
Abstract
Background
Hereditary spastic paraplegias (HSP), a group of genetically heterogeneous neurological disorders with more than 56 documented loci (SPG1-56), are described either as uncomplicated (or pure), or complicated where in addition to spasticity and weakness of lower extremeties, additional neurological symptoms are present, including dementia, loss of vision, epilepsy, mental retardation and ichthyosis. We identified a large consanguineous family of Indian descent with four affected members with childhood onset HSP (SPG54), presenting with upper and lower limb spasticity, mental retardation and agenesis of the corpus callosum.
Results
A common region of homozygosity on chromosome 8 spanning seven megabases (Mb) was identified in the affected individuals using the Illumina human cytoSNP-12 DNA Analysis BeadChip Kit. Exome sequencing identified a homozygous stop gain mutation (pR287X) in the phospholipase A1 gene DDHD2, in the affected individuals, resulting in a premature stop codon and a severely truncated protein lacking the SAM and DDHD domains crucial for phosphoinositide binding and phospholipase activity.
Conclusion
This mutation adds to the knowledge of HSP, suggests a possible founder effect for the pR287X mutation, and adds to the list of genes involved in lipid metabolism with a role in HSP and other neurodegenerative disorders.
Similar content being viewed by others

Background
Hereditary spastic paraplegias (HSP), are a heterogeneous group of rare inherited neurological disorders [1]. The primary clinical signs include progressive spasticity and weakness of lower limbs and hip muscles. Therefore, gait impairment is present in the vast majority of individuals with HSP. Urinary urgency is also a common symptom which can be an early presenting sign.
HSP is classified clinically as “uncomplicated” (pure or non-syndromic) if symptoms are limited to progressive spastic weakness in the legs, although often accompanied by urinary urgency and subtle dorsal column impairment, or “complicated” if associated with additional symptoms involving neurological or systemic abnormalities including dementia, ataxia, mental retardation, neuropathy, distal wasting, loss of vision, epilepsy or ichthyosis.
The prevalence of HSP is estimated at 3–10 cases per 100,000 populations in Europe [2], and there is considerable variation in the severity, age of onset, and degree of progression of symptoms, which can appear at any stage from infancy to 60 years old [3].
To date the more than 50 distinct genetic loci associated with distinct subtypes of HSP have been mapped [1], are associated with different modes of inheritance, including autosomal dominant, autosomal recessive and X-linked. The most common type of HSP is the autosomal dominant form that accounts for 70% of the cases which are mostly pure HSP. By contrast, complicated HSPs are mostly autosomal recessive. For autosomal recessive HSP, at least 22 different genes have been reported [4, 5]. In addition, there is considerable variation in age of onset, severity, and progression of symptoms between or even within a family owing to other factors such as modifying genes or environmental effects.
We have identified a large extended consanguineous family of Indian origin with four affected children, two in each branch of the family, sharing the same clinical phenotype of HSP (SPG54). They are all females who have difficulty in walking, and have motor delay, developmental delay, lower limb spasticity, and mental retardation. Brain MRI shows hypoplastic corpus callosum with possible agenesis of the splenium.
Methods
Human subjects and DNA samples
The subjects who participated in the study are members of a Saudi Arabia based Indian family (Figure 1). Prior to commencement of the clinical and molecular investigations, informed consent was signed by the legal guardians, the parents, on behalf of the affected children and they agreed to publish the study outcomes. Ethical approval (ref. no. 24-14), according to the Declaration of Helsinki, was obtained from the Institutional Review Board (IRB), Princess Al-Jawhara Albrahim Center of Excellence in Research of Hereditary Disorders and the Unit of Biomedical Ethics Research Committee, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia. Peripheral or venous blood from six unaffected (III-1, III-2, III-3, III-4, IV-4, IV-5) and four affected (IV-1, IV-3, IV-7, IV-8) individuals was collected in EDTA tubes and stored at 4°C. Genomic DNA was extracted using QIAamp ® mini DNA extraction kit (Qiagen, USA). The DNA was quantified by Nanodrop-2000 (Thermo Scientific, USA) spectrophotometer.

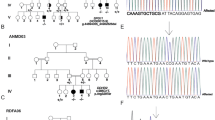
Pedigree structure of HSP family (SPG54) and sequence confirmation of the DDHD2 mutation. a Family pedigree. Squares and circles indicate males and females, respectively. Darkened symbols represent affected members, and slashes represent deceased. b Representative sequence traces of subjects and control. Sanger sequencing confirmed the homozygous nonsense mutation (c.859C >T, p.Arg287*) of the DDHD2 gene identified in the probands (IV-1, IV-3, IV-7, IV-8). The control sequence trace demonstrates the wild-type sequence. c A schematic illustration of DDHD2, showing four protein domains [WWE, lipase, coiled–coiled region (SAM), and DDHD]. The positions of all identified mutations are indicated. The mutation identified in this study is indicated in red.
Genomewide homozygosity mapping
Genomewide homozygosity mapping in four affected and six unaffected individuals was performed using Illumina iScan system with HumanCytoSNP-12v12.1 kit following the manufacturer’s protocol (http://www.support.illumina.com/array/protocols.ilmn). Loss of heterozygosity (LOH) regions were detected by analyzing the SNPs for copy number variation with GenomeStudio software.
Whole exome sequencing (WES)
To identify the causative mutation, we undertook whole exome sequence analysis in a trio samples set (III-1 father, III-2 mother, IV-1 affected daughter), using the SureSelect human All Exon kit (Agilent Technologies). This was followed by sequencing on a HiSeq2000 (Illumina) with 100 bp paired end reads. Sequence reads were aligned to the reference genome (hg19 build) using Novoalign (Novocraft Technologies Sdn Bhd). Duplicate reads, resulting from PCR clonality or optical duplicates, and reads mapping to multiple locations were excluded from downstream analysis. Depth and breadth of sequence coverage were calculated with custom scripts and the BedTools package.
Sanger sequencing and WES variants validation
Potential variants identified by whole exome sequencing were validated by Sanger sequencing of all family members. Genomic DNA was PCR amplified and screened by DNA cycle sequencing using Big Dye Terminator v3.1 Cycle Sequencing Kit, together with an ABI 3130 Genetic Analyzer (Life Technologies, USA). Ensembl genome browser (http://www.asia.ensembl.org) was used for obtaining genomic sequence and coding exons information for these genes. Primer sequences were designed for each exon using Primer3 software [6] and checked for specificity using Basic Local Alignment Search Tool (BLAST; http://www.ncbi.nlm.nih.gov/blast). Sequence variants identified were analyzed using BioEdit software (www.mbio.ncsu.edu/bioedit.html).
Results
We recruited four affected individuals (IV-1, IV-3, IV-7, IV-8) from a consanguineous Indian family with HSP with one previous miscarriage due to unknown causes in the first trimester (Figure 1). On clinical examination, the girls showed increased muscle tone and gait impairment due to progressive bilateral lower limb spasticity. The first presentation of symptoms in all affected individuals was “toe-walking”. Although all affected individuals had lower limb spasticity, the younger sisters (IV-7, IV-8) were still able to walk unsupported in contrast to the others who were having difficulty in walking and were dependent on wheelchairs (IV-1, IV3). They also presented with severe upper limbs spasticity with rigidity, unlike other affected members in the second loop. All four affected individuals were assessed clinically as having mild to moderate mental retardation, brisk tendon reflexes and an up going extensor planter response. On regular ophthalmological assessment, there were no signs of strabismus or optic-nerve hypoplasia. No sensory deficits were detected in the young sisters, but these were difficult to be assess in the older ones. No urinary incontinence was reported in the younger sister–sister pair. This was difficult to assess in the other pair because they were on diapers. The MRI for one of the young sisters (IV-7), showed hypoplastic corpus callosum with possible agenesis of the splenium.
We genotyped ten available samples from the family using the Illumina HumanCytoSNPv12.2 chip with approximately 300,000 SNPs together with Illumina iScan platform. A common 7,454,059 base long (position 32,134,461 to 39,588,519) homozygous region at chromosome 8p11.23-8p12, including more than 55 genes, was found in all four affected individuals.
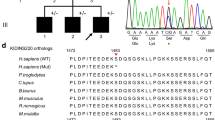
We undertook whole exome sequencing of both parents and one affected daughter. We searched the chromosome 8p region of homozygosity for non-synonymous variants or stop gain mutations. Based on the assumption that the candidate mutation is autosomal recessive, led us to concentrate on a homozygous stop gain (stop codon) mutation in the DDHD2 gene in the affected individual that was heterozygous in both parents. This mutation at position 38103270, causing a C to T substitution at c.859 (c.C859T) introduced a stop codon at amino acid position 287 (p.R287X). Initially, this variant was not found in our in-house database of over 1,000 exome sequences largely of Caucasian origin or in exome variant server (http://www.evs.gs.washington.edu/EVS/). In order to confirm this mutation and to examine its segregation within the family, Sanger sequencing was performed for all ten family members. This confirmed a homozygous transition mutation (C > T) at cDNA base position 859 in exon 8 of DDHD2 gene in all four affected individuals. The obligate carriers were heterozygous for this mutation. To further confirm that the mutation was not a common polymorphism, a panel of 192 chromosomes of ethnically matched controls were sequenced. However the variant was not identified within this panel.
Discussion
We report a large extended consanguineous family of Indian descent, with affected members of this family carrying a homozygous recessive mutation in DDHD2 (SPG54). SPG54 is characterized by psychomotor delay, cognitive impairment, progressive spasticity, early onset (before the age of 2 years), thin corpus callosum, periventricular white matter abnormalities, foot contractures, dysarthria, dysphagia, strabismus and/or optic hypoplasia. Our results confirm previously studies reporting DDHD2 mutations in SPG54, extend our clinical knowledge of this condition, give further insights into genotype-phenotype contributions, suggest a founder effect for the p.R287X mutation, and adds to growing list of lipid metabolism genes playing a role in neurodegenerative disorders.
DDHD2 (DDHD-domain-containing 2), belongs to an intracellular phospholipase A1 (iPLA1) family of proteins, (DDHD1, DDHD2 and SEC23IP) that are involved in organelle biogenesis and membrane trafficking between the endoplasmic reticulum and golgi body [7, 8]. iPLA1 family proteins encode a phospholipase that hydrolyze an acyl group and fatty acids at the sn-1ester bonds of phospholipids, and contain a DDHD domain, a WWE domain, a GxSxG lipase motif, and a sterile alpha motif (SAM). SAM and DDHD both bind phatidylinositol 4-phosphate (PI(4)P) which is important in membrane trafficking [7, 8].
Mutations in DDHD2 have been previously reported in 2 Iranian, one Dutch Filipino, one Omani, one Indian, one Canadian, and one Italian family. A wide range of mutations have been reported so far, mostly affecting the SAM and DDHD domains of DDHD2 (Figure 1) including nonsense, missense, and a small deletion mutations (Additional file 1: Table S1, Additional file 2: Table S2). Overall, these mutations result in a very similar phenotype [9–12]. However there were a few minor clinical signs which appeared different. The ages of onset in our cases were 15 and 18 months and the two patients described by Gonzalez et al. [10] were noticed at the ages of 3 and 6 years. Our patients presented severe limbs spasticity especially toe-walking in addition to mild to moderate intellectual disability as compared to those reported in the previous cases [10, 11]. Other features like hypomania, a psychiatric anomaly of extreme excitement, visual impairments like strabismus or nerve optic hypoplasia, and dysarthria, a speech articulation abnormality as reported by Schuurs-Hoeijmakers et al. [11] were not observed in our cases.
The mutation identified here in our study leads to the formation of a premature termination codon (PTC) in the open reading frame of DDHD2 mRNA at amino acid position 287. If this protein is synthesized, the mutation would cause its truncation, suggesting a loss of function mechanism associated with the mutation. Whether or not this leads to a complete or partial loss of function, that might be compensated by other members of phospholipase A-1 gene family, is not known. Other studies on DDHD2 mutations have suggested a decrease in the expression levels of DDHD2 mRNA, owing to nonsense mediated RNA decay, might also contribute to the pathogenesis [11].
The identification of pR287X mutation in two Iranian families, including one family with two affected siblings carrying this mutation, suggested a founder effect [10, 11]. However, the family we describe is of Indian origin. This raises the intriguing possibility of an ancestral mutational event common to both the Indian and Iranian families, a view supported by previous studies showing the Zoroastrians from Iran moved to India in 900 AD following the Arab invasion [13]. Alternatively, this may represent a mutation hotspot.
Conclusion
Although the precise pathogenic mechanisms involving DDHD2 are not known, mutations in genes involved in common intracellular signaling pathways involving HSP, Parkinson’s disease, amyotrophic lateral sclerosis and Alzheimer’s disease have recently been reported [5, 14] and gene knockouts in drosophila suggest a role for DDHD2 in synaptic organization and transmission [11]. The role of genes involved in lipid metabolism is of particular interest and DDHD2 joins a growing list of such genes contributing to HSP and other neurodegenerative disorders [1, 5, 9, 15].
References
Fink JK (2013) Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol 126(3):307–328
Salinas S, Proukakis C, Crosby A, Warner TT (2008) Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol 7(12):1127–1138
Rainier S, Chai JH, Tokarz D, Nicholls RD, Fink JK (2003) NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am J Hum Genet 73(4):967–971
Doi H, Ohba C, Tsurusaki Y, Miyatake S, Miyake N, Saitsu H et al (2013) Identification of a novel homozygous SPG7 mutation in a Japanese patient with spastic ataxia: making an efficient diagnosis using exome sequencing for autosomal recessive cerebellar ataxia and spastic paraplegia. Int Med Tokyo Jpn 52(14):1629–1633
Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD et al (2014) Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343(6170):506–511
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Method Mol Biol 132:365–386
Sato S, Inoue H, Kogure T, Tagaya M, Tani K (2010) Golgi-localized KIAA0725p regulates membrane trafficking from the Golgi apparatus to the plasma membrane in mammalian cells. FEBS Lett 584(21):4389–4395
Inoue H, Baba T, Sato S, Ohtsuki R, Takemori A, Watanabe T et al (2012) Roles of SAM and DDHD domains in mammalian intracellular phospholipase A1 KIAA0725p. Biochim Biophys Acta 1823:930–939
Citterio A, Arnoldi A, Panzeri E, D’Angelo MG, Filosto M, Dilena R et al (2014) Mutations in CYP2U1, DDHD2 and GBA2 genes are rare causes of complicated forms of hereditary spastic paraparesis. J Neurol 261:373–381
Gonzalez M, Nampoothiri S, Kornblum C, Oteyza AC, Walter J, Konidari I et al (2013) Mutations in phospholipase DDHD2 cause autosomal recessive hereditary spastic paraplegia (SPG54). Eur J Hum Genet EJHG 21(11):1214–1218
Schuurs-Hoeijmakers JH, Geraghty MT, Kamsteeg EJ, Ben-Salem S, de Bot ST, Nijhof B et al (2012) Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am J Hum Genet 91:1073–1081
Schuurs-Hoeijmakers JH, Vulto-van Silfhout AT, Vissers LE, van de Vondervoort II, van Bon BW, de Ligt J et al (2013) Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J Med Genet 50:802–811
Mohyuddin A, Mehdi SQ (2005) HLA analysis of the parsi (Zoroastrian) population in Pakistan. Tissue Antigens 66:691–695
Singleton AB (2014) A unified process for neurological disease. Science 343:497–498
Tesson C, Nawara M, Alih MAM, Rossingnol R, Zaki MS, Al Balwi M et al (2012) Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am J Hum Genet 97:1051–1064
Authors’ contributions
The study design was conceived by JN, NA, MJ, JYA-A and HSAM. NA, NV, SA, MS and KB contributed to the data. JN, NA, NV, MJ and HSAM contributed to writing the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Ministry of Higher Education, Royal Embassy of Saudi Arabia, Cultural Bureau, London, UK for generous financial support, and Jacob Ranson for comments on the manuscript and help with designing the Tables. This work was also funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, under Grant No. (1-287/1433 HiCi). The authors, therefore, acknowledge the DSR technical and financial support.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Table S1. List of mutations identified in DDHD2 gene causing hereditary spastic paraplegia (SPG54). Previously reported mutations known to cause the SPG54 phenotype are listed.
Additional file 2:
Table S2. Clinical comparison of three SPG54 families with pR287X mutation. The detailed clinical phenotype identified in our family is compared with two previously reported families with the same mutation in DDHD2.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Alrayes, N., Mohamoud, H.S.A., Jelani, M. et al. Truncating mutation in intracellular phospholipase A1 gene (DDHD2) in hereditary spastic paraplegia with intellectual disability (SPG54). BMC Res Notes 8, 271 (2015). https://doi.org/10.1186/s13104-015-1227-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-015-1227-4