
Abstract
Background
Emerging evidence suggests that gut microbiota plays a predominant role in Crohn’s disease (CD). However, the microbiome alterations in the early stage of CD patients still remain unclear. The present study aimed to identify dysbacteriosis in patients with early CD and explore specific gut bacteria related to the progression of CD.
Methods
This study was nested within a longitudinal prospective Chinese CD cohort, and it included 18 early CD patients, 22 advanced CD patients and 30 healthy controls. The microbiota communities were investigated using high-throughput Illumina HiSeq sequencing targeting the V3–V4 region of 16S ribosomal DNA (rDNA) gene. The relationship between the gut microbiota and clinical characteristics of CD was analyzed.
Results
Differential microbiota compositions were observed in CD samples (including early and advanced CD samples) and healthy controls samples. Notably, Lachnospiracea_incertae_sedis and Parabacteroides were enriched in the early CD patients, Escherichia/Shigella, Enterococcus and Proteus were enriched in the advanced CD patients, and Roseburia, Gemmiger, Coprococcus, Ruminococcus 2, Butyricicoccus, Dorea, Fusicatenibacter, Anaerostipes, Clostridium IV were enriched in the healthy controls [LDA score (log10) > 2]. Furthermore, Kruskal–Wallis Rank sum test results showed that Blautia, Clostridium IV, Coprococcus, Dorea, Fusicatenibacter continued to significantly decrease in early and advanced CD patients, and Escherichia/Shigella and Proteus continued to significantly increase compared with healthy controls (P < 0.05). The PICRUSt analysis identified 16 remarkably different metabolic pathways [LDA score (log10) > 2]. Some genera were significantly correlated with various clinical parameters, such as fecal calprotectin, erythrocyte sedimentation rate, C-reactive protein, gland reduce, goblet cells decreased, clinical symptoms (P < 0.05).
Conclusions
Dysbacteriosis occurs in the early stage of CD and is associated with the progression of CD. This data provides a foundation that furthers the understanding of the role of gut microbiota in CD’s pathogenesis.
Similar content being viewed by others


Background
Crohn’s disease (CD) is a chronic and relapsing gastrointestinal inflammatory disease associated with a high risk of disability [1, 2]. Its incidence has been rapidly increasing worldwide, especially in newly industrialized countries, causing a heavy health economic burden [3]. However, the course of CD is difficult to predict, as it varies from patient to patient. Also, the characteristics of this disease in early stage and the factors influencing the progression of the disease are poorly understood, significantly affecting the clinical decision-making [4]. Further investigations of the influencing factors may provide evidence for CD management, especially in the early treatment, and improves the outcomes of disease [4, 5].
The pathogenesis of CD has not been thoroughly studied. Multiple factors, such as genetics, diet, dysbiosis of gut microbiota and overactivated immune response, are involved in the onset of CD [1, 4, 5]. Among these factors, gut microbiota dysbiosis has been identified as the key factor in the pathogenesis of CD and has been observed in CD patients [6]. Generally, the gut microbiota of healthy adults consists mainly of phyla Firmicutes, Bacteroidetes, and Actinobacteria. In addition, small amounts of Proteobacteria, Verrucomicrobia, Euryarchaeota, and Fusobacteria were found in human fecal samples [7]. Compared to healthy subjects, most studies of CD patients have reported decreased bacterial diversity with an reduction of protective gut microbiota, such as Firmicutes (e.g., butyrate-producing bacteria Faecalibacterium, Roseburia, Oscillibacter and Coprococcus, etc.), Actinobacteria (e.g., Bifidobacterium) and Verrucomicrobia (e.g., the mucolytic bacteria Akkermansia muciniphila), combined with an expansion of putative inflammatory groups, such as Escherichia coli (phylum Proteobacteria), Fusobacterium spp. (phylum Fusobacteria), and the species Ruminococcus gnavus (producer of an inflammatory polysaccharide) [7,8,9,10]. As far as we know, many studies have focused on the characteristics of changes in the gut microbiota of CD patients and its predictive or monitoring value in response to CD treatment [11, 12], postoperative recurrence [13, 14], disease activity [15,16,17,18,19], etc. Moreover, studies have also found that the gut microbiota can be used as a biomarker to distinguish CD and non-CD [20]. However, whether the gut microbiota is a cause or a consequence remains unclear, and the relationship between the gut microbiota and the course of CD is still not clearly understood [7, 10, 21]. Perhaps the best way to capture these clues is to investigate the characteristics of gut microbiota changes during the biologic onset or early clinical stage of CD [4].
Although the Paris’s definition of early CD aimed at optimizing CD treatment strategies was proposed in 2012 [22], understanding of the characteristics of gut microbiota changes at this stage is still insufficient, especially in Chinese cohort. More relevant research could help to improve the understanding of CD disease course stage and disease progression and provide evidence for developing early treatment methods targeting the gut microbiota. Therefore, the objective of this study was to analyze the biodiversity of gut microbiota in early-stage and advanced-stage CD patients and healthy controls through high-throughput Illumina HiSeq sequencing targeting the V3–V4 region of 16S ribosomal DNA (rDNA) gene and to compare the differences in the composition of gut microbiota between groups. In addition, we also aimed to identify the dysbacteriosis in the patients with early CD, and find out the specific gut bacteria related to the progression of CD, thus providing new insight for the pathogenesis and early course of CD and prompting the development of clinical prevention, diagnosis and treatment targeting gut microbiota.
Results
Patient characteristics and sequencing information
A total of 40 patients with CD were enrolled between December 2018 and October 2021. Groups were matched for age and gender. The mean age of early CD group (EG) was 35.5, and 36.7 years of advanced CD group (AG). Patients with CD but with no CD-related surgical history were all in the active stage [Disease Activity Index of CD (CDAI > 150)]. The detailed clinical features of patients in EG and AG were shown in Table 1.
A total of 2,457,406 clean reads were obtained, with an average of 35,614 (29,200–38,965) effective sequences per sample, where the total number of base pairs was 1,051,888,436 bp, and the average sequence length was 414 (400–440) bp. The filtered sequences were clustered at a similarity of 0.97, and the number of OTUs was 9471 with an average of 136 per sample. OTU Venn diagram analysis showed that the OUT numbers of the healthy control groups (CG), EG, and AG were 507, 394, and 459, respectively. Among them, the unique OTU numbers of each group were 81, 11, and 48 (Additional file 1). The Rank abundance curve showed reasonable richness and homogeneity of the species composition of each group (Additional file 1).
Early CD patients has an altered gut microbiome
Early CD patients has unique microbial community characteristics
When comparing bacterial alpha diversity between EG, AG and CG, including community richness (observed species, chao1) and diversity (Shannon and Simpson), we found an overall difference in each diversity index (Fig. 1). Significant differences (P < 0.05) with respect to community richness (observed species) were observed both between EG and CG and between AG and CG. The chao 1 index of was significant differences (P < 0.05) between AG and CG, but the difference was not statistically significant (P > 0.05) between EG and CG. Moreover, the pattern of richness was found to be similar in EG and AG. When considering the species diversity of microbiota (Shannon and Simpson), the differences between AG and CG was statistically significant (P < 0.05). However, the differences between the remaining groups were not statistically significant.

Alpha diversity index box plot. The community richness between early CD groups, advanced CD groups and control groups was showed as A (observed species diversity index) and B (chao1 index) and the species diversity of microbiota was showed as C (Shannon diversity) and D (Simpson diversity index). The horizontal axis represents the sample grouping, and the vertical axis represents the Alpha diversity index value of different groups. CG control group, EG early group of CD patients, AG advanced group of CD patients
Early CD patients has unique microbial community structure
We used principal component analysis (PCoA) to investigate the community structure of microbiota in EG, AG and CG (Fig. 2). We found that samples tended to cluster together based on disease; however, to a certain extent, there was an overlap between all groups (Fig. 2A). CD samples were mostly distinct from those of normal controls, which indicated differences with respect to community structure of the microbiota between CD and controls (Anosim: EG vs CG, R = 0.146, P = 0.008; AG vs CG, R = 0.186, P = 0.001). However, samples of EG and AG were located closely, which suggested a similar bacterial community structure in the context of both early CD and advanced CD (Anosim: R = − 0.033, P = 0.866) (Fig. 2B). Our results indicated that the bacterial community structure in CD was different from that in controls; however, there was no difference with respect to the alterations of bacterial community structure in fecal samples of the early and advanced CD patients.

Microbial community structure in early CD groups (EG), advanced CD groups (AG) and control groups (CG). A UniFrac heatmap analysis. Patient Group (PG), including EG and AG. And CG means control group. B Principal component analysis (PCoA). The percentage represents the contribution rate of the principal dimension to the sample difference

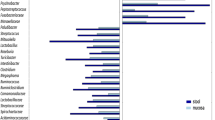
Species classification and abundance analysis of CD patients and controls. A The phylum bar graph of the intestinal flora of each group; B The genus bar graph of the intestinal flora of each group. CG control group, EG early group of CD patients, AG advanced group of CD patients
Species classification and abundance analysis of CD patients and controls
We also analyzed the relative abundance of microbes in the gut microbiome between CD patients and controls at the phylum level and genus level. At the phylum classification level, Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria accounted for more than 97% of the relative abundance of each group at the phylum classification level. Compared with CG, the abundances of Bacteroidetes increased both in EG and AG, while the abundances of Firmicutes decreased. However, no difference was observed between early and advanced CD at phylum level (Fig. 3A). At the level of genus classification, compared with the controls, the abundance of Bacteroides and Parabacteroides both increased in EG, while the abundance of Gemmiger and Dialister both decreased in EG; the abundance of Roseburia, Gemmiger and Lachnospiracea_Incertae_sedis decreased in AG. Compared with the EG, the abundance of Roseburia and Lachnospiracea_Incertae_sedis in AG both decreased (Fig. 3B).
Early CD patients harbored unique bacterial biomarkers
Next, we used LEfSe differential analysis to analyze the differences in gut microbial community structure between early and advanced stages of CD. The CG had a high proportion of genera Roseburia, Gemmiger, Coprococcus, Ruminococcus 2, Butyricicoccus, Dorea, Fusicatenibacter, Anaerostipes, Clostridium IV. A high proportion of genera Lachnospiracea_incertae_sedis and Parabacteroides was observed in EG, and a high proportion of genera Escherichia/Shigella, Enterococcus and Proteus in the advanced stage (Fig. 4A and B, LDA Score (log10) > 2).

Significant difference analysis of CD patients and controls. A and B LEfSe analysis. The figure lists bacterial communities with LDA score (log 10) > 3 and P < 0.05. p, phylum; c, class; o, order; f, family; g, genus. C Rank sum test analysis between groups (P < 0.05). At the genus level, there are a total of 17 differential bacterial genera, including: Anaerostipes, Bacteroides, Blautia, Butyricicoccus, Clostridium IV, Coprococcus, Dorea, Enterococcus, Escherichia/Shigella,Fusicatenibacter, Gemmiger, Lachnospiracea_incertae_sedis, Parabacteroides, Proteus, Pseudomonas, Roseburia, Ruminococcus 2. In particular, the abundance sum of Proteus and Pseudomonas in at least one group is 0, which cannot be shown by Boxplot. D Spearman correlation analysis of genera with significant differences among groups. E Spearman correlation heat map analysis between clinical markers and characteristic bacteria. CG control group, EG early group of CD patients, AG advanced group of CD patients
We further analyzed the KRUSkal–Wallis Rank sum test and found 17 genera (Anaerostipes, Bacteroides, Blautia, Butyricicoccus, Clostridium IV, Coprococcus, Dorea, Enterococcus, Escherichia/Shigella, Fusicatenibacter, Gemmiger, Lachnospiracea_incertae_sedis, Parabacteroides, Proteus, Pseudomonas, Roseburia, Ruminococcus 2) with significant differences among different groups (P < 0.05). Compared with CG, the abundance of Bacteroides, Enterococcus, Escherichia/Shigella, Parabacteroides, Proteus in EG was significantly increased (P < 0.05), while the abundance of Butyricicoccus, Blautia, Ruminococcus 2, Dorea, Anaerostipes, Coprococcus, Clostridium IV, Fusicatenibacter, Gemmiger was significantly decreased (P < 0.05). Compared with the EG, the abundance of Escherichia/Shigella and Proteus in AG was significantly increased (P < 0.05), while Roseburia, Lachnospiracea_incertae_sedis, Fusicatenibacter, Coprococcus, Blautia, Clostridium IV and Anaerostipes were significantly decreased (P < 0.05) (Fig. 4C).
In order to further clarify the dynamic relationship between the symbiotic flora and opportunistic pathogens, the Spearman correlation heat map was used to show the important patterns and relationships among characteristic flora. Enterococcus and Escherichia/Shigella were positively correlated with each other, while these were negatively correlated with other genus (Fig. 4D). Moreover, we exported the clinical association between the microbiota and CD characteristics [gland reduce, fecal calprotectin (FC), goblet cells decreased, sex, crypt structure changes, C-reactive protein (CRP), anti-saccharomyces cerevisiae antibody (ASCA)/perinuclear antineutrophil cytoplasmic antibody (p-ANCA), erythrocyte sedimentation rate (ESR), platelet, age, clinical symptoms (abdominal pain and diarrhea)] using Spearman correlation heat map analysis, finding a significant negative correlation between Dorea, Butyricicoccus, Roseburia and gland reduce, Roseburia and FC, Butyricicoccus and goblet cells decreased, Parabacterodies and sex, Clostridium IV and CRP, Dorea and ESR and a significant positive correlation between Escherichia/Shigella and CRP, Ruminococcus 2 and clinical symptoms (Fig. 4E).
KEGG pathways analysis gut microbiome of early CD patients
The phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) method [23, 24] was used to predict the KEGG (Kyoto Encyclopedia of Genes and Genomes database) pathways between the microbiome of CD patients and healthy subjects. A total 113 significantly different KEGG orthologues (KOs) were detected in the microbiome of these groups [LDA Score (log10) > 2, P < 0.05, Wilcoxon rank-sum test analysis, data not shown]. Among them, there were 29 KOs enriched in early stage and 19 in advanced stage. In the level 2 of KEGG, 16 of significant pathways were identified. The high functions of EG were related to the pathways of Energy Metabolism, Folding Sorting and Degradation, Metabolism of Other Amino Acids, Digestive System and Transport and Catabolism, while the high functions of microbial genes in CG were related to the pathways of Membrane Transport, Cell Motility, Environmental Adaptation and Cell Growth and Death and the high functions of AG were related to the pathways of Glycan Biosynthesis and Metabolism, Cellular Processes and Signaling, Neurodegenerative Diseases, Xenobiotics Biodegradation and Metabolism, Signaling Molecules and Interaction, Poorly Characterized and Infectious Diseases (Fig. 5). In level 3 of KEGG, 50 of significant pathways were identified (Additional file 1). Notably, the microbial gene function related to Folate biosynthesis, Thiamine metabolism, Lysosome and Peroxisome was characteristically increased in the early CD patients.

Functional predictions of microbiota present in the fecal of CD patients and healthy controls. Significant KEGG pathways of Level 2 for the microbiome of the CD and healthy groups was identified. PICRUSt Phylogenetic Investigation of Communities by Reconstruction of Unobserved States
Discussion
The balance between beneficial gut commensals and pathogens is crucial to human health. The dysbiosis of gut microbiota contributes to gut inflammation and may be closely related to the onset and progression of CD [4, 5, 7]. However, our understanding of the relationship between gut microbiota and CD is still relatively poor as it is difficult to detect and diagnose CD in the biologic onset or early stage of disease (e.g., preclinical) [5, 25]. In the present study, we explored the characteristics of gut microbiota changes in the early CD of Paris’s definition and advanced CD [22], and found that significant gut microbiota dysbiosis with unique bacterial biomarkers and metabolic pathway changes in the early stage of Chinese CD patients. The observed dysbiosis was associated with disease progression.
Our study revealed that the dysbiosis in early CD mainly manifested in the following three aspects: first, compared with the health subjects, the fecal microflora community abundance in patients with early CD decreased, however, the observed difference was not statistically significant, while that of patients with advanced CD showed a significant decrease in microbial diversity. Secondly, PCoA results could distinguish the bacterial community structure of patients with early CD from that of the health subjects with statistically significant differences. Finally, the abundance and structure of microbiomes of early and advanced CD were similar. Previous studies have shown that fecal microbiota diversity in western and Chinese patients with CD decreased compared with health subjects [19, 20, 26]. However, the gut microbiota of patients with inactive CD and those with active CD were similar in structure and could not be distinguished by PCoA [26]. Furthermore, altered microbiome composition and stability in CD were not associated with disease activity or long-term course [19]. These inconsistencies may be due to the differences in study design, disease stage, drug use, diet, etc., but the reasons for those conditions are not fully understood, and further research is needed.
We detected detailed compositional alterations in the fecal microbiota of patients with early-stage CD at different taxonomic levels and found a significant reduction in multiple short-chain fatty acid (SCFA)-producing bacteria, including Blautia, Clostridium IV, Coprococcus, Dorea, Fusicatenibacter, and a significant increase in Escherichia/Shigella, and Proteus. This trend of gut microbiota imbalance was more obvious in the advanced stage of CD, indicating that gut microbiota imbalance was closely related to the progression of CD inflammation. Although these results were similar to those of previous studies [14, 26,27,28], these studies do not reflect the role of some key microbiota (e.g., Parabacteroides, Escherichia/Shigella) in the early course and progression of CD. The present study overcame this deficiency and provides clues for further researches on the mechanism of gut microbiota–host interaction in CD patients.
In the present study, we found that Bacteroides increased in CD patients and were mainly enriched in early CD, which could cause opportunistic infections when immune dysfunction or intestinal flora was imbalanced. Previous studies indicated that the changes in the abundance of Bacteroides in CD patients were controversial. The increased abundance of Bacteroides in the CD group compared with health participates was related to the maintenance of postoperative remission [4, 29]. However, other studies provided that the abundance of Bacteroides in CD patients was reduced [27, 30]; B. thetaiotaomicron, belonging to the Bacteroides was found to prevent weight loss, colonic histopathological changes and the production of inflammatory factors in a mouse enteritis model induced by dextran sodium sulfate (DSS) [31]. Therefore, more research is needed to explore and explain the potential role of Bacteroides changes in CD patients, especially in the early stages.
In the present study, Parabacteroides were enriched in CD patients, especially patients with early CD. This result was consistent with previous studies, which have shown that Parabacteroides had an important role in the pathogenesis of intestinal inflammation, and the decreased abundance of Parabacteroides in CD patients was related to the reduction of inflammation [12, 32, 33]. Some studies have shown that certain Parabacteroides may aggravate the inflammatory response [34,35,36]. Oral administration of P. distasonis to mice with DSS-induced colitis could significantly aggravate the inflammation [35]. The strain of Parabacteroides distasonis, known as CAVFT-HAR46, isolated from microlesions of cavernous fistulas in the intestinal wall of patients with CD, may be the potential pathogenic cause of CD [35]. In addition, Parabacteroides had an important role in the pathogenesis of intestinal inflammation; the number of Parabacteroides in children with CD under high-level pressure stress was found to increases significantly, suggesting they were related to CD inflammation [36]. However, it remained unclear whether the changes in the abundance of Parabacteroides were the cause or the result of intensified or reduced intestinal inflammation.
In addition, our results showed that Enterococcus were also enriched in the feces of CD patients, indicating it may be play a key role in the disease course of CD. Enterococcus, which can produce extracellular peroxide and damage the DNA of mucosal epithelial cells, is an opportunistic human bacterial pathogen [37]. Previous studies have shown that the abundance of Enterococcus in children with CD was significantly increased [38, 39], which was closely related to postoperative recurrence in adult CD patients and was conducive to the fermentation of proteolysis and lactic acid production, while the proteolytic flora was associated with the accumulation of end products known to be toxic to colon cells [40]. In addition, the abundance of Enterococcus faecalis in the faeces of CD patients was related to the location of the lesion. Usually, the abundance of Enterococcus faecalis in patients with ileal CD is higher than that in patients with ileocolon type [20]. Therefore, we hypothesized that Enterococcus may be involved in impairing the intestinal mucosal barrier at the early stage of CD through its secretion or its metabolites, promoting the occurrence of inflammation.
Noteworthy, Escherichia/Shigella and Proteus were enriched in patients with advanced CD compared with the early patients in the present study, suggesting that these bacteria may be the key factors in the progression of the disease. Escherichia/Shigella, a gram-negative bacillus, which could spread between intestinal mucosal cells and eventually colonize, protecting itself from the destruction of innate immunity in the intestine, could cause inflammatory destruction of the intestinal epithelial barrier and lead to the apoptosis of macrophage [20, 41, 42]. Previous studies [20, 27, 43, 44] have also found that Escherichia was enriched in CD patients, and some Escherichia coli strains with adhesion and invasiveness increased in the ileal mucosa of CD patients, which indicated that Escherichia was related to the pathogenesis of CD. Moreover, a cross-sectional study of a large sample showed that Escherichia could be used as a landmark to distinguish CD from non-CD [20]. Other studies also found that Proteus was related to the severity of colitis in mice [45]. Therefore, Escherichia/Shigella and Proteus have great significance for understanding the progression and prognosis of CD; however, further research is needed. Our research finding and the above reports indicated that opportunistic pathogens such as Parabacteroides, Bacteroides, Enterococcus, Escherichia/Shigella and Proteus, which dynamically which changed in the natural course of CD, might play a key role in the occurrence and development of CD. The opportunistic pathogens are expected to become the microbial biomarkers for early CD diagnosis.
The gut microbiota in the early course of CD is characterized not only by the increased opportunistic pathogens but also by the decreased abundance of some common beneficial bacteria, such as SCFA-producing bacteria, which is consistent with the results of previous studies [46]. However, the present study also confirmed that the dynamic reduction of beneficial bacteria in gut microbiota was related to the process of intestinal inflammation and could be used as the potential marker to predict the evolution of CD. The abundance of Firmicutes in early CD patients decreased significantly compared with the controls, mainly due to the significant decrease in genera Coprococcus, Ruminococcus 2, Butyricicoccus, Dorea, Clostridium IV, etc. This decreasing trend was even more pronounced in patients with advanced CD, especially for Roseburia, Lachnospiracea_incertae_sedis, Fusobacterium, Blautia, and Anaerostipes, which in advanced CD decreased more significantly compared with patients in early CDs. Previous studies have shown that these bacteria had an important role in maintaining the dynamic balance of intestinal mucosal immune regulation through their metabolites [7, 10, 47]. Coprococcus not only produces butyrate but is also related to the dopamine metabolic pathway, and dopamine is a key brain signal in the pathogenesis of depression, which may explain the higher depression status in CD patients compared to normal people [48]. Moreover, recent studies have pointed to changes in gut microbiota composition and host processing of bacterial-derived metabolites associated with CD, particularly a reduction in the taxa of Roseburia, Lachnospiraceae, and Ruminococcus 2, etc. [49]. We also found the dynamics of characteristic flora using Spearman correlation heat maps, and our results showed that there was some unknown interaction between these characteristic microbiotas, but further studies are needed to confirm this interaction mode of microbiota in order to deeply understand the dynamic changes of intestinal microbiota in CD patients, and to provide a basis for the regulation of intestinal microbiota. Moreover, these characteristic bacteria were correlated with CRP, FC and ESR etc., which reflect the inflammatory state of CD, further suggesting that the gut microbiota dysbiosis may have cascade amplification in CD, thereby promoting the progression of CD and providing clues for exploring the application of existing CD-related biomarkers and intestinal micro-flora in the early diagnosis of CD in future. Taken together, our results further improve insights into the mechanism of gut microbiota in CD, considering that gut microbiota dysbiosis may be more reversible in the early stage of CD than in the progressed patients. Therefore, targeting intestinal microbiota in the early stage of CD may be more meaningful.
Although many bacteria and metabolites associated with CD have been identified, understanding the mechanisms through which microbes influence the occurrence and development of CD requires an extension from association to causation, and the functional analysis of gut microbiota provides a perspective. Functional analysis which has unique significance in the differentiation between groups is often more important than species composition analysis in biological value. PICRUSt (Phylogenetic Investigation of Communities by reconstruction of unobserved States) studies community phyloevolution through recessive state reconstruction, the software predicts metagenomic functional composition based on 16S rDNA and reference sequence databases [23, 24]. By comparing the results of functional analysis of metagenomic sequencing data and corresponding 16S predicted functional analysis, it is found that the accuracy of this method is 84–95%, and the functional analysis of intestinal microbiota and soil microbiota is close to 95%, which can greatly reflect the functional gene composition in the sample [24]. Our PICRUST results suggested that an imbalance in gut microbiota was involved in the progression of CD by changing their gene function, which could provide important reference information for the study of downstream microbiota interaction/response mechanism.
There were some limitations in this study. First, this was a cross-sectional study, but it was nested within a longitudinal prospective Chinese CD cohort [50], which to some extent eliminates possible confounding factors to further understand and validate the dynamic changes of gut microbiota during natural CD processes, and to develop suitable microbial biomarkers for early diagnosis. However, it should be pointed out that the Paris’s definition of early CD is a disease stage defined to optimize treatment strategies [22], which can only partly reflect the early stage of the disease course. Therefore, it is necessary to capture CD patients at an earlier stage or preclinical or even biological stage to truly reveal the natural history of CD through prospective follow-up cohort studies, and establish a biobank to disclose the dynamic changes and roles of gut microbiota in the pathogenesis and disease progression of CD. Second, the sample size of the current study was relatively small, further large-sample multi-omics studies were needed to clarify the potential mechanisms and pathways of gut microbiota imbalance involved in the occurrence and development of CD in order to provide a basis for updating the prevention and treatment strategies of CD. Third, this study only analyzed the fecal flora which was similar to the mucosal associated flora of patients with CD [8]. More tests for analyzing the intestinal mucosal associated flora are needed to further elucidate the role of gut microbiota in the biologic onset or early clinical stage of CD. Furthermore, despite the widespread use of the 16s rDNA sequencing method, it was often challenged that use of different sequence processing pipelines may bring ambiguous results, such as different alignment methods, OTU binning procedures, different kits used to extract DNA, the 16S rDNA regions amplified, and reference databases, etc. [51]. Recently, discussions of standards and pipelines have helped researchers improve the data quality, such as the standard for human fecal sample processing [52], normalization strategies for data characteristics [53] and the future use of metagenomic sequencing as a replacement. In this study, we have used the state-of-the-art pipeline to ensure the reliable interpretation of the 16S sequencing data, and we also look forward to more remarkable finding in our future studies with metagenomic sequencing and integrated multi-omics strategies.
Despite these limitations, the present study demonstrated that the progressive consumption of SCFAs producing bacteria during the course of CD significantly affects the functional metabolism, which is not conducive to the maintenance of intestinal epithelial integrity and the regulation of inflammatory response, and may be a potential factor of etiology in the early stage of CD. In addition, the progressive rise of opportunistic pathogens exacerbates gut dysbiosis during CD, which in turn affects disease progression.
Conclusions
Our study reported that the gut microbiota of patients with early-stage CD is characterized by a significantly decreased of SCFA-producing bacteria, including Blautia, Clostridium IV, Coprococcus, Dorea, Fusicatenibacter and a significantly increased of Escherichia/Shigella and Proteus compared to healthy controls. This trend was particularly evident with the progression of the disease, and Escherichia/Shigella may be the key genera in CD disease progression; however, this should be further studied.
Methods
Subject recruitment
This cross-sectional study was nested within a longitudinal prospective Chinese CD cohort at the Seventh Medical Center of Chinese PLA General Hospital (Clinical Trials: ChiCTR-1900022728) and was conducted in accordance with the principles of good clinical practice. As reported in our previous study, we achieved early diagnosis of CD by prospectively following up patients with unexplained ileocecal inflammatory lesions for a median of 27 months [50]. These subjects were all from early-diagnosed CD patients (December 2018 to October 2021). The diagnosis of CD is based on standard clinical, endoscopic, radiological and histological criteria [54]. Patients were divided into the early group (EG) and the advanced group (AG) according to the consensus of international experts in Paris [22]. Inclusion criteria included: age 18–75 years; meeting the diagnostic criteria for CD [54]; the patients of EG without complications were all newly diagnosed cases and meet the Paris expert consensus [22]; the prebiotics, probiotics, antibiotics, glucocorticoids, immunosuppressants, biological agents, etc. has not been used in all participates for at least 2 months. Exclusion criteria included: Patients with other immune-related diseases such as ankylosing, spondylitis and psoriasis; Patients with other basic diseases such as diabetes, hypertension and cardiovascular diseases; Patients with suspected of infectious enteritis by stool culture or other etiological examination. Thirty healthy people who received physical examination in our hospital during the same period were included.
The study was approved by the Ethics Committee of the Seventh Medical Center of the Chinese PLA General Hospital (#2016-45, #2017-46), and informed consent was obtained from all participates. The sequencing data have uploaded to the GenBank (Bio project ID: PRJNA784251, http://www.ncbi.nlm.nih.gov/bioproject/784251). All authors had access to the study data and have reviewed and approved the manuscript.
Sample collection and DNA extraction
Fresh fecal samples were collected from the recruited subjects and then transported to the laboratory with an ice pack within 2 h. All samples were frozen immediately and stored at – 80 °C for further analyses. DNA was extracted from fecal samples using based on the Manual of QIAamp Fast DNA Stool Mini Kit (Qiagen, Germany).
16S rDNA gene sequencing
The V3–V4 region of the bacteria 16S ribosomal RNA genes was amplified by PCR reaction (95 °C for 3 min, followed by 30 cycles at 98 °C for 20 s, 58 °C for 15 s, and 72 °C for 20 s and a final extension at 72 °C for 5 min) using barcoded primers: 341F 5′-CCTACGGGRSGCAGCAG-3′ and 806R 5′-GGACTACVVGGGTATCTAATC-3′. A total of 30 uL mixture containing 15 uL of 2× KAPA Library Amplification ReadyMix, 1 uL of each primer (10 uM), 50 ng of template DNA, and ddH20 in each sample were performed using PCR reactions.
DNA extraction and amplification were performed on the negative controls consisting of an empty sterile storage tube, using the same procedures and reagents as the fecal sample. There was no detectable amplification in the negative controls. Amplicons were extracted from 2% agarose gels, purified using the AxyPrep DNA GelExtraction Kit (Axygen Biosciences, Union City, CA, U.S.) according to the manufacturer’s instructions and quantified using Qubit 2.0 (Invitrogen, U.S.). All quantified amplicons were pooled to equalize the concentrations for sequencing using Illumina MiSeq/HiSeq (Illumina, Inc., CA, USA). The paired end reads of 250 bp were overlapped on their 3 ends for concatenation into original longer tags by using PANDAseq (https://github.com/neufeld/pandaseq, version 2.9). DNA extraction, library construction and sequencing technology were conducted in Realbio Genomics Institute (Shanghai, China).
Process of sequencing data
Assembled tags, trimmed of barcodes and primers were further checked to clarify their rest lengths and average base quality. 16S tags were restricted between 220 and 500 bp in order to restrict the average Phred score of bases more than 20(Q20) and less than 3 ambiguous N. The copy number of tags was enumerated and redundancy of repeated tags was removed. Only the tags with frequency > 1, which indicated to be more reliable, was clustered into Operational Taxonomic Units (OTUs). OTUs were clustered with 97% similarity using UPARSE (http://drive5.com/uparse/) and chimeric sequences were identified and removed using Usearch (version 7.0.1090). Each representative tags were assigned to a taxon by RDP Classifier (http://rdp.cme.msu.edu/) according to the RDP database (http://rdp.cme.msu.edu/) using a confidence threshold of 0.8. OTU profiling table and alpha diversity analyses were also achieved by python scripts of QIIME (version 1.9.1).
Statistical analysis
Data of clinical characteristics are expressed as the mean ± standard deviation (SD). Categorical data is represented by the number of cases and percentage. These analyses were performed using the SPSS 26.0 software (IBM Corporation, Armonk, NY).
Photorefractive curves were created using R software to ensure sufficient sequencing depth for each sample. To assess the significance of differences between groups, the relative abundance of each bacterial group in R-3.2.3 (http://cran.r-project.org) was tested by Chaol index, Simpson index and Shannon index, all used for Alpha diversity. Weighted UniFrac and Principal Co-ordinates Analysis (PCoA) were used to analyze differences in beta diversity. Wilcoxon rank sum test was performed using SPSS 26.0 software to identify the top 20 differentially abundant species in the early CD group and the advanced CD group. The rank sum test was used for continuous variables to analyze factors such as clinical indicators; the χ2 test was used for categorical variables; and Fisher’s exact test was used for discrete variables. P < 0.05 was considered statistically significant.
Availability of data and materials
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Abbreviations
- CD:
-
Crohn’s disease
- EG:
-
Early CD group
- AG:
-
Advanced CD group
- CG:
-
Healthy controls group
- rDNA:
-
Ribosomal DNA
- CDAI:
-
Disease Activity Index of CD
- PCoA:
-
Principal component analysis
- PICRUSt:
-
Phylogenetic investigation of communities by reconstruction of unobserved states
- FC:
-
Fecal calprotectin
- CRP:
-
C-reactive protein
- ASCA:
-
Anti-saccharomyces cerevisiae antibody
- p-ANCA:
-
Perinuclear antineutrophil cytoplasmic antibody
- ESR:
-
Erythrocyte sedimentation rate
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes database
- KOs:
-
KEGG orthologues
- SCFA:
-
Short-chain fatty acid
- SD:
-
Standard deviation
- DSS:
-
Dextran sodium sulfate
References
Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1785–94. https://doi.org/10.1053/j.gastro.2011.01.055.
Ramadas AV, Gunesh S, Thomas GA, Williams GT, Hawthorne AB. Natural history of Crohn’s disease in a population-based cohort from Cardiff (1986–2003): a study of changes in medical treatment and surgical resection rates. Gut. 2010;59(9):1200–6. https://doi.org/10.1136/gut.2009.202101.
Ng SC, Kaplan GG, Tang W, Banerjee R, Adigopula B, Underwood FE, et al. Population density and risk of inflammatory bowel disease: a prospective population-based study in 13 countries or regions in Asia-Pacific. Am J Gastroenterol. 2019;114(1):107–15. https://doi.org/10.1038/s41395-018-0233-2.
Sorrentino D, Nguyen VQ, Chitnavis MV. Capturing the biologic onset of inflammatory bowel diseases: impact on translational and clinical science. Cells. 2019;8(6):548. https://doi.org/10.3390/cells8060548.
Torres J, Burisch J, Riddle M, Dubinsky M, Colombel JF. Preclinical disease and preventive strategies in IBD: perspectives, challenges and opportunities. Gut. 2016;65(7):1061–9. https://doi.org/10.1136/gutjnl-2016-311785.
Sartor RB, Wu GD. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology. 2017;152(2):327-39.e4. https://doi.org/10.1053/j.gastro.2016.10.012.
Simões CD, Maganinho M, Sousa AS. FODMAPs, inflammatory bowel disease and gut microbiota: updated overview on the current evidence. Eur J Nutr. 2022;61(3):1187–98. https://doi.org/10.1007/s00394-021-02755-1.
Pittayanon R, Lau JT, Leontiadis GI, Tse F, Yuan Y, Surette M, et al. Differences in gut microbiota in patients with vs without inflammatory bowel diseases: a systematic review. Gastroenterology. 2019. https://doi.org/10.1053/j.gastro.2019.11.294.
Alam MT, Amos G, Murphy A, Murch S, Wellington E, Arasaradnam RP. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020;12:1. https://doi.org/10.1186/s13099-019-0341-6.
Lee M, Chang EB. Inflammatory Bowel Diseases (IBD) and the microbiome-searching the crime scene for clues. Gastroenterology. 2021;160(2):524–37. https://doi.org/10.1053/j.gastro.2020.09.056.
Rajca S, Grondin V, Louis E, Vernier-Massouille G, Grimaud JC, Bouhnik Y, et al. Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn’s disease. Inflamm Bowel Dis. 2014;20(6):978–86. https://doi.org/10.1097/MIB.0000000000000036.
Schäffler H, Herlemann DP, Klinitzke P, Berlin P, Kreikemeyer B, Jaster R, et al. Vitamin D administration leads to a shift of the intestinal bacterial composition in Crohn’s disease patients, but not in healthy controls. J Dig Dis. 2018;19(4):225–34. https://doi.org/10.1111/1751-2980.12591.
Dey N, Soergel DA, Repo S, Brenner SE. Association of gut microbiota with post-operative clinical course in Crohn’s disease. BMC Gastroenterol. 2013;13:131. https://doi.org/10.1186/1471-230X-13-131.
De Cruz P, Kang S, Wagner J, Buckley M, Sim WH, Prideaux L, et al. Association between specific mucosa-associated microbiota in Crohn’s disease at the time of resection and subsequent disease recurrence: a pilot study. J Gastroenterol Hepatol. 2015;30(2):268–78. https://doi.org/10.1111/jgh.12694.
Seksik P, Rigottier-Gois L, Gramet G, Sutren M, Pochart P, Marteau P, et al. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut. 2003;52(2):237–42. https://doi.org/10.1136/gut.52.2.237.
Scanlan PD, Shanahan F, O’Mahony C, Marchesi JR. Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in Crohn’s disease. J Clin Microbiol. 2006;44(11):3980–8. https://doi.org/10.1128/JCM.00312-06.
Andoh A, Kobayashi T, Kuzuoka H, Tsujikawa T, Suzuki Y, Hirai F, et al. Characterization of gut microbiota profiles by disease activity in patients with Crohn’s disease using data mining analysis of terminal restriction fragment length polymorphisms. Biomed Rep. 2014;2(3):370–3. https://doi.org/10.3892/br.2014.252.
Wills ES, Jonkers DM, Savelkoul PH, Masclee AA, Pierik MJ, Penders J. Fecal microbial composition of ulcerative colitis and Crohn’s disease patients in remission and subsequent exacerbation. PLoS ONE. 2014;9(3): e90981. https://doi.org/10.1371/journal.pone.0090981.
Galazzo G, Tedjo DI, Wintjens D, Savelkoul P, Masclee A, Bodelier A, et al. Faecal microbiota dynamics and their relation to disease course in Crohn’s disease. J Crohns Colitis. 2019;13(10):1273–82. https://doi.org/10.1093/ecco-jcc/jjz049.
Pascal V, Pozuelo M, Borruel N, Casellas F, Campos D, Santiago A, et al. A microbial signature for Crohn’s disease. Gut. 2017;66(5):813–22. https://doi.org/10.1136/gutjnl-2016-313235.
De Musis C, Granata L, Dallio M, Miranda A, Gravina AG, Romano M. Inflammatory bowel diseases: the role of gut microbiota. Curr Pharm Des. 2020;26(25):2951–61. https://doi.org/10.2174/1381612826666200420144128.
Peyrin-Biroulet L, Billioud V, D’Haens G, Panaccione R, Feagan B, Panés J, et al. Development of the Paris definition of early Crohn’s disease for disease-modification trials: results of an international expert opinion process. Am J Gastroenterol. 2012;107(12):1770–6. https://doi.org/10.1038/ajg.2012.117.
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–21. https://doi.org/10.1038/nbt.2676.
Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38(6):685–8. https://doi.org/10.1038/s41587-020-0548-6.
Nguyen VQ, Jiang D, Hoffman SN, Guntaka S, Mays JL, Wang A, et al. Impact of diagnostic delay and associated factors on clinical outcomes in a U.S. inflammatory bowel disease cohort. Inflamm Bowel Dis. 2017;23(10):1825–31. https://doi.org/10.1097/MIB.0000000000001257.
Ma HQ, Yu TT, Zhao XJ, Zhang Y, Zhang HJ. Fecal microbial dysbiosis in Chinese patients with inflammatory bowel disease. World J Gastroenterol. 2018;24(13):1464–77. https://doi.org/10.3748/wjg.v24.i13.1464.
Wright EK, Kamm MA, Teo SM, Inouye M, Wagner J, Kirkwood CD. Recent advances in characterizing the gastrointestinal microbiome in Crohn’s disease: a systematic review. Inflamm Bowel Dis. 2015;21(6):1219–28. https://doi.org/10.1097/MIB.0000000000000382.
Vester-Andersen MK, Mirsepasi-Lauridsen HC, Prosberg MV, Mortensen CO, Träger C, Skovsen K, et al. Increased abundance of proteobacteria in aggressive Crohn’s disease seven years after diagnosis. Sci Rep. 2019;9(1):13473. https://doi.org/10.1038/s41598-019-49833-3.
Rehman A, Lepage P, Nolte A, Hellmig S, Schreiber S, Ott SJ. Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients. J Med Microbiol. 2010;59(Pt 9):1114–22. https://doi.org/10.1099/jmm.0.021170-0.
Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53(5):685–93. https://doi.org/10.1136/gut.2003.025403.
Delday M, Mulder I, Logan ET, Grant G. Bacteroides thetaiotaomicron ameliorates colon inflammation in preclinical models of Crohn’s disease. Inflamm Bowel Dis. 2019;25(1):85–96. https://doi.org/10.1093/ibd/izy281.
Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11:7. https://doi.org/10.1186/1471-2180-11-7.
Zitomersky NL, Atkinson BJ, Franklin SW, Mitchell PD, Snapper SB, Comstock LE, et al. Characterization of adherent bacteroidales from intestinal biopsies of children and young adults with inflammatory bowel disease. PLoS ONE. 2013;8(6):e63686. https://doi.org/10.1371/journal.pone.0063686.
Dziarski R, Park SY, Kashyap DR, Dowd SE, Gupta D. Pglyrp-regulated gut microflora Prevotella falsenii, parabacteroides distasonis and Bacteroides eggerthii enhance and Alistipes finegoldii attenuates colitis in mice. PLoS ONE. 2016;11(1):e0146162. https://doi.org/10.1371/journal.pone.0146162.
Yang F, Kumar A, Davenport KW, Kelliher JM, Ezeji JC, Good CE, et al. Complete genome sequence of a Parabacteroides distasonis strain (CavFT hAR46) isolated from a gut wall-cavitating microlesion in a patient with severe crohn’s disease. Microbiol Resour Announc. 2019. https://doi.org/10.1128/MRA.00585-19.
Mackner LM, Hatzakis E, Allen JM, Davies RH, Kim SC, Maltz RM, et al. Fecal microbiota and metabolites are distinct in a pilot study of pediatric Crohn’s disease patients with higher levels of perceived stress. Psychoneuroendocrinology. 2020;111:104469. https://doi.org/10.1016/j.psyneuen.2019.104469.
Huycke MM, Abrams V, Moore DR. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis. 2002;23(3):529–36. https://doi.org/10.1093/carcin/23.3.529.
Quince C, Ijaz UZ, Loman N, Eren AM, Saulnier D, Russell J, et al. Extensive modulation of the fecal metagenome in children with Crohn’s disease during exclusive enteral nutrition. Am J Gastroenterol. 2015;110(12):1718–29. https://doi.org/10.1038/ajg.2015.357. (quiz 1730).
Kowalska-Duplaga K, Gosiewski T, Kapusta P, Sroka-Oleksiak A, Wędrychowicz A, Pieczarkowski S, et al. Differences in the intestinal microbiome of healthy children and patients with newly diagnosed Crohn’s disease. Sci Rep. 2019;9(1):18880. https://doi.org/10.1038/s41598-019-55290-9.
Cummings JH, Macfarlane GT. The control and consequences of bacterial fermentation in the human colon. J Appl Bacteriol. 1991;70(6):443–59. https://doi.org/10.1111/j.1365-2672.1991.tb02739.x.
Phalipon A, Sansonetti PJ. Shigellosis: innate mechanisms of inflammatory destruction of the intestinal epithelium, adaptive immune response, and vaccine development. Crit Rev Immunol. 2003;23(5–6):371–401. https://doi.org/10.1615/critrevimmunol.v23.i56.20.
Iebba V, Aloi M, Civitelli F, Cucchiara S. Gut microbiota and pediatric disease. Dig Dis. 2011;29(6):531–9. https://doi.org/10.1159/000332969.
Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115(6):1405–13. https://doi.org/10.1016/s0016-5085(98)70019-8.
Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127(2):412–21. https://doi.org/10.1053/j.gastro.2004.04.061.
Hold GL, Smith M, Grange C, Watt ER, El-Omar EM, Mukhopadhya I. Role of the gut microbiota in inflammatory bowel disease pathogenesis: what have we learnt in the past 10 years. World J Gastroenterol. 2014;20(5):1192–210. https://doi.org/10.3748/wjg.v20.i5.1192.
Takahashi K, Nishida A, Fujimoto T, Fujii M, Shioya M, Imaeda H, et al. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in crohn’s disease. Digestion. 2016;93(1):59–65. https://doi.org/10.1159/000441768.
Gasaly N, de Vos P, Hermoso MA. Impact of bacterial metabolites on gut barrier function and host immunity: a focus on bacterial metabolism and its relevance for intestinal inflammation. Front Immunol. 2021;12:658354. https://doi.org/10.3389/fimmu.2021.658354.
Valles-Colomer M, Falony G, Darzi Y, Tigchelaar EF, Wang J, Tito RY, et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat Microbiol. 2019;4(4):623–32. https://doi.org/10.1038/s41564-018-0337-x.
Metwaly A, Reitmeier S, Haller D. Microbiome risk profiles as biomarkers for inflammatory and metabolic disorders. Nat Rev Gastroenterol Hepatol. 2022. https://doi.org/10.1038/s41575-022-00581-2.
Ma XZ, Lu XJ, Jin P, et al. Follow-up of ileocecal inflammatory lesions and its significance in early diagnosis of Crohn’s disease. Chin J Dig. 2020;40(5):306–13. https://doi.org/10.3760/cma.j.cn311367-20200121-00030.306-13.
Church DL, Cerutti L, Gürtler A, Griener T, Zelazny A, Emler S. Performance and application of 16S rRNA gene cycle sequencing for routine identification of bacteria in the clinical microbiology laboratory. Clin Microbiol Rev. 2020;33(4):e00053-19. https://doi.org/10.1128/CMR.00053-19.
Costea PI, Zeller G, Sunagawa S, Pelletier E, Alberti A, Levenez F, et al. Towards standards for human fecal sample processing in metagenomic studies. Nat Biotechnol. 2017;35(11):1069–76. https://doi.org/10.1038/nbt.3960.
Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5(1):27. https://doi.org/10.1186/s40168-017-0237-y.
Kucharzik T, Ellul P, Greuter T, Rahier JF, Verstockt B, Abreu C, et al. ECCO guidelines on the prevention, diagnosis, and management of infections in inflammatory bowel disease. J Crohns Colitis. 2021;15(6):879–913. https://doi.org/10.1093/ecco-jcc/jjab052.
Acknowledgements
We thank Professor Shirong Li, Department of Gastroenterology, Seventh Medical Center of PLA General Hospital, for the support in establishing pre-clinical and early CD patients and for the guidance in study design. We thank Dr. Yongsheng Teng of the 940th Hospital of Joint Logistics Support Force of PLA for the support in drafting the manuscript. We thank Dr. Tingting Wang and Dr. Wangfang Li for their support in data analysis and paper revision.
Funding
This work was supported by Capital’s Funds for Health Improvement and Research (Grant No. 2018-1-5091). However, the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
XZM, XJL, JQS conceived and designed the study. XZM participated in data acquisition. XZM, ZWY, LY performed data analysis and interpretation. XZM, XJL, LY, DZW, JFX, YJ, XW, HX, SL, MJZ, YQH participated in biological sample collection. XZM, WYZ participated in preparing the manuscript. JQS, PJ, YQH, MXZ, WYZ reviewed and edited the manuscript. JQS, PJ, YQH, XJL supervised the study. All authors critically revised the manuscript for important intellectual property and approved submitting this manuscript. The manuscript was revised and corrected for the English presentation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the Seventh Medical Center of the PLA General Hospital (#2016-45, #2017-46), and informed consent was obtained from all participates. All methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Figure S1. Sequencing information. A. OTUs Venn diagram analysis; B. Rank abundance curve. The X-axis represents the Rank of OTUs Abundance, and the Y-axis represents the corresponding OTUs Abundance. The Rank-Abundance curve can intuitively reflect the classified Abundance and evenness contained in the sample, that is, in the horizontal direction, the higher the value of the curve on the horizontal axis, the higher the Abundance. In the vertical direction, the flatter the curve, the more uniform the species distribution. Figure S2. Functional predictions of microbiota present in the fecal of CD patients and healthy controls. Significant KEGG pathways of Level 3 for the microbiome of the CD and healthy groups was identified. PICRUSt, Phylogenetic Investigation of Communities by Reconstruction of Unobserved States.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ma, X., Lu, X., Zhang, W. et al. Gut microbiota in the early stage of Crohn’s disease has unique characteristics. Gut Pathog 14, 46 (2022). https://doi.org/10.1186/s13099-022-00521-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13099-022-00521-0