. For example, studies show that the hyper-methylation of certain CpG loci is associated with increased cancer risk via reduced expression of cancer-suppressor genes [41,42]. It has been proposed that age-related changes in DNA methylation play a similar role in increasing the risk of obesity, T2D, and MetS, but the specific genes involved and the specific changes in their functioning are yet to be determined [43-45].
Studies of genome-wide DNA methylation can be conducted using various populations, and each design has its advantages and disadvantages. For example, monozygotic twins are genetically identical, so epigenetic differences found in twin pairs are likely to be either stochastic or environmentally induced, rather than genetically inherited [46]. In studies using both mono- and dizygotic twins, it is possible to estimate the relative effect of genetic versus environmental influences to some extent. But since twin pairs are of the same age, the differential effect of age cannot be compared within the pairs and the comparison of twins of different ages does not offer any special advantage over comparing two unrelated individuals of different ages. Studies using unrelated subjects have the advantage that large numbers of subjects are relatively easy to recruit, but the epigenetic landscape of unrelated subjects can be influenced by population structure and systematic differences in environmental exposures that may not be easy to identify and that may thus confound the results. In a family-based cohort, we can reduce the confounding effect of genetic variation and population structure and, when large extended families live close to each other and follow similar traditions, they share many environmental factors as well, thus reducing the confounding effect of group differences such as in diet and geographical location. Because such family-based cohorts include related individuals of different ages in the same cross-sectional study, it is possible to examine changes associated with age against a relatively stable genetic and environmental background. Another advantage of using large extended pedigrees is that it may be possible to identify epigenetic patterns that are associated with disease risk specifically within that family and not in the general population. A family-based cohort can thus be an especially powerful tool for identifying age-related methylation changes, including changes that are universally associated with aging and those that are specific to families with shared genetic and environmental risk factors for particular chronic diseases.
While no epigenome-wide study of extended, multi-generational families has yet been published, a recent study on a combination of twin and their nuclear family members examined the role of genetic features on DNA methylation [23]. These authors suggest that the majority of transgenerational similarity in DNA methylation can be explained by shared genetic effects and that epigenetic inheritance (incomplete erasure of epigenetic modifications across generations) has a relatively limited role in the observed inherited risk of various chronic disorders. This observation still needs to be confirmed in other studies and in populations at high risk for particular chronic disorders.
We assembled a cohort comprising several large extended families of Northern European descent that is enriched for obesity, central adiposity, and obesity-associated MetS traits. To identify genomic regions whose methylation status changes with aging, we conducted a genome-wide survey of peripheral blood DNA methylation and interrogated more than 485,000 CpG sites in 192 subjects from seven extended families living in two US Midwestern states, Wisconsin and Illinois.
Results
The TOPS family study of epigenetics
The TOPS Family Study of Epigenetics (TFSE) was designed to study the role of epigenetic mechanisms in linking genes and the environment using related subjects of large extended pedigrees. The average age of the cohort is 36.2 (±18.8) years, 28% of the subjects were 18 years and younger at ascertainment and 55% are females. As the subjects were selected from families that are part of a previous genetic study on the metabolic risk complications of obesity [47], the cohort is enriched for obesity and MetS traits (Table 1) with 73% of the adults being overweight or obese (based on body mass index (BMI)), 52% with waist circumference above MetS thresholds (>102 cm in men; >88 cm in women), 31.9% with evidence of insulin resistance (based on homeostatic model of assessment (HOMA) > 3.5) [48,49], 20.3% with hypertriglyceridemia (>150 mg/dl), and 65.7% with high-density lipoprotein (HDL) below MetS thresholds (<40 mg/dL in males and <50 mg/dl in females). Overall, 23.7% of the adults of our cohort met the ATPIII definition of having MetS [50]. In the pediatric subjects, the prevalence of overweight and obesity (BMI >85th percentile) was 32%. All analyses accounted for the relatedness of family members by conditioning the fixed effects of methylation status on the expected genetic similarity of relatives (Table 2) [51].
The genome-wide autosomal map of age-associated DNA methylation in the TFSE cohort
We have implemented a data cleaning procedure aiming to retain only the informative CpG probes for downstream analyses (see Additional file 1). Of a total of 485,512 CpG sites that were assayed on the Illumina HM450 panel, a total of 137,168 autosomal CpG sites passed our data cleaning procedure and were entered into our statistical analysis pipeline for age association tests.
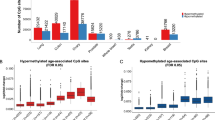
Using methylation status represented by M values (see the ‘Methods’ section), we tested each epigenetic marker for association with age in linear mixed models that included the random effect of kinship using SOLAR [52] (see the ‘Methods’ section). Our models also accounted for the fixed effects of sex and blood cell subtype proportions. Figure 1 shows a Manhattan plot of the CpG sites whose methylation status was associated with age. Of these, 22,122 age-associated CpG sites in our cohort surpassed the genome-wide significance threshold (P α=0.05 = 3.65 × 10−7 after correction for multiple testing). The characteristics of these age-associated CpG sites are shown in Figures 2 and 3. The percentage of genome-wide age-associated sites per probes on each chromosome is shown in Figure 2A. We observed that 39% of these age-associated sites are located within potential regulatory regions of genes (from 5′ UTR to the first exon, Figure 2B). The effect of age on DNA methylation at each individual CpG site is shown as regression coefficients of normalized M value per year of age in Figure 2C. We found that 14,155 of these genome-wide significant CpG sites are positively associated with age while 7,967 sites are negatively associated. This gives a ratio of 1.8 for epigenetic loci that exhibit increasing methylation over time versus those that show decreasing methylation. Examples of genome-wide significant age-associated sites include CpG loci located in the promoter region of the obesity gene LEP [53], the childhood obesity gene OLFM4 [53], the T2D gene IRS2 [54], and the newly identified MetS gene TFAP2B [55] (Figure 3A–D).

Strength of associations of genome-wide autosomal CpG methylation status with age in our TFSE cohort. Manhattan plot shows the significance level of age association of each CpG locus by chromosomal position. Each grey dot represents an individual CpG site. The red line depicts the genome-wide significance threshold after correction for Bonferroni multiple testing, P α=0.05 = 3.65 × 10−7. Colored dots represent epigenetic regions belonging to genes previously associated with obesity, T2D, and MetS. Blue dots obesity-related genes in adults, red dots obesity related genes in children, orange dots obesity related genes in children and adults, green dots T2D genes, yellow dots MetS genes. Selected genes are labeled. Genes found to be within an aDMC are shown in bold and italicized.

Characteristics of age-associated CpG sites. (A) Methylation sites are shown based on their chromosomal location (x-axis). The percentage of sites was determined by the number of age-associated sites on each chromosome over the number of sites in the analysis on each chromosome (n = 137,168). (B) Histogram shows the distribution of genome-wide age-associated sites in relation to gene architecture. ‘Age-related’ sites are the 22,122 sites that were found to be genome-wide age-associated and ‘All’ are the locations of all the sites run in the analysis (n = 137,168). (C) Histogram of regression coefficients for 22,122 loci with genome-wide significant association with age.

DNA methylation changes with age in candidate genes related to obesity, MetS, and T2D. (A) Methylation of CpG site cg12782180 located in the TSS1500 of LEP with respect to age. (B) Methylation of CpG site cg08119452 located in the TSS1500 of OLFM4 with respect to age. (C) Methylation of CpG site cg05404236 located in the first exon of IRS2 with respect to age. (D) Methylation of CpG site cg24366557 located in the body of TFAP2B with respect to age. (E) Pyrosequencing validation of differential methylation with age identified by arrays. Scatter plot shows the overlay of age-associated methylation changes at CpG site cg14956327 (DDO) probed by the Illumina HM450 array (black dots) and pyrosequencing (green dots). Only children and adolescents samples were used in validation tests. The insert shows direct comparisons between the Illumina array and pyrosequencing validation.
To validate the methylation results obtained using the HM450 array, we quantitatively assessed the methylation status of CpG locus cg14956327 in 48 samples ranging from 6–21 years of age using pyrosequencing (Figure 3E). The graph within Figure 3E also shows a direct comparison of 13 samples between the Illumina array and pyrosequencing validation.
We also assessed the coverage of our data by comparing our study to several recent studies of age-associated epigenetic markers (Additional files 2 and 3). We found that the current study has an exceptionally broad age range (with subjects ranging from 6 to 85 years in age) and was able to identify from 38% to 100% of previously identified age-associated CpG sites depending on the study in comparison. For example, we have identified significant age-associations at CpG sites in the genes ELOVL2, FHL2, KLF14, and PENK that had been repeatedly reported as strongly age-associated methylation markers (Additional file 3) [39,56,57]. Our study also identified 21,078 additional genome-wide significant CpG sites that have not been reported in these previous studies.
Bioinformatic analysis of genome-wide significant age-associated DNA methylation sites in the TFSE cohort
We hypothesized that in our subjects, whose family history (genetic and environmental) makes them susceptible to developing obesity and MetS, age-associated DNA methylation is enriched in genes and pathways involved in metabolic homeostasis. Changes caused by aging in these epigenetic states might lead to the malfunctions that underlie the increased prevalence of clinical symptoms of obesity and MetS in the aging population [1,3]. As the subjects we interrogated for genome-wide DNA methylation profiles were from families at high risk for developing MetS, we looked at genes with prior evidence for involvement in obesity and T2D and checked if there were CpG sites above genome-wide significance level associated with any of them.
Known genes for obesity
We found that 20 genes out of the 36 listed in a gene list based on genetic studies of human obesity [53] have one or more methylation sites that is significantly associated with age in our families (Figure 1, Additional file 4). These genes include a number of well-studied obesity genes such as LEP, POMC, PPARG, and CNR1, as well as previous obesity GWAS candidates with unclear roles in obesity etiology, such as SIM1, IRX3, and SLC6A11. Furthermore, four genes identified in GWAS for childhood obesity, such as SDCCAG8, TNKS/MSRA, OLFM4, and HOXB5, were found to be epigenetically age-associated as well.
Genes known to be associated with type 2 diabetes
Of the 20 T2D susceptibility loci recently identified in GWAS [58], ten were found to be epigenetically modified by age in our analysis (Figure 1, Additional file 5). These include PPARG, HNF1B(TCF2), TCF7L2, IGF2BP2, HHEX/IDE , KCNQ1, MTNR1B, ADAMTS9, THADA, and JAZF1. In one of the most recent studies of T2D genes using genome-wide trans-ancestry meta-analysis, seven novel loci were identified [59]. Of these seven loci, we found evidence for age-associated differential methylation at SSR1/RREB1 and LPP. We also looked for any age-associated epigenetic evidence for genes established through approaches other than GWAS and found that the following known T2D genes are under epigenetic regulation by age: IRS1 [60], IRS2 [54], AKT [61], ABCC8 [62], HNF4A [63], IPF-1(PDX1) [64], NeuroD1 [65], and GCK [66].
Pleiotropic genes known to be associated with MetS
Of the 25 genes that have been shown to play pleiotropic roles in MetS and inflammation [55], we found eight genes with one or more methylation sites significantly associated with age in our families (Figure 1; Additional file 6). These genes include GRB14, KIAA0754, MACF1, MLXIPL, SKIV2L, STK19, TFAP2B, and TRIB1.
The map of genomic locations of dense age-associated differentially methylated clusters in the TFSE cohort
We then applied a modified ‘bump-hunting’ algorithm to identify clusters of age-related methylated CpG sites. Using this algorithm, we generated a map of epigenetic ‘hot-spots’ of age-associated genomic segments. In our search algorithm, modified from a previously published method [67,68], we defined a group of sites as a dense age-associated CpG cluster when at least 50% of no less than ten sites are associated with age at genome-wide significance, and the distance between any pair of age-associated sites is no greater than 10 kb. Additional file 7 shows the autosomal map of age-associated differentially methylated CpG clusters (aDMCs) throughout the autosomal genome. We identified 290 aDMCs, of which 207 are positively associated with age, 9 are negatively associated, and 74 have sites associated in either direction. The detailed characteristics of these identified aDMCs are depicted in Figure 4. The distribution of the sizes of these aDMCs showed one peak in the aDMCs with sizes around 5 kb and another around 10 kb (Figure 4A). To study the chromosomal distribution of these aDMCs, we divided the total number of age-associated clusters on each chromosome by the total number of clusters that would be generated if the same algorithm was applied to all loci that survived our data cleaning procedure. We found that 3.8% of all clusters generated by this algorithm are age-associated and they are mostly evenly distributed across the genome, with a few chromosomes being modestly over-represented (Figure 4B). The sizes of identified aDMCs range from 457 to 69,237 bp, and we found that 80 of these aDMCs span over more than one gene (up to a maximum of four genes). When we filter genes known for obesity, T2D, and MetS based on our clustering criteria, three obesity genes, two T2D genes, and one MetS gene each contains at least one aDMC. For instance, we found an aDMC that spans a 2.1-kb region of the gene insulin receptor substrate 2 (IRS2) on chromosome 13. This cluster begins within the promoter region (1,500 bp before transcription starting site (TSS)), 5′ untranslated region (UTR) and ends in the first exon (Figure 1; Additional file 5).

Characteristics of aDMCs defined in our cohort. (A) Histogram shows the size distribution (bp) of the 290 aDMCs. (B) aDMCs are shown based on their chromosomal location (x-axis). The percentage of aDMCs was determined by the number of aDMCs on each chromosome over total number of aDMCs on each chromosome (n = 7,590).
Gene network and pathway analyses of aDMCs in our family cohort
To identify pathways that are enriched in aDMCs associated with age in our cohort in an unidirectional manner, we utilized FatiGO [69] which queried the vast amount of knowledge deposited in the Kyoto Encyclopedia of Genes and Genomes (KEGG) [70,71] and Gene Ontology (GO) databases [72]. We found a number of KEGG pathways and GO terms that are significantly enriched after adjustment in both positively and negatively age-associated clusters. There are a total of 241 genes in these 207 positively age-associated clusters and 16 genes in the 9 negatively associated clusters. As shown in Additional file 8, three KEGG pathways were found to be significantly enriched in clusters positively associated with age, including the hedgehog signaling pathway (adjusted P = 3.96 × 10−3; Figure 5A) and the maturity-onset diabetes of the young (MODY) pathway (adjusted P = 6.26 × 10−3; Figure 5B) and neuroactive ligand-receptor interaction (adjusted P = 1.58 × 10−2). A total of 387 GO terms for biological processes were significantly enriched within the aDMC genes (Additional file 9), as were 55 GO terms for molecular functions (Additional file 10). Type I diabetes mellitus was found to be the most significant pathway enriched in epigenetic clusters negatively associated with age (Figure 5C, Additional file 11). There was no significant enrichment of biological processes and molecular function GO terms within the negative clusters. The biological processes that were significantly enriched in positively associated aDMC genes include development of a multitude of organs and systems from embryonic phases to adulthood, stem cell development and maintenance, cell recognition, motility and migration, regulation of cell differentiation and proliferation and cell cycle, response to temperature and other abiotic stimulus, response to dietary excess, cytokine and insulin secretion, metabolism homeostasis, adult behavior, and aging. The top four GO terms enriched in age-associated epigenetic loci are all related to development of the brain/nervous system. The molecular functions of these genes are also highly enriched in sequence-specific DNA binding.

Top Kegg pathways enriched with aDMCs identified by the FatiGO analysis. (A) The MODY pathway with identified genes of aDMCs and age-associated DNA methylation sites. Genes found with age-associated sites are shown with a blue background, while genes within an aDMC are shown with a green background. (B) The Hedgehog signaling pathway with identified aDMCs and age-associated DNA methylation sites. Genes found with age-associated sites are shown with a blue background, while genes within an aDMC are shown with a green background. (C) Type 1 diabetes pathway with identified aDMCs and age-associated DNA methylation sites.
Analysis of differential age effects on CpG methylation at known MetS candidate loci in subjects with MetS and those without
We tested the hypothesis that the aging rates for CpG methylation at MetS genes are different in subjects expressing MetS symptoms as compared to subjects without MetS. There are 127 CpG sites which belong to 11 genes previously implicated in MetS genetics (55) in our dataset after QC. Of these, 23 CpG loci were genome-wide significantly associated with age in our cohort. We examined the aging rate of each of these 23 epigenetic markers in the two groups of adult subjects that were separated using the ATPIII definition of MetS (24% with MetS) and found that four sites of four different genes showed nominal differentiation between the two groups (Table 3). Remarkably, we found that not only do the aging rates differ in these two groups by 2.6–5.1 fold but their methylation also changes over time in opposite directions. As shown in Figure 6A, CpG site cg06117072 exhibited increased methylation over age in non-MetS subjects but decreased over time in MetS subjects. In other sites such as cg10092878 in the MetS gene MLX interacting protein-like (MLXIPL), the methylation aging rates did not show any significant difference between the two groups of subjects (Figure 6B).

MetS status and aging rate of candidate CpG site methylation. (A) Methylation of CpG site cg06117072 located in the gene body of TFAP2B, a known MetS gene, with respect to age, in subjects with MetS and those without. (B) Methylation of CpG site cg10092878 in the MetS gene MLXIPL with respect to age, in subjects with MetS and those without.
Discussion
We report here the first survey of age-associated peripheral blood DNA methylation in a cohort of Northern European origin comprised of large extended families. Using families with extended pedigrees to study the association between age and epigenetic modifications of each interrogated CpG locus can be more powerful than designs using unrelated [34,73], identical and fraternal twins [36,37], or small nuclear pedigrees [74]. It has been shown that genetic effects determine the majority of transgenerational similarity in DNA methylation in humans [23], and by using extended pedigrees, we may have more power to detect age-affected epigenetic signals as compared to designs using unrelated subjects because these families will have less of the epigenetic variation that can be caused by differences in the genetic makeup of unrelated individuals (Additional file 2). By comparing our study with several recent genome-wide age-association studies, we found that the current study has one of the broadest age sample ranges, including children and adolescents, and we were able to identify the majority of previously identified age-associated CpG sites [35,39-41,56,57,75-77] as well as more than 20,000 novel age-associated CpG sites.
Since our study utilized an obesity-prone cohort, it is possible that some of the methylation changes we see in the older subjects are due to the effect of obesity and not due to aging per se. We cannot exclude this possibility, but since we found a large number of loci that have also been discovered in other studies of age-related methylation changes (Additional file 2) that were not related to obesity, we believe that our findings are more likely to be age-related than obesity-related.
We examined the age-associated methylation loci that surpassed genome-wide significance level (after Bonferoni correction, P α = 0.05 = 3.65 × 10−7) in genomic regions densely packed with age-associated CpG sites. We have named these clusters as aDMCs (Additional file 7). We found a total of 290 aDMCs across the autosomal genome with the majority of clusters containing unidirectional, positively associated CpG sites. Only nine clusters were found with CpG sites all negatively associated with age. The sizes of these clusters range from less than 0.5 Kb in a transcription promoter to a nearly 70 Mb region that can span up to four genes.
The families used in the current study all live in rural or semi-urban regions of the states of Wisconsin (six families) and Illinois (one family) and are mostly categorized as working and/or middle class families (TOPS Club Inc., personal communication). There are likely to be broadly similar household routines of dietary intake and activity within these large families. We therefore expect that by using these families to study epigenetic changes over age, we reduce systematic differences in external environmental factors such as geographical location and dietary and lifestyle patterns.
The probands of our TFSE cohort were recruited based on the presence of at least two obese and one never-obese sibling(s) in each family, thus raising the likelihood of having obesity-prone genetic patterns in these families. We hypothesize that since our subjects have an obesity-prone genetic background and live in an obesity-inducing environment, the genomic regions that are strongly associated with age are also enriched in gene groups and pathways involved in metabolism-related cascades.
To find these loci, we first searched in our list of age-associated differentially methylated CpG sites and clusters for any gene(s) previously characterized for a role in the etiology of obesity, T2D, or MetS and inflammation. We found that the majority of previously established obesity, T2D, or MetS and inflammation genes overlap with one or more of our age-associated differentially methylated clusters (Figure 1, Additional files 4, 5, and 6). When we filter these genes based on our clustering criteria, three obesity genes, two T2D genes, and one MetS gene each contains at least one aDMC (Figure 1, Additional files 4, 5, and 6). Some of these genes have been well-characterized previously. For example, the product of gene IRS2 works as a signaling mediator between cytoplasmic receptor kinases and downstream effectors including PI3 Kinase, Akt, and mTOR [78] and is an important adaptor in cascades regulated by insulin, insulin-like growth factor 1, interleukin 4 (IL-4), and other cytokines [79]. Diseases associated with disrupted functions of IRS2 include fatty liver disease and glucose intolerance, which is a precursor to MetS [80,81].
Single-minded 1 (SIM1) is a helix-loop-helix PAS domain transcription factor. Sim, its homologue in Drosophila, is a key factor in determining the differentiation of central nervous system (CNS) midline cells [82]. Losing one functional copy of Sim1 in mice leads to early-onset obesity, hyperinsulinemia, and hyperleptinemia [83]. In humans, disruption of SIM1 gene locus has been found to have caused profound early-onset obesity [84]. In our analysis, SIM1 has an aDMC that spans 22,914 bp covering the 5′-untranslated region to exon 8 containing 44 CpG sites that were significantly associated with age including five sites situated in the promoter region (TSS1500 to first exon).
A number of genes known to work in the regulation of food intake and energy balance are found to have one or more CpG markers modifiable by age. These include leptin and related genes LEP, LEPR, CNR1, DRD2, and SLC6A4 as well as the hypothalamic factor POMC and its processor PCSK1 and neurotropic factor BDNF and its receptor NTRK2 [53]. Clinically, disruption of POMC, PCSK1, BDNF, and NTRK2 is associated with severe early-onset obesity [85,86]. PPARG is one of the most replicated diabetes and obesity genes, with sequence variants strongly associated with obesity [87] and T2D [88]. We have found two age-associated differentially methylated sites inside the body of the PPARG gene.
Multiple obesity and T2D candidate genes identified by the GWAS approach were found to have age-associated DNA methylation associations in our analysis. These genes include extensively replicated genes such as BDNF, which is not only shown to be associated with adult obesity but also with childhood obesity [89]. Furthermore, our results include four other genes for childhood obesity (identified by GWAS) that were differentially methylated according to age: SDCCAG8 and TNKS/MSRA [90] were first identified in Northern European populations for early-onset extreme obesity whereas OLFM4 and HOXB5 are two recently discovered candidate genes for common childhood obesity that were found by combining 14 existing GWAS datasets [91]. Interestingly, we found that the gene SDCCAG8 contains epigenetic markers that alter their methylation more rapidly in children than in adults (data not shown), suggesting a possible explanation for these genes having more profound effect on clinical phenotypes in children than in later life.
Pathway enrichment analysis reveals that in our family cohort, age-associated DNA methylation sites are densely packed around genes working in pathways such as the hedgehog signaling pathway, the MODY pathway, and T1D pathway (Additional files 8 and 10). These findings suggest intriguing connections between epigenomic profiles and the high risk and high prevalence of obesity and MetS traits in our study subjects. Hedgehog signaling is not only a key regulator of development in both invertebrate and vertebrate systems but it also plays a critical and conserved role in fat formation [92], fat storage, and brown fat and muscle cell metabolism reprograming in animals [93]. Blocking Hh signaling by an antibody in adult mice fed on a high fat diet protects them from gaining weight and developing liver steatosis [94]. The molecular cascade of Hh signaling involves the initial binding of one of the Hh proteins Sonic Hh (SHH), Indian Hh (IHH), and Desert Hh (DHH), with receptor PACH1 and PACH2 that release its inhibition on the membrane receptor Smoothened (SMO). Released SMO then activates a complex signaling cascade, which leads to nuclear translocation of transcription factors of the Gli (GLI) family and the resultant activation or repression of downstream genes [95-97]. In our results, age-associated differential methylation was found to be associated with Hh genes IHH and DHH, Hh receptor genes PACH1 and PACH2, and SMO and the downstream effector genes GLI2 and GLI3, and several other regulators of the Hh pathways including SUFU and PKA (Figure 5). Densely packed age-associated CpG sites, defined as aDMCs, are present in the regulatory region and/or the body of genes of transcription factors WNT, BMPs, and Zic2 (Figure 5). Our findings suggest a novel mechanism in which the process of aging influences genomic regulatory marks of a group of genes that work in pathways critical for fat cell fate determination, fat storage, cellular metabolism reprogramming, and diet-dependent regulation of mammalian body weight and lipid metabolism.
Diabetes mellitus, like obesity, is a chronic condition that increases in its prevalence as people age [1,3], but the mechanism underlying this age-dependent risk is not clear. The ‘acquired’ form of diabetes, T2D, is a complex syndrome whose onset is determined by multiple genes and their interactions with the environment [98]. T2D affects an estimated 350 million people in the world today and is caused by complete or partial malfunction in a body’s ability to respond to blood glucose through production of insulin [99,100]. MODY is a type of diabetes that is defined collectively by clinical symptoms caused by mutations in any one of the six genes that work in the MODY pathway. Five of these six genes (HNF4A, HNF1A, PDX1, HNF1B, NeuroD1) encode transcription factors that regulate β-cell homeostasis and/or insulin production and secretion, and gene GK is a glucokinase that is involved in beta cell sensing of blood glucose levels [101]. In our epigenetic study of obesity-prone families, the MODY gene pathway was the second most significant pathway with dense enrichment of DNA methylation CpG sites/clusters strongly associated with age. This finding suggests a novel mechanism in which epigenetic changes affect outcomes of acquired obesity-induced beta-cell and insulin unresponsiveness that leads to T2D. As the patterns in DNA methylation of these loci change over age, it may explain the polygenic, complex, and subtle features that are observed in the development of obesity and T2D and their subsequent course.
In this study, we demonstrated the first evidence of differential methylation aging in MetS genes in MetS subjects as compared with non-MetS subjects. Our results showed an interesting pattern in which the epigenetic changes over age are slower in MetS subjects, and its directionality is the opposite to that in non-MetS subjects in all four of these identified loci (Table 3; Figure 6). Our results suggest that the age effects on epigenetic changes are both genomic locus- and MetS status-dependent. The four nominal MetS-differentiated aging markers are situated at genes that have been previously shown to be metabolism and MetS traits-relevant in genetic or cell biology studies. But the epigenetic implications of these genes in MetS have not been reported. Transcription factor activating enhancer binding protein 2 beta (TFAP2B) (cg06117072) encodes a transcription factor from the AP-2 family. This gene has been implicated in dietary weight maintenance [102,103]. Growth factor receptor-bound protein 14 (GRB14) (cg04926134) encodes an adaptor protein that binds with insulin receptors and insulin-like growth factor receptors that may have an inhibitory effect on insulin signaling and may play important roles in metabolic homeostasis and growth regulation [104-107]. Tribbles pseudokinase-1 (TRIB1) (cg14683125), a mitogen-activated protein kinase (MAPK) activation modulator, was found to control migration and proliferation of smooth muscle cells [108] and has been implicated in lipid metabolism [109-111]. Microtubule-actin crosslinking factor 1 (MACF1) (cg22697325) is a member of protein family that form bridges between different cytoskeletal elements [112].
With the size of our sample, we do not have enough statistical power to detect all existing signals for MetS-specific aging methylation. We expect that with a larger sample size, one can discover many more disease state-dependent epigenetic markers not only for MetS but also for other aging relevant conditions such as obesity, T2D, dementia, and cancer. This may eventually lead to new clinical approaches in screening and diagnosing people with differential ‘epigenetic risks’ for developing diseases as they age.
We examined the epigenetic changes associated with aging in DNA obtained from peripheral blood, a tissue type that is routinely used as a surrogate for mapping age-related DNA methylation changes [35] because even though DNA methylation (unlike the DNA sequence) is tissue specific, there is a significant portion of the epigenome with patterns of DNA methylation common to multiple tissues [113], and this may be especially true of age-related methylation patterns [40,75]. In addition, a study using peripheral blood has the potential to identify changes in DNA methylation that can be practically used as a diagnostic test in clinic, where blood is much more likely to be available for testing as compared to other tissue samples. However, based on our findings, it will be interesting in a future study to profile some of our age-associated candidate sites in target tissues such as beta cells and to relate these patterns with beta cell function and insulin gene expression.
We conducted our study in a family cohort of Northern European descent. Generalization of our findings requires validation in distinct cohorts with similar pedigree structures. It will also be a valuable expansion of our cross-sectional study if we can recall some of our subjects to obtain longitudinal data on the epigenetic changes as well as on the functional status of metabolic pathways over time in the same individuals. Due to the scope and the focus of the current study, we have not looked at the associations of particular methylation loci with phenotypes in our subjects as this will be approached in the future. The connection between epigenetic status and gene expression in blood and certain target tissue types also warrants further investigation.
Although our mapping utilized one of the array-based epigenetic platforms that gave the highest available genomic site coverage, it has not nearly exhausted the epigenome. A next-generation sequencing-based approach such on methyl-binding domain-isolated genomic sequencing (MiGS), MeDIP-seq, or bisulphite-sequencing [114] will be a way to improve the coverage of all possible epigenetic sites with age.
Conclusions
We have conducted one of the first genome-wide surveys of age-associated DNA methylation in a family cohort with large extended pedigrees. In families at high risks for developing obesity-related metabolic disorders, we found age-associated genomic loci densely situate near genes that function in the hedgehog signaling pathway and in MODY. These findings suggest a novel mechanism underlying the gradual deleterious effects of multiple genes and their interactions with nutrition over time, which may contribute to obesity and its complications. The results from this study shed light on the relationship between aging and increased prevalence of obesity, T2D, and related abnormalities and thus may lead to novel approaches for early detection and prevention of these health-endangering conditions.
Methods
Samples
The study cohort consists of 192 individuals ranging in age from 6 to 85 years old representing seven families. Of these, 53 subjects were 18 years and younger at ascertainment and 106 are females. Details of recruitment and phenotyping procedures have been described previously [47,115]. Briefly, each nuclear family was recruited through an obese proband (BMI ≥ 30) who was a member of TOPS Club with the minimal requirement of the availability of two obese siblings, a least one, preferably both, of the parents and one never-obese (BMI ≤ 27) sib and/or parent. A subsequent extension included the ascertainment of all biologically related members over the age of 18 including aunts, uncles, grandparents, and adult children and their accompanying parent(s). Recently, this cohort was further enhanced by the ascertainment of their children and adolescent descendants (aged 6–18 years) with their accompanying parent(s). Clinical phenotypes for all subjects included weight, height, BMI, waist circumference (WC), hip circumference (HC), waist to hip ratio (WHR), fasting glucose (FG), fasting insulin (FI), insulin to glucose ratio (IGR), homeostasis model assessment (HOMA), plasma triglycerides (TG), total cholesterol (TC), low density lipoprotein cholesterol (LDL-c), high density lipoprotein cholesterol (HDL-c), systolic and diastolic blood pressure (sBP and dBP), and pulse. Total abdominal fat, visceral fat (VF), and subcutaneous fat (SubQF) were measured by computed tomography scans of the fourth lumbar spine [116] in adults and by magnetic resonance imaging at the same level in children and adolescents; circulating levels of adiponectin and leptin were measured by a double antibody equilibrium radioimmunoassay (RIA) (Millipore Corporation, Billerica, MA) and TNF-alpha, interleukin-1beta (IL-1β), and interleukin-6 (IL-6) levels that were measured as previously described [117]. Adult Treatment Panel III (ATPIII) criteria was used to identify adults with MetS. Informed consent was obtained from the participating subjects. All study procedures for adults, adolescents, and children were approved by the Institutional Review Boards of the Medical College of Wisconsin (HRRC#325-94 and HRRC#013-00) and Children’s Hospital of Wisconsin (CHW 04/87), respectively.
Illumina Methylation 450 k data production
Genomic DNA was isolated from peripheral blood after an overnight fast on the same day when each subject was assayed for obesity and MetS phenotypes; thus, the CpG methylation states profiled from these samples reflect the epigenetic status associated with that individual’s current state of body composition and metabolism. One microgram of human genomic DNA was sodium bisulfite-treated for cytosine (C) to thymine (T) conversion using the EZ DNA Methylation kit (Zymo Research) according to the manufacturer’s guidelines. The converted DNA was purified and prepped for analysis on the Illumina HumanMethylation450 microarray following the manufacturer’s guidelines. The Illumina HumanMethylation450k microarray measures the methylation levels of more than 485,000 methylation sites. It includes CpG sites surrounding the transcription start sites (−200 to −1,500 bp, 5′UTRs and exon 1) for 99% of RefSeq genes, CpG sites within non-coding RNAs, intergenic regions identified in genome-wide association studies as well as CpG islands/shores/shelves and open sea of the genome. CpG annotations (chromosomal location, reference gene, etc.) were identified using the Illumina manifest 1v2.GenomeStudio software and Methylation Module (Illumina) was used to generate final reports containing signal intensities and detection P values excluding X and Y chromosomes. No background subtraction or control normalization was applied with GenomeStudio.
Genomic CpG methylation data QC and processing
For initial quality control preparation of the Infinium Human Methylation 450 K data, we used the Lumi: QN + BMIQ pipeline described previously [118]. Raw signal intensities and detection P values of 22 autosomal chromosomes were extracted from GenomeStudio and loaded into Lumi. Next, quality control of the data resulted in the removal of CpG sites with detection P value ≥ 0.01 in more than 5% of the samples (471,473 sites left). All samples had at least 99% CpG sites with detection P value ≥ 0.01; thus no samples were removed. Recently, multiple groups have reported that this array contains cross-reacting probes that cannot be distinguished between multiple chromosomal positions and that therefore need to be excluded from downstream analysis [119]. Furthermore, studies including ours (Y.Z., unpublished data) have shown that a significant proportion of genomic CpG loci are common polymorphic locations where both C or G or the dinucleotides are changed to a different code, thus abolishing the ability of being methylated in that genome [119]. Considering the inaccuracy these single nucleotide polymorphisms (SNPs) may cause in the quantification of methylation status of these CpG sites, we therefore excluded all known polymorphic CpG sites.
Color bias adjustment (Col.Adj) and quantilenormilzation (QN) were performed on signal intensities as implemented in Lumi. Briefly, the QN works on total signal intensity, assuming that the distributions of the pooled methylated and unmethylated probes are similar for different samples. Intensities were then used to generate Beta values. Within Lumi, ‘β’ values are defined as follows:
where I M and I U represent the fluorescence intensity originating from methylated or unmethylated CpG locus and α is a constant. Beta mixture quantile dilation (BMIQ) was then performed on β values of QNed data to account for probe type bias. As the Illumina platforms have been shown to discriminate beta values that differ as little as 17% [120,121], we excluded from analysis probes that ranged <0.17 in β values (n = 243,711) to ensure probes analyzed might exert a significant biological change. After these steps, a total of 137,168 CpG sites for all 192 samples were imported into data analysis. BMIQ’ed β values for probes with ≥0.17 variation were then converted to M values for data analysis. Lumi defines M values as:
All analyses were run using M values, which are more statistically valid for analysis of differential methylation levels owing to its more homoscedastic nature [122].
Peripheral blood cell subtype estimation
To estimate cell-type proportions, we used the R minfi package and estimateCellCounts function [123,124]. This method estimated the proportions of six cell types (monocytes, granulocytes, CD8+ T-cells, CD4+ T-cells, NK cells, and B cells) for each individual based on their genome-wide methylation signatures, using an external reference inferred from sample profiles of cell-specific methylation [125]. For each regression test, five of the six proportions were used as covariates.
Validation by pyrosequencing
DNA methylation at selected sites was validated in a subset of the original cohort by the bisulfite pyrosequencing. This subset consisted of 47 male subjects ages 6 to 21. One microgram of human genomic DNA was sodium bisulfite converted using the EZ DNA Methylation kit (Zymo Research) according to the manufacturer’s guidelines. Pyrosequencing was performed using the PyroMark MD system (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Briefly, the PCR was performed with 10 μM primers, one of which was biotinylated for later purification by Streptavidin Sepharose (VWR). The oligonucleotide primers were purchased from IDT and used for the amplified region of DDO: the forward primer, TGTTTAGGAGAAAGGAGTAAGTGATT; the reverse biotinylated primer, ACCCATTATTCACCATACCTACAA; and the pyrosequencing primer, TTTTATGGAGTTGTTTTTGTTAAG. Sepharose beads containing the PCR product were washed and purified using 0.2 M NaOH and the Pyrosequencing Vacuum Prep Tool (QIAGEN). Five microliters of the PCR products was sequenced, and methylation was quantified using the provided software (QIAGEN).
Statistical analysis
Analysis of age-associated CpG loci
The quantitative genetic analyses program SOLAR [52] was used to analyze DNA methylation differences associated with age in the whole cohort. SOLAR is a software package designed to perform tests of genetic and epigenetic association in family data. Parameter estimation by maximum likelihood is performed for both random and fixed effects; in the present context, the random effect of expected allele sharing given pedigree relationships is estimated to account properly for the non-independence of related individuals. For each individual i, the value of a trait Y is modeled as:
where μ is the trait mean, X i is a vector of fixed effects measured on individual i, β is a corresponding vector of regression coefficients, and g i and ε i are, respectively, a random additive genetic effect and an error term. The covariance of the trait in any two individuals i, j is decomposed as:
where 2ϕ i,j is a kinship coefficient (representing the expected proportion of alleles shared identical by descent for two individuals of a given relationship class) and σ 2 g and σ 2 ε are, respectively, additive genetic and residual components of variance. Inclusion of the random effect terms appropriately conditions the estimates of the fixed effect parameters on the relatedness of study subjects. Analyses were performed for each CpG site separately, using M values, where M was modeled as a linear function of age with models that included the random effect of kinship. Sex and cell type composition were included as covariates in all models to account for systematic differences in methylation between men and women. Bonferroni correction for multiple testing, P α=0.05 = 3.65 × 10−7.
Analysis of MetS status on aging rate in candidate loci
A candidate CpG site-based regression analysis against MetS status in each subject was performed to determine if there is differential aging of DNA methylation in subjects with metabolic syndrome compared to those without. In this model, two tests were done: in one, the slopes and intercepts of the regression lines are allowed to differ by MetS status, and in another, a null is forced to be the same. The test of statistically equal intercepts asks whether methylation differs by MetS status for the measured span of ages. The test of equal slopes asks whether MetS impacts change in methylation with age.
Identification of genomic clusters of age-associated CpGs
The R package bump hunter [67] was used to identify genomic clusters of age-associated CpG sites. The clusterMaker function within the bump hunter package was applied to the genome-wide age-associated CpGs, and clusters are formed if two positions are within 10 kb of each other. Each chromosome is clustered independently from each other.
To account for array bias, we took the minimum and maximum position of each cluster and looked at the total number of probes from that region that were originally implemented into the analysis. To further define our aDMCs, at least ten CpG sites had to be in the original data set which was implemented into the analysis. At least 50% of those sites had to be genome-wide significantly associated with age. Through this method, we identified a total of 246 aDMCs throughout the autosomal genome. We further looked at the direction of effect age has on each CpG within the identified clusters. If 100% of the CpGs in the cluster had the same direction of effect, it was labeled as ‘positive’ or ‘negative’. If there was variable direction of effects within the cluster, it was labeled as ‘varying’.
Gene ontologies and pathway analysis of aDMCs
Gene Ontology analysis was done with the FatiGO tool [69], which uses Fisher’s exact test to detect significant overrepresentation of GO terms and disease pathways. FatiGO pools multiple databases, such as the Gene Ontology (GO) terms [72] and Kyoto Encyclopedia of Genes and Genomes (KEGG) [70,71]. In our study, the set of one direction cluster genes were analyzed for overrepresentation against the rest of the genome. Multiple test correction to account for the multiple hypothesis tested (one for each term) is applied to reduce false positives. GO terms and KEGG pathways with adjusted P value < 0.05 are considered significant.
References
Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–128.
Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103(2):137–49.
Ward BW, Schiller JS. Prevalence of multiple chronic conditions among US adults: estimates from the National Health Interview Survey, 2010. Prev Chronic Dis. 2013;10:120203. DOI: http://dx.doi.org/10.5888/pcd10.120203.
Hanson M, Gluckman P. Developmental origins of noncommunicable disease: population and public health implications. Am J Clin Nutr. 2011;94(6 Suppl):1754S–8. doi: 10.3945/ajcn.110.001206.
Santos MS, Joles JA. Early determinants of cardiovascular disease. Best Pract Res Clin Endocrinol Metab. 2012;26(5):581–97.
Kelishadi R, Poursafa P. A review on the genetic, environmental, and lifestyle aspects of the early-life origins of cardiovascular disease. Curr Probl Pediatr Adolesc Health Care. 2014;44(3):54–72.
Heerwagen MJ, Miller MR, Barbour LA, Friedman JE. Maternal obesity and fetal metabolic programming: a fertile epigenetic soil. Am J Physiol Regul Integr Comp Physiol. 2010;299(3):R711–22.
Suter MA, Ma J, Vuguin PM, Hartil K, Fiallo A, Harris RA, et al. In utero exposure to a maternal high-fat diet alters the epigenetic histone code in a murine model. Am J Obstet Gynecol. 2014;210(5):463. e1-463.e11.doi: 10.1016/j.ajog.2014.01.045.
Delcuve GP, Rastegar M, Davie JR. Epigenetic control. J Cell Physiol. 2009;219(2):243–50.
Wang G, Walker SO, Hong X, Bartell TR, Wang X. Epigenetics and early life origins of chronic noncommunicable diseases. J Adolesc Health. 2013;52(2 Suppl 2):S14–21.
Tammen SA, Friso S, Choi SW. Epigenetics: the link between nature and nurture. Mol Aspects Med. 2013;34(4):753–64.
Barros SP, Offenbacher S. Epigenetics: connecting environment and genotype to phenotype and disease. J Dent Res. 2009;88(5):400–8.
van Otterdijk SD, Mathers JC, Strathdee G. Do age-related changes in DNA methylation play a role in the development of age-related diseases? Biochem Soc Trans. 2013;41(3):803–7.
Hauser MT, Aufsatz W, Jonak C, Luschnig C. Transgenerational epigenetic inheritance in plants. Biochim Biophys Acta. 2011;1809(8):459–68.
Morgan HD, Sutherland HE, Martin DIK, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Gen. 1999;23:314–8.
Cropley JE, Suter CM, Beckman KB, Martin DIK. Germ-line epigenetic modification of the murine Avy allele by nutritional supplementation. Proc Natl Acad Sci U S A. 2006;103(46):17308–12.
Waterland RA, Travisano M, Tahiliani KG. Diet-induced hypermethylation at agouti viable yellow is not inherited transgenerationally through the female. FASEB J. 2007;21(12):3380–5.
Waterland RA, Travisano M, Tahiliani KG, Rached MT, Mirza S. Methyl donor supplementation prevents transgenerational amplification of obesity. Int J Obesity. 2008;32:1373–9.
Csaba G, Karabélyos C, Inczefi-Gonda A, Pállinger E. Three-generation investigation on serotonin content in rat immune cells long after β-endorphin exposure in late pregnancy. Horm Metab Res. 2005;37(3):172–7.
Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014;157(1):95–109.
Hocher B. More than genes: the advanced fetal programming hypothesis. J Reprod Immunol. 2014. doi: 10.1016/j.jri.2014.03.001.
Soubry A, Hoyo C, Jirtle RL, Murphy SK. A paternal environmental legacy: evidence for epigenetic inheritance through the male germ line. Bioessays. 2014;36(4):359–71. doi: 10.1002/bies.201300113. Epub 2014 Jan 16.
McRae AF, Powell JE, Henders AK, Bowdler L, Hemani G, Shah S, et al. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 2014;15(5):R73. doi: 10.1186/gb-2014-15-5-r73.
Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature. 2010;467:963–6.
Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20. doi: 10.1038/nrg3354.
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–9.
Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–53.
Reynolds RM, Jacobsen GH, Drake AJ. What is the evidence in humans that DNA methylation changes link events in utero and later life disease? Clin Endocrinol (Oxf). 2013;78(6):814–22.
Szyf M, Bick J. DNA methylation: a mechanism for embedding early life experiences in the genome. Child Dev. 2013;84(1):49–57.
Gomes MV, Pelosi GG. Epigenetic vulnerability and the environmental influence on health. Exp Biol Med. 2013;238(8):859–65.
Drake AJ, McPherson RC, Godfrey KM, Cooper C, Lillycrop KA, Hanson MA, et al. An unbalanced maternal diet in pregnancy associates with offspring epigenetic changes in genes controlling glucocorticoid action and fetal growth. Clin Endocrinol. 2012;77(6):808–15.
Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011;60(5):1528–34.
Ziller MJ, Gu H, Müller F, Donaghey J, Tsai LT, Kohlbacher O, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500(7463):477–81.
Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8(4):e1002629. doi: 10.1371/journal.pgen.1002629.
Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, et al. Age-associated DNA methylation in pediatric populations. Genome Res. 2012;22:623–32.
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–9.
Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11(4):694–703.
Wong CC, Caspi A, Williams B, Craig IW, Houts R, Ambler A, et al. A longitudinal study of epigenetic variation in twins. Epigenetics. 2010;5(6):516–26.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–67.
Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, et al. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013;14:R102. http://genomebiology.com//2013/14/9/R102.
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20(4):440–6.
Gautrey HE, van Otterdijk SD, Cordell HJ; Newcastle 85+ Study Core Team, Mathers JC, Strathdee G. DNA methylation abnormalities at gene promoters are extensive and variable in the elderly and phenocopy cancer cells. FASEB J. 2014. [Epub ahead of print].
Barres R, Zierath JR. DNA methylation in metabolic disorders. Am J Clin Nutr. 2011;93(4):897S–900.
de Mello VD, Pulkkinen L, Lalli M, Kolehmainen M, Pihlajamäki J, Uusitupa M. DNA methylation in obesity and type 2 diabetes. Ann Med. 2014;46(3):103–13.
vanDijk SJ, Molloy PL, Varinli H, Morrison JL, Muhlhausler BS, members of EpiSCOPE. Epigenetics and human obesity. Int J Obes. 2014. doi:10.1038/ijo.2014.34. [Epub ahead of print].
Bell JT, Spector TD. A twin approach to unraveling epigenetics. Trends Genet. 2011;27(3):116–25.
Kissebah AH, Sonnenberg GE, Myklebust J, Goldstein M, Broman K, James RG, et al. Quantitative trait loci on chromosomes 3 and 17 influence phenotypes of the metabolic syndrome. Proc Natl Acad Sci U S A. 2000;97:14478–83.
Esteghamati A, Ashraf H, Khalilzadeh O, Zandieh A, Nakhjavani M, Rashidi A, et al. Optimal cut-off of homeostasis model assessment of insulin resistance (HOMA-IR) for the diagnosis of metabolic syndrome: third national surveillance of risk factors of non-communicable diseases in Iran (SuRFNCD-2007). Nutr Metab. 2010;7:26. doi: 10.1186/1743-7075-7-26.
Keskin M, Kurtoglu S, Kendirci M, Atabek ME, Yazici C. Homeostasis model assessment is more reliable than the fasting glucose/insulin ratio and quantitative insulin sensitivity check index for assessing insulin resistance among obese children and adolescents. Pediatrics. 2005;115(4):e500–3.
Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735–52.
Almasy L, Blangero JT. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62(5):1198–211.
Blangero J, Williams JT, Almasy L. Variance component methods for detecting complex trait loci. Adv Genet. 2001;42:151–81.
Xia Q, Grant SFA. The genetics of obesity. Ann N Y Acad Sci. 2013;1281:178–90.
Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391(6670):900–4.
Kraja AT, Chasman DI, North KE, Reiner AP, Yanek LR, Kilpeläinen TO, et al. Pleiotropic genes for metabolic syndrome and inflammation. Mol Genet Metab. 2014. http://dx.doi.org/10.1016/j.ymgme.2014.04.007 [epub ahead of print].
Garagnani P, Bacalini MG, Pirazzini C, Gori D, Giuliani C, Mari D, et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell. 2012;11(6):1132–4.
Florath I, Butterbach K, Müller H, Bewerunge-Hudler M, Brenner H. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet. 2014;23(5):1186–201.
McCarthy MI, Zeggini E. Genome-wide association studies in type 2 diabetes. Curr Diab Rep. 2009;9(2):164–71.
DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium; Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; Mexican American Type 2 Diabetes (MAT2D) Consortium; Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium, Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Gen. 2014;46:234–44.
Rung J, Cauchi S, Albrechtsen A, Shen L, Rocheleau G, Cavalcanti-Proença C, et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet. 2009;41(10):1110–5.
Kim YB, Nikoulina SE, Ciaraldi TP, Henry RR, Kahn BB. Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J Clin Invest. 1999;104(6):733–41.
Florez JC, Jablonski KA, Kahn SE, Franks PW, Dabelea D, Hamman RF, et al. Type 2 diabetes-associated missense polymorphisms KCNJ11 E23K and ABCC8 A1369S influence progression to diabetes and response to interventions in the Diabetes Prevention Program. Diabetes. 2007;56(2):531–6.
Holmkvist J, Almgren P, Lyssenko V, Lindgren CM, Eriksson KF, Isomaa B, et al. Common variants in maturity-onset diabetes of the young genes and future risk of type 2 diabetes. Diabetes. 2008;57(6):1738–44.
Hani EH, Stoffers DA, Chèvre JC, Durand E, Stanojevic V, Dina C, et al. Defective mutations in the insulin promoter factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J Clin Invest. 1999;104(9):R41–8.
Gonsorcíková L, Průhová S, Cinek O, Ek J, Pelikánová T, Jørgensen T, et al. Autosomal inheritance of diabetes in two families characterized by obesity and a novel H241Q mutation in NEUROD1. Pediatr Diabetes. 2008;9(4 Pt 2):367–72.
Vionnet N, Stoffel M, Takeda J, Yasuda K, Bell GI, Zouali H, et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356(6371):721–2.
Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012;41(1):200–9.
Martino D, Loke YJ, Gordon L, Ollikainen M, Cruickshank MN, Saffery R, et al. Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013;14(5):R42.
Al-Shahrour F, Diaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics. 2004;4:578–80.
Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30.
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2005;25(1):25–9.
Almén MS, Nilsson EK, Jacobsson JA, Kalnina I, Klovins J, Fredriksson R, Schiöth HB. Genome-wide analysis reveals DNA methylation markers that varies with both age and obesity. Gene. 2014. doi: 10.1016/j.gene.2014.07.009.
Gentilini D, Mari D, Castaldi D, Remondini D, Ogliari G, Ostan R, et al. Role of epigenetics in human aging and longevity: genome-wide DNA methylation profile in centenarians and centenarians’ offspring. Age. 2013;35(5):1961–73.
Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13(10):R97.
Horvath S. DNA methylation age of human tissues and cell types. Genome Boil. 2013;14(10):R115.
Bocklandt S, Lin W, Sehl ME, Sanchez FJ, Sinsheimer JS, Horvath S, et al. Epigenetic predictor of age. PLoS One. 2011;6(6):e14821.
Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol. 2014;220(2):T1–23.
Schmitz-Peiffer C, Whitehead JP. IRS-1 regulation in health and disease. IUBMB Life. 2003;55(7):367–74.
Fritsche L, Weigert C, Häring HU, Lehmann R. How insulin receptor substrate proteins regulate the metabolic capacity of the liver—implications for health and disease. Curr Med Chem. 2008;15(13):1316–29.
Valverde AM, González-Rodríguez A. IRS2 and PTP1B: two opposite modulators of hepatic insulin signalling. Arch Physiol Biochem. 2011;117(3):105–15.
Crews ST, Thomas JB, Goodman CS. The Drosophila single-minded gene encodes a nuclear protein with sequence similarity to the per gene product. Cell. 1988;52:143–51.
Michaud JL, Boucher F, Melnyk A, Gauthier F, Goshu E, Lévy E, et al. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet. 2001;10(14):1465–73.
Holder Jr JL, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101–8.
Philippe J, Stijnen P, Meyre D, De Graeve 1, Thuillier D, Delplanque J, Gyapay G, Sand O, Creemers JW, Froguel P, Bonnefond A. A nonsense loss-of-function mutation in PCSK1 contributes to dominantly inherited human obesity. Int J Obes. 2014. doi:10.1038/ijo.2014.96. [Epub ahead of print].
Dubern B, Clement K. Leptin and leptin receptor-related monogenic obesity. Biochimie. 2012;94(10):2111–5.
Deeb SS, Fajas L, Nemoto M, Pihlajamäki J, Mykkänen L, Kuusisto J, et al. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet. 1988;20(3):284–7.
Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;2000(26):76–80.
Zhao J, Bradfield JP, Zhang H, Sleiman PM, Kim CE, Glessner JT, et al. Role of BMI-associated loci identified in GWAS meta-analyses in the context of common childhood obesity in European Americans. Obesity. 2011;19(12):2436–9.
Scherag A, Dina C, Hinney A, Vatin V, Scherag S, Vogel CI, et al. Two new loci for body-weight regulation identified in a joint analysis of genome-wide association studies for early-onset extreme obesity in French and German study groups. PLoS Genet. 2010;6(4):e1000916.
Bradfield JP, Taal HR, Timpson NJ, Scherag A, Lecoeur C, Warrington NM, et al. A genome-wide association meta analysis identifies new childhood obesity loci. Nat Genet. 2012;44(5):526–31.
Suh JM, Gao X, McKay J, McKay R, Salo Z, Graff JM. Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab. 2006;3(1):25–34.
Teperino R, Amann S, Bayer M, McGee SL, Loipetzberger A, Connor T, et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell. 2012;151:414–26.
Buhman KK, Wang LC, Tang Y, Swietlicki EA, Kennedy S, Xie Y, et al. Inhibition of hedgehog signaling protects adult mice from diet-induced weight gain. J Nutr. 2004;134:2979–84.
Nybakken K, Perrimon N. Hedgehog signal transduction: recent findings. Curr Opin Genet Dev. 2002;12:503–11.
Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–87.
Cohen Jr MM. The hedgehog signaling network. Am J Med Genet. 2003;123A:5–28.
Gupta D, Krueger CB, Lastra G. Over-nutrition, obesity and insulin resistance in the development of β-cell dysfunction. Curr Diabetes Rev. 2012;8(2):76–83.
Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet. 2014;383(9922):1068–83.
Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2 · 7 million participants. Lancet. 2011;378(9785):31–40.
Gat-Yablonski G, Shalitin S, Phillip M. Maturity onset diabetes of the young—review. Pediatr Endocrinol Rev. 2006;3:514–20. Erratum in: PediatrEndocrinol Rev. 2007, 5(1):470.
Stocks T, Ängquist L, Hager J, Charon C, Holst C, Martinez JA, et al. TFAP2B-dietary protein and glycemic index interactions and weight maintenance after weight loss in the DiOGenes trial. Hum Hered. 2013;75(2–4):213–9.
Stocks T, Angquist L, Banasik K, Harder MN, Taylor MA, Hager J, et al. TFAP2B influences the effect of dietary fat on weight loss under energy restriction. PLoS One. 2012;7(8):e43212.
Depetris RS, Hu J, Gimpelevich I, Holt LJ, Daly RJ, Hubbard SR. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Mol Cell. 2005;20(2):325–33.
Desbuquois B, Carré N, Burnol AF. Regulation of insulin and type 1 insulin-like growth factor signaling and action by the Grb10/14 and SH2B1/B2 adaptor proteins. FEBS J. 2013;280(3):794–816.
Goenaga D, Hampe C, Carré N, Cailliau K, Browaeys-Poly E, Perdereau D, et al. Molecular determinants of Grb14-mediated inhibition of insulin signaling. Mol Endocrinol. 2009;23(7):1043–51.
Balogh K, Asa SL, Zheng L, Cassol C, Cheng S, Ezzat S. The insulin resistance Grb14 adaptor protein promotes thyroid cancer ret signaling and progression. Oncogene. 2012;31(36):4012–21.
Sung HY, Guan H, Czibula A, King AR, Eder K, Heath E, et al. Human tribbles-1 controls proliferation and chemotaxis of smooth muscle cells via MAPK signaling pathways. J Biol Chem. 2007;282(25):18379–87.
Ollila HM, Utge S, Kronholm E, Aho V, Van Leeuwen W, Silander K, et al. TRIB1 constitutes a molecular link between regulation of sleep and lipid metabolism in humans. Transl Psychiatry. 2012;2(3):e97.
Aung LH, Yin RX, Wu DF, Li Q, Yan TT, Wang YM, et al. Association of the TRIB1 tribbles homolog 1 gene rs17321515 A > G polymorphism and serum lipid levels in the Mulao and Han populations. Lipids Health Dis. 2011;10:230. doi: 10.1186/1476-511X-10-230.
Garcia-Rios A, Perez-Martinez P, Mata P, Fuentes F, Lopez-Miranda J, Alonso R, et al. Polymorphism at the TRIB1 gene modulates plasma lipid levels: insight from the Spanish familial hypercholesterolemia cohort study. Nutr Metab Cardiovasc Dis. 2011;21(12):957–63.
Sun D, Leung CL, Liem RK. Characterization of the microtubule binding domain of microtubule actin crosslinking factor (MACF): identification of a novel group of microtubule associated proteins. J Cell Sci. 2001;114(Pt 1):161–72.
Xu Z, Taylor JA. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis. 2014;35(2):356–64.
Bock C. Analysing and interpreting DNA methylation data. Nat Gen. 2012;13:705–19.
Ali O, Cerjak D, Kent Jr JW, James R, Blangero J, Zhang Y. Obesity, central adiposity and cardiometabolic risk factors in children and adolescents: a family-based study. Pediatr Obes. 2014;9(3):e58–62.
Peiris AN, Hennes MI, Evans DJ, Wilson CR, Lee MB, Kissebah AH. Relationship of anthropometric measurements of body fat distribution to metabolic profile in premenopausal women. Acta Med Scand Suppl. 1988;723:179–88.
Dupont NC, Wang K, Wadhwa PD, Culhane JF, Nelson EL. Validation and comparison of luminex multiplex cytokine analysis kits with ELISA: determinations of a panel of nine cytokines in clinical sample culture supernatants. J Reprod Immunol. 2005;66:175–91.
Marabita F, Almgren M, Lindholm ME, Ruhrmann S, Fagerström-Billai F, Jagodic M, et al. An evaluation of analysis pipelines for DNA methylation profiling using the Illumina HumanMethylation450 BeadChip platform. Epigenetics. 2013;8(3):333–46.
Chen Y, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–9.
Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, et al. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS One. 2009;4(8):e6767.
Carless MA, Kulkarni H, Kos MZ, Charlesworth J, Peralta JM, Göring HHH, et al. Genetic effects on DNA methylation and its potential relevance for obesity in Mexican Americans. PLoS One. 2013;8(9):e73950.
Zhuang J, Widschwendter M, Teschendorff AE. A comparison of feature selection and classification methods in DNA methylation studies using the Illumina Infinium platform. BMC Bioinformatics. 2012;13:59. doi:10.1186/1471-2105-13-59.
Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361.
Acknowledgements
This work is supported by grants from the NIH (DK071895-03 and DK65598-01) for A.H.K. and in part with federal funds from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN275201100014C (OA and YZ). Quantitative genetic methods development and analysis was supported in part by NIH-National Mental Health Institute grant MH059490 (JB). This work has been also supported by grants by TOPS Club, Inc. to A.H.Kissebah and Y. Zhang. The late Dr. Ahmed H. Kissebah was the principal investigator on the various NIH funded studies that led to the collection of this unique family-based cohort.
We also acknowledge the contribution of Jacqueline Marks (Medical College of Wisconsin) for her supervision of all biochemical procedures and Ruth Gielow (TOPS Club, Inc.) and her assistants in the recruitment process and finally to all the members of TOPS Club, Inc. and their families who volunteered for this study. We appreciate the technical support provided by Glenn M. Bushee at the Clinical and Translational Science Institute of Southeast Wisconsin. The authors thank Dr. Howard Jacob for reviewing the early drafts of this manuscript.
Accession numbers
The NCBI Gene Expression Omnibus accession number for the methylation data generated from the 192 samples reported in this paper is under the Gene Series GSE60132.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
OA assisted in the phenotyping procedures, contributed to the discussion, and wrote and revised the manuscript. DC performed statistical and bioformatic analyses. JWK contributed to the data analysis and discussion and revised the manuscript. RJ contributed to subject recruitment and phenotyping. JB contributed to the study design. MAC contributed to the study design, provided technical support, and contributed to discussion and revision of the manuscript. YZ conceived the study, led data collection and analyses, contributed to the discussion, and wrote and revised the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Data cleaning pipeline for probed Illumina 450 k methylation signals.
Additional file 2:
Summary of age-associated DNA methylation signals identified in the current and previous studies.
Additional file 3:
Comparison of age-associated CpG sites identified in this study with sites identified in previous studies.
Additional file 4:
Previously known obesity genes with CpG sites found to be age associated in TFSE.
Additional file 5:
Previously known type 2 diabetes genes with CpG sites found to be age associated in TFSE.
Additional file 6:
Previously known MetS genes with CpG sites found to be age associated in TFSE.
Additional file 7:
Autosomal map of age-associated differentially methylated CpG clusters (aDMCs).
Additional file 8:
KEGG pathways significantly enriched in positive aDMCs identified by the FatiGO analysis.
Additional file 9:
GO Biological Processes significantly enriched in positive aDMCs.
Additional file 10:
GO Molecular Functions significantly enriched in positive aDMCs.
Additional file 11:
KEGG pathways significantly enriched in negative aDMCs identified by the FatiGO analysis.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Ali, O., Cerjak, D., Kent, J.W. et al. An epigenetic map of age-associated autosomal loci in northern European families at high risk for the metabolic syndrome. Clin Epigenet 7, 12 (2015). https://doi.org/10.1186/s13148-015-0048-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-015-0048-6