
Abstract
Epigenetics is currently one of the hottest topics in basic and biomedical research. However, to date, most of the studies have been descriptive in nature, designed to investigate static distribution of various epigenetic modifications in cells. Even though tremendous amount of information has been collected, we are still far from the complete understanding of epigenetic processes, their dynamics or even their direct effects on local chromatin and we still do not comprehend whether these epigenetic states are the cause or the consequence of the transcriptional profile of the cell. In this review, we try to define the concept of synthetic epigenetics and outline the available genome targeting technologies, which are used for locus-specific editing of epigenetic signals. We report early success stories and the lessons we have learned from them, and provide a guide for their application. Finally, we discuss existing limitations of the available technologies and indicate possible areas for further development.
Similar content being viewed by others


Review
Epigenetic landscape and cell fate
The human body is built from more than 200 different cell types organized in various tissues. It is fascinating that although all these cells stem from a single cell (the zygote) and contain exactly the same DNA sequence (except for the antibody-forming B cells of the immune system [1]), they express different sets of genes and have diametrically different functions, phenotypes and behaviour in the body. To explain how these phenotypic differences arise during embryonic development, in 1957 in his book “The Strategy of the Genes”, Waddington defined the famous concept of the epigenetic landscape, in which cells can be imagined as marbles rolling down towards a hill’s bottom. The marbles (cells) compete for the grooves on the slope, which define their developmental trajectories, and come to rest at the base of the hill in defined positions. These defined positions demarcate eventual cell fates, meaning the tissue types which the cells adapt [2].
The human genome comprises approximately three billion base pairs, which represent a large repository of information. The fact that different cells contain basically the same DNA but show very distinct phenotypes indicates that regulated access to this information is key to understanding cell identity and, therefore, human development and health. The last 60 years of research in the field of epigenetics was focused mostly on elucidating the molecular mechanisms and the enzymatic machinery responsible for epigenetic control of gene expression, as well as the distribution of various epigenetic marks in different cell types from healthy and diseased tissues and organisms. Progress in the next-generation sequencing and proteomic approaches allowed systematic analysis and identification of novel epigenetic marks and their distribution across the genome and nucleus. Few large-scale collaborative projects, like ENCODE (http://www.genome.gov/encode/) [3,4], Roadmap Epigenomics Mapping consortium (http://www.roadmapepigenomics.org/) [5] and Blueprint (http://www.blueprint-epigenome.eu/) [6] provided a validated repository of epigenetic states of various tissues.
Despite the tremendous progress in understanding the epigenetic signalling pathways, as well as characterization of the epigenetic marks (DNA and histone modifications) and the enzymatic machinery that can write, read and remove these marks, many fundamental questions still remain unresolved, mainly due to technological limitations. For example, are various epigenetic signals the cause or the consequence of the cell transcriptional profile? What is the sequential order of epigenetic transitions between the repressed and activated states? Are epimutations drivers or merely by-products of a diseased state, and finally, what is the contribution of epigenetics to disease (cancer) development [7,8]?
In this review, we try to define the area of synthetic epigenetics and outline the available genome targeting technologies, which are used for targeted editing of epigenetic signals. We report early success stories and the lessons we have learned from them, as well as potential biomedical applications and existing limitations of the available technologies.
Synthetic epigenetics
We define synthetic epigenetics as the design and construction of novel specific artificial epigenetic pathways or the redesign of existing natural biological systems, in order to intentionally change epigenetic information of the cell at desired loci. In this broad definition view, somatic cell nuclear transfer experiments (SCNT) [9,10], direct cell fate conversion (also known as transdifferentiation) [11,12], generation of induced pluripotent stem cells (iPS cells) [13,14] and targeted epigenome editing by programmable DNA binding domains fused to epigenetic modifiers (epigenetic editing) [15] all constitute synthetic epigenetic phenomena. Due to their random nature of introduced epigenome modification (in terms of locus or sequence specificity), we do not consider epigenetic drugs (like azacytidine or trichostatin A [16,17]) as synthetic epigenetics tools.
Importantly, there is a qualitative difference between the nuclear transfer and induced pluripotency experiments when compared to epigenetic editing. In the experiments with somatic cell nuclear transfer and during generation of induced pluripotent stem cells, genome-wide changes in the epigenetic state of the cells are triggered by a mixture of defined oocyte-specific factors or forced expression of a selection of transcription factors, respectively. This is achieved through the restoration of robust pluripotency promoting transcription factor networks with self-enforcing feedback mechanisms, which rewire the transcriptional profile of the cell [18,19], and the epigenome is progressively adjusted during this process [20-22]. Interestingly, overexpression of some epigenetic modifiers, like ten-eleven translocation (TET) enzymes, can catalyse and enhance the dedifferentiation process [23,24]. Therefore, in these experiments, the observed epigenetic changes are rather a necessary product than the initial trigger of the cell conversion [25], which allow efficient reaching and maintenance of the de-differentiated state. This strategy can be viewed as an indirect (or top-down) approach to change the epigenome by rewiring the transcriptional profile of the cell. Somatic cell nuclear transfer and generation of induced pluripotent stem cells have been extensively covered in numerous excellent reviews [26-30].
On the other hand, in a bottom-up approach, a local direct change to the epigenome can be introduced by targeting an effector domain to the desired locus. In this epigenetic editing [15] approach, programmable DNA binding domains (DBDs) target a selected epigenetic modifier to desired loci and ensure deposition of the corresponding epigenetic mark at nearby chromatin. Consequently, local changes to the epigenome can be observed, and transcriptional and epigenetic response to these changes can be studied in a defined context.
Existing technologies used to modulate transcription of desired genes, like RNAi [31], gene knock-out experiments or expression of recombinant proteins (for example, overexpression of cDNA constructs), as well as programmable activators and silencers [32-36] require constitutive expression of the constructs to maintain the effect [34] or introduce irreversible changes to the genome. In contrast, epigenetic editing offers the possibility that the epigenetic signal and the corresponding change in the gene’s expression status are heritably maintained by cellular machinery over multiple cell divisions even after the initial epigenetic editing construct is cleared from the cells [34]. Therefore, a transient introduction of the construct can lead to a persistent modulation of gene expression without any genomic damage being introduced, hence making epigenetic editing safer and more suitable for therapeutic use. In this article, we will focus on the currently blooming and exciting field of targeted epigenome modification.
The epigenetic editing tools are perfectly suited to treat and study the molecular mechanisms underlying epigenetic diseases like cancer, chronic diseases or imprinting defects. For example, DNA methylation has been already used to silence overexpressed oncogenes [37-39] and could be further used to repress hypoxia-inducible factors [40], possibly leading to cancer regression. On the other hand, silenced tumour suppressor genes [41] could be reactivated using targeted DNA demethylation. Likewise, imprinting defects (for example, Beckwith-Wiedemann syndrome) could be reverted or their effects could be weakened by specific alteration of the epigenetic state of the affected imprinting control regions [42-44]. Chronic diseases are very often correlated with abnormal epigenetic changes [45-48]. With epigenetic editing technology, one could attempt to reprogram these disease-promoting epigenetic states and therefore restore normal functioning of the cell. An interesting new approach would also be to specifically change the differentiation state of pluripotent or differentiated cells, by rewiring their epigenetic profile towards another cell type.
Bottom-up synthetic epigenetics or epigenetic editing
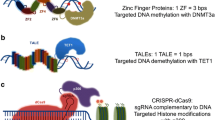
The concept of bottom-up synthetic epigenetics (epigenetic editing) relies on the combination of an artificial DNA binding domain, which can directly bind a unique sequence found within the desired locus, with an effector domain able to edit the epigenetic state of that locus (Figure 1). To date, various genome targeting domains and epigenetic modifiers have been used to direct activating or repressing marks to desired loci (reviewed in [15,49-51]). Discovery of novel programmable genome binders, like TALE and CRISPR/Cas9 systems, as well as progress in understanding of epigenetic enzymes has spurred new interest and brought excitement to the epigenetic editing field [52,53]. In 2014, Nature Methods has distinguished epigenome editing as a “Method to Watch” [54]. In the next paragraphs, we review the available technologies for epigenome targeting and lessons learned from the published success stories of epigenetic editing, and provide a guide for perspective researchers aiming to develop novel methods or apply existing ones for their own research.

The concept of epigenetic editing. Targeting device, a sequence-specific DNA binding domain which can be redesigned to recognize desired sequences is fused to an effector domain, which can modify the epigenetic state of the targeted locus, leading to a persistent biological effect (gene activation or repression). Green lollipops represent introduced modification of either DNA bases or histone tails.
Genome targeting proteins
For most of the known DNA interacting proteins, no simple DNA recognition code exists that could connect individual amino acid residues with the corresponding DNA base [55]. For this reason, it was impossible for many years to redesign DNA interacting proteins for novel pre-defined specificities [56,57]. C2H2 zinc fingers were the first example of modular and predictable DNA recognition modules in which one zinc finger unit binds to three base pairs [58]. More recently, two more programmable genome binders were discovered: the TAL effector arrays [59] and CRISPR/Cas9 systems [60], which are discussed below.
Zinc finger arrays
C2H2 zinc fingers were the first example of predictable DNA interaction domains amenable to rational protein design (reviewed in [58,61]), and until very recently, they were the domains of choice for sequence-specific genome targeting. Natural and engineered zinc finger arrays are composed of tandem repeated zinc finger modules. Each unit comprises around 30 amino acid residues that form a compact structure stabilized by zinc ions bound to two invariable cysteine and two histidine residues [62]. Separate zinc finger units were systematically modified and selected for recognition of various trinucleotides. Repositories of possible zinc fingers recognizing particular trinucleotide sequences are readily available [63,64]. Typical custom-made zinc finger arrays comprise between three and six individual zinc finger modules and can consequently bind target sites ranging from 9 to 18 base pairs in length. Arrays with six or longer zinc finger motifs are particularly interesting, as they can recognize target sites that are long enough to potentially address a unique sequence in the context of a mammalian genome.
Currently, two main methods are used to generate engineered zinc finger arrays: context-dependent modular assembly [65] and bacterial selection systems [66]. Context-dependent modular assembly (CoDA) relies on combining smaller zinc finger units of known specificity into larger arrays. Generation of custom zinc finger arrays with the use of a bacterial selection system has the potential to deliver highly efficient DNA binding modules. Because the zinc finger arrays, after their assembly from a large combinatorial library of shorter zinc finger modules, are tested right away in the cellular environment, the chances of obtaining well-functioning combinations of modules for a particular sequence are greatly increased. However, this process can be tedious and time consuming to have the arrays selected and validated [67]. On the other hand, CoDA can provide a large number of zinc finger arrays relatively quickly (within 1–2 weeks) [65], on the expense of the design’s success rate. Online tools for design of zinc finger arrays are available (http://zifit.partners.org/ZiFiT/ [68,69]).
TAL effector arrays
Another class of customizable DNA binding domains, the transcription activating-like effectors (TALEs) are important virulence factors initially isolated from the bacterial plant pathogen Xanthomonas [70]. Members of the TALE family are composed of tandemly arranged and highly similar 34 amino acid repeats. Each repeat recognizes a single base pair, and the recognition specificity of TALEs correlates with the amino acid composition of the repeat variable di-residues (RVD) localized in positions 12 and 13 of each repeat [71]. Due to the modularity of TALEs and the restriction of base specificity to the RVDs, an elegant code governing the DNA recognition specificity was elucidated [59,72]. Because of the simplicity of the recognition code, the readout being restricted to a single base pair per repeat unit and the lack of neighbour effects, the custom TALE arrays can be efficiently generated by modular assembly [73]. However, both C2H2 zinc finger and TALE arrays need to be redesigned for each particular DNA sequence of interest, which is time and resource consuming.
CRISPR/Cas9 system
The newest exciting addition to the genome targeting toolbox repository is the CRISPR/Cas9 system [74]. CRISPR (clustered regularly interspaced short palindromic repeats) functions as a prokaryotic adaptive immune system that confers resistance to exogenous genetic elements such as plasmids and phages [75]. In the natural system, short segments of foreign DNA (spacers) are incorporated into the genome between CRISPR repeats and serve as an adaptive memory of previous exposures [76]. CRISPR spacers are transcribed into non-coding precursor RNAs and further processed into mature CRISPR RNAs (crRNAs), which in turn guide CRISPR-associated (Cas) proteins to recognize and cleave invading genetic elements containing matching sequences [77]. Many CRISPR systems have been found in bacteria and archaea; they were categorized into three distinct types. Type II (referred here as CRISPR/Cas9) is the simplest, as it requires only one protein component for genome targeting, given that the appropriate guide RNA is provided. This system has been harnessed for genome engineering in a broad range of organisms [60,74]. The CRISPR/Cas9 protein requires a Cas9-specific protospacer-adjacent motif (PAM) sequence being present at the 3′-end of the targeted sequence for efficient binding and cleavage. CRISPR/Cas9 proteins recognize their targets based on Watson/Crick base pairing and rely on complementarity of the recognized DNA and the guide RNA sequences. Therefore, re-targeting of the guide RNA-Cas9 nuclease complex to a new locus only requires introduction of a new guide RNA sequence complementary to the new target sequence. In addition, orthologous Cas9 systems (isolated from diverse bacterial species) [78] fused to a selection of different epigenetic modifiers could be simultaneously used in a single experiment to target various epigenetic modifications to selected loci (same or different). These properties make the guide RNA/Cas9 system the most promising genome targeting approach available so far. For targeting epigenetic modification, a catalytically inactive Cas9 variant has to be used, which still can recognize and bind the target sequence, but cannot cleave it [78-80].
Selection of the genome targeting proteins
Each of the available programmable genome targeting devices discussed above offers unique advantages and disadvantages (summarized in Table 1). When choosing the appropriate targeting domain for synthetic epigenetics application, few important properties should be taken into consideration: specificity of the target recognition, sensitivity to the state of DNA modification, ease of design and construct generation, as well as possibility for multiplexing (as discussed below).
Unpredictable off-site targeting can result in the modification of epigenetic information at different loci than anticipated and thereby influence the obtained biological outcome, leading to false conclusions of the studies. Therefore, specificity of target recognition is of pivotal importance because strong binding to an off-target site, resulting from binding of the targeting device, could also lead to a stable, but unwanted epigenome modification around that region. On the other hand, the excess of epigenetic editors present in the cell is not expected to be deleterious. These surplus proteins could either be recruited by natural interaction partners to their native target sites and contribute to regular cellular processes or they could modify random sites on the genome, causing only minimal fluctuations of epigenetic signals that would be efficiently counteracted.
The specificity of zinc finger arrays will likely differ for each particular design [81]. TALE arrays are simpler in their DNA recognition code and assembly; therefore, the possible off-target sequences are more predictable. TALEs were also shown to be very selective in target binding [50,82]. It was demonstrated recently that the CRISPR/Cas9 system suffers from relaxed sequence specificity [83]. This is because not all the positions of the recognized sequence are read equally stringently [84], leading to frequent off-target binding [33,85]. However, successful attempts to improve the specificity have been reported as well [84,86]. Additionally, the requirement for the PAM motif limits the genomic sequences that can be targeted. Nevertheless, the recent solution of the crystal structure of the Cas9 protein bound to guide RNA and substrate DNA showing the mode of interaction between the PAM site and the Cas9 protein will hopefully facilitate directed evolution studies to re-engineer the PAM sequence requirement or even to completely remove the PAM dependency [87,88].
Both zinc fingers and TALEs read the sequence in the major groove of DNA [89,90]. Importantly, the known mammalian DNA modifications, 5-methylcytosine (5mC) and its oxidation products are also presented in the major groove of DNA and therefore can influence the binding of these domains to DNA [91,92]. There are examples of both natural and synthetic zinc finger proteins that recognize 5-methylated cytosine embedded in a specific DNA context [93]. Interestingly, Isalan and Choo have in vitro evolved zinc finger Zif268 protein to recognize and specifically bind HhaI (GCGC) and HaeIII (GGCC) methylated sites in a specific context, by using rounds of selection of phage displayed zinc finger randomized libraries that employed M.HhaI- and M.HaeIII-methylated DNA as baits [94]. Such methylation-specific zinc finger arrays could be employed for selectively targeting modified parts of the genome, for example, methylated gene promoters for targeted DNA demethylation and gene activation, provided that the exact methylation state of the promoter is known. In contrast, DNA sequence recognition of CRISPR/Cas9 relies on the Watson-Crick base pairing of the RNA guide and DNA sequence; therefore, it is not affected by DNA modifications found in mammalian genomes [95]. The modification state sensitivity of the zinc finger and TALE arrays should be taken into consideration while designing experiments, especially when the methylation status of the targeted region is not known. Additionally, certain TALE repeats have been reported to be insensitive to 5mC modification and therefore could be employed to overcome this limitation [91].
Overall, the CRISPR/Cas9 systems offer substantial advantages over zinc finger and TALE arrays, like simplicity of target design, possibility for multiplexing (targeting two or more sites at the same time) and independence of DNA modifications. Moreover, orthologous CRISPR/Cas9 proteins isolated from various bacteria could be employed for simultaneous targeting of different functionalities to the same or different loci [78]. However, relatively relaxed target specificity and potentially adverse immunogenicity might hamper their use in the clinic. In addition, the CRISPR/Cas9 system has not been employed for epigenetic editing yet (what is likely to be shown in the nearest future). On the other hand, zinc fingers and TALEs can offer superior target specificity and could be better tolerated by the immune system of a potential patient [96].
Epigenetic modification domains (effector domains)
Selection of the appropriate effector domain is based on the planned application and intended effect on transcription and epigenetic state. Nature provides numerous possible modification domains, which can be employed for the particular function. Various effector modifiers have been already fused to zinc fingers, TALE arrays and other DNA binding domains to target their activities to endogenous or genome integrated targets, reporter plasmids or viral DNA [15,97]. Examples include DNA methyltransferases (bacterial M.SssI targeted in vitro to synthetic DNA [98], M.HpaII targeted to reporter plasmids and integrated viral DNA [99,100] and eukaryotic Dnmt3a catalytic domain and full-length proteins, Dnmt3a-Dnmt3L single-chain constructs targeted to endogenous loci [38,101]), ten-eleven translocation DNA demethylases (targeted to endogenous loci [102,103]), thymine DNA glycosylase (targeted to endogenous locus [104]), histone methyltransferases (G9a—targeted to integrated Gal4 binding site [105], G9a [106] and Suv39H1 [106,107] targeted to endogenous locus, Ezh2 targeted to reporter construct [108]) and histone demethylase (Lsd1 targeted to endogenous locus [109]), as well as histone deacetylases (targeted to reporter plasmid [110]), which could either activate or repress targeted genes. Interestingly, a light-controlled TALE system was used by the Feng Zhang and George Church laboratories to target histone modifiers and VP64 transcription activating domain. Even though the observed effects regarding histone modifications are modest (oscillating between 1.5-fold and 3-fold), this method in principle allows introduction of the mark not only at the desired locus, but also at a desired time [111].
Many (if not most) of the targeted epigenome modifications studies were focused on manipulating the DNA methylation state (either to specifically methylate or demethylate gene promoters), in order to repress active oncogenes or activate silenced tumour suppressor genes, respectively. This might be explained by the fact that in contrast to the not yet fully understood mechanism of maintenance of histone modifications during mitotic division [112], the mechanisms of setting up and inheritance of DNA methylation have been thoroughly studied (reviewed in [113]).
Methylation of gene promoters around transcription start sites and first exons is strongly correlated with gene repression [113,114]. Once DNA methylation is established, it is inherited after semiconservative DNA replication by the action of hemi-methylation-specific DNA methyltransferase Dnmt1 [115,116]. Therefore, targeted DNA methylation can provide a unique opportunity to heritably switch off gene expression (loss of function) [37-39]. It could be used for example to silence the overexpressed oncogenes by DNA methylation in cancer cells. On the other hand, targeted DNA demethylation, as shown in recent publications [102-104], offers an interesting way to activate the desired gene expression from its native locus (gain of function). Straightforward applications of these approaches for clinical studies could be, for example, to demethylate the promoters and thereby activate the expression of tumour suppressor genes, which are commonly silenced in cancer cells [117-120]. Moreover, new functions to differentiated cells could be conveyed by activating genes, which are normally not expressed in that cell type.
No matter which effector domain is selected for targeting, the applicability of that domain should include the evaluation of the extent and stability of the introduced modification and its biological effect, as discussed in the next sections.
Stability of introduced epigenetic modifications
Despite the dynamic nature of epigenetic information, the overall cell state and global epigenetic states are heritable and maintained remarkably stably during multiple mitotic divisions. This is mainly due to the cooperation and redundancy of the multiple epigenetic signals (DNA and histone modifications), as well as to the transcriptional activity of each particular gene [121], which can reinforce the preservation of the current state. Thus, in contrast to the transient overexpression of transcription factors, modification of the epigenetic signal at selected promoters, through depositing of either activating or inactivating epigenetic marks, promises heritable maintenance of the induced state throughout multiple cell divisions. It is poorly investigated, however, if this assumption indeed holds true. Until now, most publications addressing directed epigenome modification did not investigate the long-term stability of the introduced mark, with few noble exceptions. Heritable DNA methylation and gene repression were observed after targeting zinc finger fused with the M.HpaII F35H mutant to the genomically integrated minimal thymidine kinase promoter governing expression of the CAT reporter gene construct. The introduced DNA methylation and the repressive effect were observed even 17 days post transfection of the zinc finger fusion constructs, when the expression of the ZF-HpaII F35H construct was no longer detectable neither at mRNA nor at protein levels [100]. However, in this report, an artificially introduced genomic locus was targeted; this study might therefore not represent a proof of concept for the stability of targeted DNA methylation at a natural genomic locus. Repression of a native tumour suppressor gene MASPIN locus was observed after its targeting by a zinc finger fused to Dnmt3a catalytic domain (CD) or KRAB transcription repression domain. Interestingly, after clearance of the construct from the cells, both the stable DNA methylation (up to 50 days post transfection) and the gene repression were maintained only in the case of ZF-Dnmt3a CD, but were lost in the case of ZF-KRAB transcription repression domain [34]. Even though the mechanisms of histone modification maintenance have not been completely understood at a molecular level, histone marks also seem to be stably maintained in the cells. In support of this, H3K27 methylation introduced by targeting Ezh2 next to the Gal4 binding site was maintained 4 days post clearance of the targeting construct [108]. Spreading and long-term stability (over multiple cell divisions) of H3K9me3 triggered by recruitment of HP1α to Oct4 promoter was also observed [122].
These studies illustrate that targeted epigenome modification can indeed withstand numerous cell divisions and is superior over effects introduced with transient targeting of repressors. Nonetheless, the introduced epigenetic modification might not be stable in all genomic contexts and will depend on the pre-existence of other activating or repressive epigenetic marks, the location of the locus in the euchromatic or heterochromatic region, as well as on the extent of the introduced modification and the nature of the mark itself. It is likely that small and local changes to the epigenome may not be efficiently maintained, and therefore, the locus would return to the initial state before perturbation happened [114,115]. Therefore, in order to achieve a stable effect, it might be beneficial to modify as big part of the region of interest as possible, either by employing a spreading mechanism or by targeting the fusion construct to multiple places within the same locus to achieve a cumulative effect [102]. In addition, simultaneous targeting of multiple epigenetic modifiers to the same locus, which reinforce the same effect, might strengthen the stability of the enforced new state.
Spreading of the mark across the genome
Since the epigenetic modifier is tethered to the DNA binding domain, which binds tightly to its recognition sequence, the length of the region which can be directly modified is limited. The extent of the linker between the targeting device and the epigenetic modifier is the main determinant of the possible reach distance. In the majority of the studies, the most efficient introduction of modification was observed in the nearest vicinity of the binding site of the targeting domain (10–40 bp [98,102,123]), which is in line with the typical distance that the linker region between the DBD and effector domain can provide. This is illustrated in Figure 2, which shows models of possible epigenetic targeting constructs with the DNA sequence and nucleosomes drawn to scale. Of course, if extensive DNA bending and looping are considered, the modification could reach further.

Structural models of possible epigenetic editing devices. The structural models of the proteins were taken from PDB repository (zinc finger [PDB:1P47], TALE [PDB:2YPF], CRISPR/Cas9 [PDB:4OO8], M.HhaI [PDB:5MHT], Dnmt3a/3L [PDB:2QRV], TET2 [PDB:4NM6], nucleosome [PDB:1AOI], a 21 amino acid linker was generated in PyMol, and 60 bp DNA sequence was generated with the make-na server (http://structure.usc.edu/make-na/server.html)). The models are drawn to scale and should provide an idea of the architecture of the synthetic constructs used for epigenetic editing. Modelling was done manually in PyMol. Zinc finger, DNA-bound zinc finger array fused to M.HhaI; TALE, synthetic TALE array fused to the catalytic domain of human TET2; CRISPR/Cas9, Cas9 protein fused to a Dnmt3a/Dnmt3L hetero-tetramer. The distances in base pairs and angstroms are indicated.
Due to mechanical constraints of the targeted epigenetic editing machinery, it is unlikely that a single and even very stable binding event would lead to a widespread modification at larger distances. Interestingly, histone modification [106] and DNA methylation spreading was observed beyond the expected distance that could be reached when considering the provided domain linker. We and the others observed the deposition of DNA methylation marks up to 300 bp and more from the targeted site when using the catalytic domain of Dnmt3a (or the Dnmt3a-Dnmt3L single-chain construct) to human EpCAM and VEGFA promoters [37,38]. Theoretically, this observation can be explained by extensive looping of the DNA in this region, which in turn would allow the tethered DNA methyltransferase to reach more distant regions of the DNA sequence. In a more interesting hypothesis, the spreading might be explained by a polymerization of Dnmt3a on the EpCAM and VEGFA promoters. Indeed, it has been shown that Dnmt3a cooperatively polymerases along the DNA molecule [124-126] and that its methylation activity is stimulated by the filament formation [127]. In this model, the targeted molecule of Dnmt3a would recruit additional molecules of the enzyme (maybe even the endogenous protein) to the modified region, leading to an efficient methylation of a larger genomic region adjacent to the targeted sequence (nucleation point). Whether the experimentally observed broad modification of these regions is due to DNA looping and nucleosome wrapping or the proposed spreading mechanism needs to be further investigated.
Whereas most of the studies were designed to achieve widespread DNA methylation of the targeted locus, Chaikind and colleagues developed an opposite strategy to selectively methylate a single CpG site in the genome using a split enzyme approach [128,129]. Two inactive parts of a DNA methyltransferase (M.SssI, M.HhaI) are separately directed to sites flanking the selected CpG, where they assemble to an active enzyme and methylate that target site, thus allowing to study the epigenetic consequences of a single methylation event. Additionally, the split enzyme approach would limit the extent of off-target effects, because the functional enzyme is only reconstituted at the targeted locus.
Delivery of programmable epigenetic editors for epigenetic therapy
The efficiency of introducing epigenetic change and therefore possible biomedical application depend in big part on the vehicle used for delivery of the constructs. So far, only cultured cells and not whole organisms were used for epigenetic editing applications. However, similar delivery methods as used for genome engineering can be applied for this purpose as well. Besides the traditional approaches, like transfections of transient expression plasmids, transduction of adeno-associated viral (AAV) [130], lenti-, retro- or adeno-viral vectors, new ways to deliver the gene or protein cargo to the cells have been discovered. One intrinsic problem with the aforementioned viral delivery systems is their limited insert size capacity (in particular AAV and lenti-viruses), which can become a restricting factor because the epigenome editing constructs tend to be very large (in particular Cas9 fusions) and targeting of multiple loci at once may become difficult. To overcome these constraints, new delivery strategies were developed. An interesting approach is the hydrodynamic injection method, in which the plasmids encoding the targeting constructs are injected directly into the bloodstream of an animal. Subsequently, the cells can internalize the DNA and express the protein, which can in turn exert the desired effect in the cells [131]. Another interesting novel approach is to deliver purified proteins to the cells by attaching cell-penetrating peptides to the purified protein constructs or RNA:protein complexes (in the case of CRISPR/Cas9), therefore allowing spontaneous uptake by the cells [132-134]. Likewise, zinc finger arrays were shown to be intrinsically cell permeable and therefore could be easily delivered [135]. However, not all the proteins are intrinsically cell permeable. To overcome this limitation, in the most recent report, the authors have tethered TALE-VP64 proteins with negatively supercharged domains (containing large amounts of acidic residues) to deliver the proteins using poly-cationic transfection reagents. Interestingly, Cas9 when complexed with guide RNA could be delivered without this additional domain [136].
Conclusions
From the time when the field of epigenetics was defined by Waddington until nowadays, we have learned a lot about epigenetic regulation of gene expression and maintenance of cellular identity. Substantial progress was achieved in identification and understanding the role of various epigenetic marks, their distribution in healthy and diseased tissues and the enzymatic machinery responsible for depositing, reading and removing these marks. This progress was accompanied or rather preceded by technological development, like chromatin immunoprecipitation (ChIP), bisulfite sequencing, high-throughput proteomic and whole genome approaches. The bottom-up synthetic epigenetics (targeted epigenome editing), although still in its infancy, constitutes an area of extensive research. New technological developments will likely increase the specificity of targeting devices and the efficiency of effector domains in setting the desired epigenetic marks and will supply engineered systems for spreading the modification across the whole locus, providing efficient and reliable tools for stable modification of the epigenome. The most exciting progress is expected from the CRISPR/Cas9 system, as it allows the biggest flexibility and ease of design of new targets and the possibility to construct target libraries [137,138], which will allow unprecedented control of the epigenetic states at desired loci.
Synthetic epigenetics has the potential to address so far unapproachable areas of basic and clinical research. It provides tools and methods, which allow dissection of the epigenetic signalling cascades and to identify the driver and passenger modifications. It can widen our understanding of epigenetic dynamics and the basis of signal inheritance. Top-down synthetic epigenetic approaches have already made a big contribution in generating disease model cell lines. Epigenetic editing will further foster biomedical research by addressing the epigenetic contribution to complex and simple diseases, through discovery and validation of disease-promoting epimutations and provide means for reverting them. It also offers tools to probe factors responsible for cell identity and allows intelligent control of the cell fate.
Abbreviations
- 5mC:
-
5-Methylcytosine
- Cas9:
-
CRISPR-associated protein 9
- ChIP:
-
Chromatin immunoprecipitation
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- DBDs:
-
DNA binding domains
- Dnmt:
-
DNA methyltransferase
- EpCAM:
-
Epithelial cellular adhesion molecule
- Ezh2:
-
Enhancer of zeste 2
- iPS cells:
-
Induced pluripotent stem cells
- KRAB domain:
-
Krüppel-associated box domain
- PAM:
-
Protospacer adjacent motif
- RNP:
-
Ribonucleoprotein
- RVD:
-
Repeat variable di-residues
- SCNT:
-
Somatic cell nuclear transfer
- TALE:
-
Transcription activating-like effector
- TET:
-
Ten-eleven translocation
- VEGFA:
-
Vascular endothelial growth factor A
- ZF:
-
Zinc finger
References
Gurdon J. Nuclear reprogramming in eggs. Nat Med. 2009;15(10):1141–4.
Waddington CH. The strategy of the genes; a discussion of some aspects of theoretical biology. London: Allen & Unwin; 1957.
Consortium EP. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306(5696):636–40.
Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74.
Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol. 2010;28(10):1045–8.
Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A, et al. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol. 2012;30(3):224–6.
Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17(3):330–9.
Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–63.
Gurdon JB, Elsdale TR, Fischberg M. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature. 1958;182(4627):64–5.
Gurdon JB. Adult frogs derived from the nuclei of single somatic cells. Dev Biol. 1962;4:256–73.
Graf T. Historical origins of transdifferentiation and reprogramming. Cell Stem Cell. 2011;9(6):504–16.
Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51(6):987–1000.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72.
de Groote ML, Verschure PJ, Rots MG. Epigenetic editing: targeted rewriting of epigenetic marks to modulate expression of selected target genes. Nucleic Acids Res. 2012;40(21):10596–613.
Bauman J, Verschraegen C, Belinsky S, Muller C, Rutledge T, Fekrazad M, et al. A phase I study of 5-azacytidine and erlotinib in advanced solid tumor malignancies. Cancer Chemother Pharmacol. 2012;69(2):547–54.
Yang G, Tian J, Feng C, Zhao LL, Liu Z, Zhu J. Trichostatin a promotes cardiomyocyte differentiation of rat mesenchymal stem cells after 5-azacytidine induction or during coculture with neonatal cardiomyocytes via a mechanism independent of histone deacetylase inhibition. Cell Transplant. 2012;21(5):985–96.
Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122(6):947–56.
Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134(3):521–33.
Maza I, Hanna JH. Hijacked by an oocyte: hierarchical molecular changes in somatic cell nuclear transfer. Mol Cell. 2014;55(4):507–9.
Liang G, Zhang Y. Embryonic stem cell and induced pluripotent stem cell: an epigenetic perspective. Cell Res. 2013;23(1):49–69.
Jullien J, Miyamoto K, Pasque V, Allen GE, Bradshaw CR, Garrett NJ, et al. Hierarchical molecular events driven by oocyte-specific factors lead to rapid and extensive reprogramming. Mol Cell. 2014;55(4):524–36.
Gao Y, Chen J, Li K, Wu T, Huang B, Liu W, et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell. 2013;12(4):453–69.
Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471(7336):68–73.
Simonsson S, Gurdon J. DNA demethylation is necessary for the epigenetic reprogramming of somatic cell nuclei. Nat Cell Biol. 2004;6(10):984–90.
Ooi J, Liu P. Pluripotency and its layers of complexity. Cell Regen. 2012;1(1):7.
Krishnakumar R, Blelloch RH. Epigenetics of cellular reprogramming. Curr Opin Genet Dev. 2013;23(5):548–55.
Fulka Jr J, Langerova A, Loi P, Ptak G, Albertini D, Fulka H. The ups and downs of somatic cell nucleus transfer (SCNT) in humans. J Assist Reprod Genet. 2013;30(8):1055–8.
Maherali N, Hochedlinger K. Guidelines and techniques for the generation of induced pluripotent stem cells. Cell Stem Cell. 2008;3(6):595–605.
Ogura A, Inoue K, Wakayama T. Recent advancements in cloning by somatic cell nuclear transfer. Philos Trans R Soc Lond Ser B Biol Sci. 2013;368(1609):20110329.
Deng Y, Wang CC, Choy KW, Du Q, Chen J, Wang Q, et al. Therapeutic potentials of gene silencing by RNA interference: principles, challenges, and new strategies. Gene. 2014;538(2):217–27.
Lund CV, Blancafort P, Popkov M, Barbas 3rd CF. Promoter-targeted phage display selections with preassembled synthetic zinc finger libraries for endogenous gene regulation. J Mol Biol. 2004;340(3):599–613.
Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32(7):670–6.
Rivenbark AG, Stolzenburg S, Beltran AS, Yuan X, Rots MG, Strahl BD, et al. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics. 2012;7(4):350–60.
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–51.
Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10(10):977–9.
Nunna S, Reinhardt R, Ragozin S, Jeltsch A. Targeted methylation of the epithelial cell adhesion molecule (EpCAM) promoter to silence its expression in ovarian cancer cells. PLoS One. 2014;9(1):e87703.
Siddique AN, Nunna S, Rajavelu A, Zhang Y, Jurkowska RZ, Reinhardt R, et al. Targeted methylation and gene silencing of VEGF-A in human cells by using a designed Dnmt3a-Dnmt3L single-chain fusion protein with increased DNA methylation activity. J Mol Biol. 2013;425(3):479–91.
Stolzenburg S, Rots MG, Beltran AS, Rivenbark AG, Yuan X, Qian H, et al. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic Acids Res. 2012;40(14):6725–40.
Thirlwell C, Schulz L, Dibra H, Beck S. Suffocating cancer: hypoxia-associated epimutations as targets for cancer therapy. Clin Epigenetics. 2011;3:9.
Tapia T, Smalley SV, Kohen P, Munoz A, Solis LM, Corvalan A, et al. Promoter hypermethylation of BRCA1 correlates with absence of expression in hereditary breast cancer tumors. Epigenetics. 2008;3(3):157–63.
Tee L, Lim DH, Dias RP, Baudement MO, Slater AA, Kirby G, et al. Epimutation profiling in Beckwith-Wiedemann syndrome: relationship with assisted reproductive technology. Clin Epigenetics. 2013;5(1):23.
Ibrahim A, Kirby G, Hardy C, Dias RP, Tee L, Lim D, et al. Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin Epigenetics. 2014;6(1):11.
Weksberg R, Smith AC, Squire J, Sadowski P. Beckwith-Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet. 2003;12 Spec No 1:R61–8.
Dwivedi RS, Herman JG, McCaffrey TA, Raj DS. Beyond genetics: epigenetic code in chronic kidney disease. Kidney Int. 2011;79(1):23–32.
Begin P, Nadeau KC. Epigenetic regulation of asthma and allergic disease. Allergy Asthma Clin Immunol. 2014;10(1):27.
Dayeh T, Volkov P, Salo S, Hall E, Nilsson E, Olsson AH, et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014;10(3):e1004160.
Wang Y, Shu Y, Xiao Y, Wang Q, Kanekura T, Li Y, et al. Hypomethylation and overexpression of ITGAL (CD11a) in CD4 T cells in systemic sclerosis. Clin Epigenetics. 2014;6(1):25.
Jurkowska RZ, Jeltsch A. Silencing of gene expression by targeted DNA methylation: concepts and approaches. Methods Mol Biol. 2010;649:149–61.
Guilinger JP, Pattanayak V, Reyon D, Tsai SQ, Sander JD, Joung JK, et al. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Methods. 2014;11(4):429–35.
Jeltsch A, Jurkowska RZ, Jurkowski TP, Liebert K, Rathert P, Schlickenrieder M. Application of DNA methyltransferases in targeted DNA methylation. Appl Microbiol Biotechnol. 2007;75(6):1233–40.
Blancafort P, Jin J, Frye S. Writing and rewriting the epigenetic code of cancer cells: from engineered proteins to small molecules. Mol Pharmacol. 2013;83(3):563–76.
Voigt P, Reinberg D. Epigenome editing. Nat Biotechnol. 2013;31(12):1097–9.
Rusk N. CRISPRs and epigenome editing. Nat Methods. 2014;11(1):28.
Slattery M, Zhou T, Yang L, Dantas Machado AC, Gordan R, Rohs R. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci. 2014;39(9):381–99.
Suzuki M. A framework for the DNA-protein recognition code of the probe helix in transcription factors: the chemical and stereochemical rules. Structure. 1994;2(4):317–26.
Pingoud A, Jeltsch A. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001;29(18):3705–27.
Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29:183–212.
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326(5959):1509–12.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21.
Pabo CO, Peisach E, Grant RA. Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem. 2001;70:313–40.
Laity JH, Lee BM, Wright PE. Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol. 2001;11(1):39–46.
Choo Y, Klug A. Toward a code for the interactions of zinc fingers with DNA: selection of randomized fingers displayed on phage. Proc Natl Acad Sci U S A. 1994;91(23):11163–7.
Fu F, Sander JD, Maeder M, Thibodeau-Beganny S, Joung JK, Dobbs D, et al. Zinc Finger Database (ZiFDB): a repository for information on C2H2 zinc fingers and engineered zinc-finger arrays. Nucleic Acids Res. 2009;37(Database issue):D279–83.
Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods. 2011;8(1):67–9.
Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an ‘open-source’ protocol for making customized zinc-finger arrays. Nat Protoc. 2009;4(10):1471–501.
Sander JD, Yeh JR, Peterson RT, Joung JK. Engineering zinc finger nucleases for targeted mutagenesis of zebrafish. Methods Cell Biol. 2011;104:51–8.
Sander JD, Zaback P, Joung JK, Voytas DF, Dobbs D. Zinc Finger Targeter (ZiFiT): an engineered zinc finger/target site design tool. Nucleic Acids Res. 2007;35(Web Server issue):W599–605.
Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK, Dobbs D. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res. 2010;38(Web Server issue):W462–8.
Boch J, Bonas U. Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol. 2010;48:419–36.
Scholze H, Boch J. TAL effector-DNA specificity. Virulence. 2010;1(5):428–32.
Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501.
Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012;30(5):460–5.
Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–78.
Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60(2):174–82.
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–12.
Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960–4.
Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10(11):1116–21.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152(5):1173–83.
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31(9):833.
Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8(9):765–70.
Juillerat A, Dubois G, Valton J, Thomas S, Stella S, Marechal A, et al. Comprehensive analysis of the specificity of transcription activator-like effector nucleases. Nucleic Acids Res. 2014;42(8):5390–402.
Fu YF, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822.
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–84.
Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol. 2014;32(7):677–83.
Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32(6):569–76.
Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513(7519):569–73.
Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507(7490):62–7.
Bochtler M. Structural basis of the TAL effector-DNA interaction. Biol Chem. 2012;393(10):1055–66.
Jamieson AC, Miller JC, Pabo CO. Drug discovery with engineered zinc-finger proteins. Nat Rev Drug Discov. 2003;2(5):361–8.
Kubik G, Schmidt MJ, Penner JE, Summerer D. Programmable and highly resolved in vitro detection of 5-methylcytosine by TALEs. Angew Chem Int Ed Engl. 2014;53(23):6002–6.
Liu Y, Toh H, Sasaki H, Zhang X, Cheng X. An atomic model of Zfp57 recognition of CpG methylation within a specific DNA sequence. Genes Dev. 2012;26(21):2374–9.
Sasai N, Defossez PA. Many paths to one goal? The proteins that recognize methylated DNA in eukaryotes. Int J Dev Biol. 2009;53(2–3):323–34.
Isalan M, Choo Y. Engineered zinc finger proteins that respond to DNA modification by HaeIII and HhaI methyltransferase enzymes. J Mol Biol. 2000;295(3):471–7.
Dupuis ME, Villion M, Magadan AH, Moineau S. CRISPR-Cas and restriction-modification systems are compatible and increase phage resistance. Nat Commun. 2013;4:2087.
Maier DA, Brennan AL, Jiang S, Binder-Scholl GK, Lee G, Plesa G, et al. Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the HIV co-receptor CCR5. Hum Gene Ther. 2013;24(3):245–58.
Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–55.
Xu GL, Bestor TH. Cytosine methylation targetted to pre-determined sequences. Nat Genet. 1997;17(4):376–8.
Smith AE, Ford KG. Specific targeting of cytosine methylation to DNA sequences in vivo. Nucleic Acids Res. 2007;35(3):740–54.
Smith AE, Hurd PJ, Bannister AJ, Kouzarides T, Ford KG. Heritable gene repression through the action of a directed DNA methyltransferase at a chromosomal locus. J Biol Chem. 2008;283(15):9878–85.
Li F, Papworth M, Minczuk M, Rohde C, Zhang Y, Ragozin S, et al. Chimeric DNA methyltransferases target DNA methylation to specific DNA sequences and repress expression of target genes. Nucleic Acids Res. 2007;35(1):100–12.
Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013;31(12):1137–42.
Chen H, Kazemier HG, de Groote ML, Ruiters MH, Xu GL, Rots MG. Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. 2014;42(3):1563–74.
Gregory DJ, Zhang Y, Kobzik L, Fedulov AV. Specific transcriptional enhancement of inducible nitric oxide synthase by targeted promoter demethylation. Epigenetics. 2013;8(11):1205–12.
Osipovich O, Milley R, Meade A, Tachibana M, Shinkai Y, Krangel MS, et al. Targeted inhibition of V(D)J recombination by a histone methyltransferase. Nat Immunol. 2004;5(3):309–16.
Falahi F, Huisman C, Kazemier HG, van der Vlies P, Kok K, Hospers GA, et al. Towards sustained silencing of HER2/neu in cancer by epigenetic editing. Mol Cancer Res. 2013;11(9):1029–39.
Snowden AW, Gregory PD, Case CC, Pabo CO. Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr Biol. 2002;12(24):2159–66.
Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, et al. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10(11):1291–300.
Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, et al. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol. 2013;31(12):1133–6.
Yang WM, Yao YL, Sun JM, Davie JR, Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J Biol Chem. 1997;272(44):28001–7.
Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500(7463):472–6.
Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20(3):259–66.
Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. 2011;12(2):206–22.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–92.
Jeltsch A, Jurkowska RZ. New concepts in DNA methylation. Trends Biochem Sci. 2014;39(7):310–8.
Song J, Rechkoblit O, Bestor TH, Patel DJ. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science. 2011;331(6020):1036–40.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–9.
Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T, et al. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121(3):893–904.
Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16 Spec No 1:R50–9.
Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21(2):163–7.
Kaneko S, Son J, Bonasio R, Shen SS, Reinberg D. Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev. 2014;28(18):1983–8.
Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, Crabtree GR. Dynamics and memory of heterochromatin in living cells. Cell. 2012;149(7):1447–60.
McNamara AR, Hurd PJ, Smith AE, Ford KG. Characterisation of site-biased DNA methyltransferases: specificity, affinity and subsite relationships. Nucleic Acids Res. 2002;30(17):3818–30.
Jeltsch A, Jurkowska RZ. Multimerization of the dnmt3a DNA methyltransferase and its functional implications. Prog Mol Biol Transl Sci. 2013;117:445–64.
Rajavelu A, Jurkowska RZ, Fritz J, Jeltsch A. Function and disruption of DNA methyltransferase 3a cooperative DNA binding and nucleoprotein filament formation. Nucleic Acids Res. 2012;40(2):569–80.
Jurkowska RZ, Rajavelu A, Anspach N, Urbanke C, Jankevicius G, Ragozin S, et al. Oligomerization and binding of the Dnmt3a DNA methyltransferase to parallel DNA molecules: heterochromatic localization and role of Dnmt3L. J Biol Chem. 2011;286(27):24200–7.
Emperle M, Rajavelu A, Reinhardt R, Jurkowska RZ, Jeltsch A. Cooperative DNA binding and protein/DNA fiber formation increases the activity of the Dnmt3a DNA methyltransferase. J Biol Chem. 2014,289(43):29602–13.
Chaikind B, Kilambi KP, Gray JJ, Ostermeier M. Targeted DNA methylation using an artificially bisected M.HhaI fused to zinc fingers. PLoS One. 2012;7(9):e44852.
Chaikind B, Ostermeier M. Directed evolution of improved zinc finger methyltransferases. PLoS One. 2014;9(5):e96931.
Senis E, Fatouros C, Grosse S, Wiedtke E, Niopek D, Mueller AK, et al. CRISPR/Cas9-mediated genome engineering: an adeno-associated viral (AAV) vector toolbox. Biotechnol J. 2014;9(11):1402–12.
Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514(7522):380–4.
Ramakrishna S, Kwaku Dad AB, Beloor J, Gopalappa R, Lee SK, Kim H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014;24(6):1020–7.
Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24(6):1012–9.
Liu J, Gaj T, Patterson JT, Sirk SJ, Barbas 3rd CF. Cell-penetrating peptide-mediated delivery of TALEN proteins via bioconjugation for genome engineering. PLoS One. 2014;9(1):e85755.
Gaj T, Liu J, Anderson KE, Sirk SJ, Barbas 3rd CF. Protein delivery using Cys2-His2 zinc-finger domains. ACS Chem Biol. 2014;9(8):1662–7.
Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015,33(1):73–80.
Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343(6166):80–4.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–7.
Acknowledgements
We would like to thank Dr. Renata Jurkowska for the critical reading of the manuscript and valuable discussions. We would like to apologize to all the colleagues whose work could not be mentioned due to manuscript length constraints.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have written and edited the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Jurkowski, T.P., Ravichandran, M. & Stepper, P. Synthetic epigenetics—towards intelligent control of epigenetic states and cell identity. Clin Epigenet 7, 18 (2015). https://doi.org/10.1186/s13148-015-0044-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-015-0044-x