
Abstract
Background
A Sri Lankan girl with hereditary hemorrhagic telangiectasia (Osler–Weber–Rendu syndrome) is described.
Case presentation
She presented with recurrent spontaneous epistaxis, pulmonary arterio venous malformation and oral telangiectasia. A diagnosis of Hereditary hemorrhagic telangiectasia (Osler–Weber–Rendu syndrome) was made based on the presence of three Curacao criteria (out of four). Evaluations of her jaundice revealed chronic parenchymal liver disease with multiple nodules in the liver with early portal hypertension. She had a muscular build, with elevated serum testosterone but low serum dehydroepiandrosterone sulphate levels. This could be attributed to impaired sulfation of dehydroepiandrosterone due to portocaval shunting of blood, leading to hyperandrogenemia.
Conclusions
Hyperandorogenemia due impaired sulfation of dehydroepiandrosterone as a result of portocaval shunting is seen in Hereditary haemorrhagic telangiectasia.
Similar content being viewed by others


Case description
Background
A 16-year old girl from a remote village in Sri Lanka is described.
Case presentation
She was referred from the Paediatric Cardiology Unit to the University Unit of Lady Ridgeway Hospital for Children (LRH) for further evaluation of jaundice. She was the only child born to non consanguineous healthy parents and was well till 14 years of age, when she sought medical advice from the local hospital for exertional dyspnoea. At presentation to the local hospital she was detected to have central cyanosis and clubbing but otherwise clinically normal. Her haemoglobin was 18 g/dl and the packed cell volume was 54%. Her platelet count was 73 × 109/l and SpO2 82%. The echocardiogramme revealed a pulmonary arteriovenous malformation (AVM). The ultra sound scan (USS) of the abdomen showed a normal liver and no splenomegaly.
She was referred to the cardiology unit of LRH for further evaluation. At LRH contrast echocardiogramme was performed which showed evidence of pulmonary arteriovenous fistula. The finding at cardiac catheterization was compatible with bilateral diffuse pulmonary AVMs. Due to the diffuse nature of the pulmonary AVM she was managed conservatively.
Two years later, at the age of 16 years she developed spontaneous recurrent epistaxis. She was also noted to have jaundice. Examination revealed deep jaundice, plethora, central cyanosis and clubbing. There were no other peripheral stigmata of chronic liver disease. She had coarse facial appearance. There was no cutaneous telangiectasia. Abdominal examination revealed a firm hepatomegaly of two cm, but no splenomegaly or ascites. Rest of the abdominal examination was normal. Cardiovascular system examination revealed a systolic murmur best heard over the pulmonary area. Blood pressure was normal. Rest of the examination including respiratory system and central nervous systems were clinically normal. Ultra sound scan examination of abdomen at sixteen years revealed heterogeneous linear (Liver) echo texture, increased peri portal echogenicity with splenomegaly and varices compatible with chronic liver disease and early portal hypertension. A focal lesion was seen in segment Vl of the liver. Magnetic resonance imaging (MRI) revealed a dilated splenic vein draining separately through the diaphragmatic hiatus between the inferior vena cava and the aorta possibly into the superior vena cava through the azygous vein. She continued to be managed conservatively.
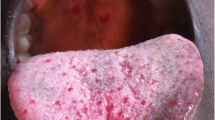
At nineteen years of age she remained jaundiced. There were telangiectases over the oral mucosa and conjunctiva. Eye referral revealed telangiectatic vessels in the right optic disc. Hepatomegaly was unchanged.
She had attained menarche at 16 year of age but had amenorrhea for several years, and now has her menstrual periods infrequently. Although she has a masculine habitus her secondary sexual characteristics were feminine. There was no clitoromegaly. Endocrinological evaluation was done to investigate for her masculine built and irregular menstruation. There was no family history suggestive of polycystic ovarian syndrome. At 19 years of age, she had an elevated serum testosterone level of 15.97 nmol/L (normal adult female range 0.52–2.43), but serum DHEAS (dehydroepiandrosterone sulfate) level were low (1.0 μmol/L, normal range 1.7–13.4). Serum 17 OHP, FSH, LH and serum estradiol levels were normal. She also had marginally low free T4, with normal TSH. USS abdomen repeated at 19 years of age was compatible with chronic liver disease with early portal hypertension. She had normal uterus and normal ovaries with no evidence of poly cystic ovaries.
MRI abdomen (Fig. 1) showed changes of chronic parenchymal liver disease with multiple nodules in both lobes of the liver. The nodules were hyperplastic with low T2 signal intensity. A 47 × 45 mm size nodule was seen in the right lobe of the liver. A liver biopsy was not done due to the risk of bleeding in the presence of multiple AVMs. MRI brain done to identify any cerebral vascular malformation was normal.

Magnetic resonance imaging showing multiple nodules in the liver
A diagnosis of hereditary hemorrhagic telangiectasia (Osler–Weber–Rendu syndrome) was made based on the presence of three out of four Curacao criteria [1]. The three criteria she fulfilled were recurrent spontaneous epistaxis, visceral (pulmonary) arterio venous malformation and telangiectasia in the oral mucosa [1]. The patient along with the family was counselled regarding the diagnosis and the prognosis.
Conclusion
This 19 year old girl has Hereditary haemorrhagic telangiectasia with chronic liver disease and pulmonary AVM. She has a masculine appearance due to impaired sulfation of dehydroepiandrosterone (DHEA) as a result of portocaval shunting.
Hereditary hemorrhagic telangiectasia—HHT (Osler–Weber–Rendu syndrome) was first described in the nineteenth century as a familial disorder causing nose bleeds, gastrointestinal bleeding and abnormal vascular structures. It is inherited as an autosomal dominant trait. A diagnosis of HHT could be made based on Curacao’s criteria, which are; Spontaneous recurrent epistaxis, multiple telangiectases at characteristic sites such as lips, oral cavity, fingers, nose, visceral lesions such as gastrointestinal telangiectasia, pulmonary AVM, hepatic AVM, Cerebral AVM and spinal AVM, a family history of a first degree relative with HHT. The presence of three criteria makes it a definite diagnosis of HHT, while two make the diagnosis possible. Our patient had spontaneous recurrent epistaxis, visceral AVM in the form of pulmonary AVM and telangiectasia in the oral mucosa. There was no family history of HHT. Based on these three criteria a definitive diagnosis of HHT was made [1]. The diagnosis was made at 19 years of age as the telangiectasia of the oral mucosa appeared only at nineteen years.
Spontaneous recurrent nose bleeds from telangiectasia of the nasal mucosa are the most common manifestations of HHT and occurs in about 95% [2]. Intervention for epistaxis should be considered if there is anaemia attributable to nose bleeding or if the frequency and duration of bleeding interferes with normal activities [2]. Our patient required no treatment as the epistaxis was self-limiting. Telangiectases of the skin and buccal mucosa also occur in about 95% of patients, typically presenting later in life than epistaxis [2]. It usually presents in about the third decade of life and gradually increases in number and size. They mostly occur in the face, lips, tongue, buccal mucosa and finger tips and can occur elsewhere. Our patient had telangiectases over the oral mucosa and conjunctivae.
Pulmonary AVMs are thin walled abnormal vessels that replace normal capillaries between pulmonary arterial and venous circulation. In one study 37% of patients with HHT demonstrated pulmonary AVM on high resolution computed tomography (CT). The safest method for treatment is transcatheter embolisation. Since our patient had bilateral diffuse pulmonary AVM conservative management was planned. Cerebral AVMs occur in approximately 10% of patients with HHT [2]. They can cause headache, seizures, ischaemia of the surrounding tissue, or haemorrhage. Our patient did not have cerebral involvement.
Hepatic involvement is variable in HHT. In one study only 8% showed symptomatic involvement [2]. In HHT, the predominant liver involvement seen was shunting of blood from hepatic artery to hepatic veins with shunting of metabolites from portal circulation to systemic circulation. A shunt from hepatic artery to portal vein leads to portal hypertension and increased blood flow can lead to nodularity of the liver due to increased deposition of fibrous tissue. The liver undergoes nodular transformation also known as “pseudo-cirrhosis” [3]. In our patient there was no vascular malformation of the liver however nodular hyperplasia was seen. She did not undergo liver biopsy as there is a risk of bleeding. Liver transplantation is currently the only curative option for symptomatic liver disease in HHT. Except for hepatocellular and biliary necrosis which requires urgent liver transplantation consensus on the most appropriate timing of the transplantation is lacking [4].
To our knowledge, elevated serum testosterone levels have not previously been reported in females with HHT. However, there have been a few reported cases of elevated serum testosterone in association with porto-systemic shunting in young females [5, 6]. The initial observation was reported in two adolescent girls with congenital porto-systemic shunting, who presented with virilisation and amenorrhea, and had elevated testosterone [5]. More recently, Bas et al. [6] reported on two 7-year old girls with congenital portosystemic shunts, presenting with premature pubarche. These females had elevated serum testosterone and other downstream androgens in the presence of low DHEAS, as seen in our patient [6]. It is postulated that the mechanism of hyperandrogenemia, could be impaired conversion of DHEA to its inactive sulphate ester DHEAS in the liver, due to bypassing of DHEA to the systemic circulation [6]. This would lead to low DHEAS and elevated DHEA in the circulation. As DHEA is a precursor for the synthesis of more potent downstream androgens such as androstenedione and testosterone, elevated DHEA would lead to elevated serum testosterone and features of hyperandrogenism [6].
This is a classical case of HHT with hyperandrogenaemic features. This probably could be the first reported case with hyperandrogenaemia secondary to hepatic AVM leading to shunting of blood leading to underclearance of proandrogenic metabolites from the circulation.
Abbreviations
- LRH:
-
Lady Ridgeway Hospital for Children
- AVM:
-
arteriovenous malformation
- USS:
-
ultra sound scan
- DHEAS:
-
dehydroepiandrosterone sulfate
- DHEA:
-
dehydroepiandrosterone
- HHT:
-
hereditary hemorrhagic telangiectasia
- MRI:
-
magnetic resonance imaging
References
Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kieldsan AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu–Osler–Weber syndrome). Am J Med Genet. 2000;91(1):66–7.
Mc Donald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management and pathogenesis. Genet Med. 2011;13(7):607–16.
Garchia-Tsao G, Korzenik JR, Young L, Henderson KJ, Jain D, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. NEJM. 2000;343(13):931–6.
Coremans L, Van den Bossche B, Colle I. Hepatic involvement in hereditary hemaorrhagic telangiectasia (HHT) or Rendu–Osler–Weber syndrome. Acta Gastroenterol Belg. 2015;78(3):319–26.
Satoh M, Yokoya S, Hachiya Y, Hachiya M, Fujisawa T, Hoshino K, et al. Two hyperandrogenic adolescent girls with congenital portosystemic shunt. Eur Pediatr. 2001;160(5):307–11.
Bas S, Guran T, Atay Z, Haliloglu B, Abali S, Turan S, et al. Premature pubarche, hyperinsulinemia and hypothyroxinemia: novel manifestations of congenital portosystemic shunts (abernethy malformation) in children. Hormone Res Paediatr. 2015;83(4):282–7.
Authors’ contributions
RD: Consultant Paediatrician attending to patient and wrote up the case report. KPKYMDS: Literature search and managed the patient, reviewed case report. SNS: Consultant Paediatric Endocrinologist-Wrote the endocrine related sections of the manuscript and reviewed case report. SNP: Consultant Paediatric Cardiologist provided cardiology follow up of patient, reviewed case report. MUJF: Consultant Radiologist provided radiology follow up, reviewed case report. PW: Consultant Paediatrician in charge of patient, considered writing the case report, wrote manuscript, reviewed case report. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent has been obtained from the patient as well as the patient’s mother for publication of this Case Report and any accompanying images. A copy of the written consent is available for review by the Editor-In Chief of this journal.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dissanayake, R., Wickramarathne, K.P.K.Y.M.D.S., Seneviratne, S.N. et al. Hereditary hemorrhagic telangiectasia, liver disease and elevated serum testosterone (Osler–Weber–Rendu syndrome): a case report. BMC Res Notes 10, 58 (2017). https://doi.org/10.1186/s13104-017-2397-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-017-2397-z