
Abstract
Background
Human norovirus (HuNoV) is the major cause of viral acute gastroenteritis for all age groups in various countries. HuNoV GII in particular accounted for the majority of norovirus outbreaks, among which GII.4 caused repeated outbreaks for a long time. Besides GII.4, other norovirus genotypes, GII.2, GII.6, and GII.17, have also been prevalent in various contexts in recent years, but few detailed epidemiological studies of them have been performed and are poorly understood. We thus conducted an epidemiological analysis of HuNoV GII in Ibaraki Prefecture, Japan, by performing surveillance in the six seasons from September 2012 to August 2018.
Results
HuNoV GI occurred almost sporadically for all genotypes; however, each genotype of GII exhibited its typical epidemiological characteristics. Although the number of outbreaks of GII.4 decreased season by season, it reemerged in 2017/2018 season. The timing of the epidemic peak in terms of number of cases for GII.17 differed from that for the other genotypes. The patients age with GII.2 and GII.6 were younger and outbreak of GII.17 occurred frequently as food poisoning. Namely, the primarily infected outbreak group differed for each genotype of HuNoV GII. Moreover, the viral load of patients differed according to the genotype.
Conclusions
Various HuNoV genotypes including GII.2, GII.4, GII.6, and GII.17 were shown to be associated with various types of outbreak sites (at childcare and educational facilities, involving cases of food poisoning, and at elderly nursing homes) in this study. These genotypes emerged in recent years, and their prevalence patterns differed from each other. Moreover, differences in outbreak sites and viral load of patients among the genotypes were identified.
Similar content being viewed by others
Background
Human norovirus (HuNoV) belongs to the family Caliciviridae, genus Norovirus. It is a leading causative agent of acute gastroenteritis in people of all ages [1]. Many reports have suggested that the HuNoV genome can evolve rapidly, resulting in many different genotypes [2, 3]. At present, HuNoV is further classified into two genogroups (genogroups I and II) and over 30 genotypes (GI.1–GI.9 and GII.1–GII.22), as revealed by detailed genetic analyses of the capsid gene [4].
Previous molecular epidemiological studies showed that some genotypes of HuNoV, including GII.2, GII.4, GII.6, and GII.17, were particularly prevalent in gastroenteritis cases worldwide during the last 10 years [5,6,7]. Among these, GII.4 variant strains suddenly emerged and caused pandemics of gastroenteritis in many regions including Japan during 2006–2014 [4, 8]. This genotype has been associated with not only gastroenteritis in infants but also food poisoning in adults in various countries [9], while the prevalence of the virus may have declined during the last three seasons [10,11,12]. Another new genotype, GII.P17–GII.17, also suddenly emerged and caused large outbreaks in some countries [13], including large food poisoning-related outbreaks in Japan [14]. Furthermore, GII.2 variant strains reemerged in the 2016/2017 season and caused pandemics in various countries including Germany, China, and Japan [15,16,17]. However, there is still no comprehensive understanding of the local molecular epidemiology of HuNoV in Japan.
Therefore, we studied the relationships between the prevalent NoV genotypes associated with gastroenteritis outbreaks and epidemiologic data in Ibaraki Prefecture, Japan, during the 2012–2018 seasons to better understanding the molecular epidemiology in a domestic area.
Results
Relationships among HuNoV genotype, season, patient age, and outbreak site of infection
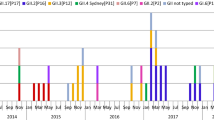
A total of 4588 clinical fecal specimens collected from September 2012 to August 2018 were examined to detect HuNoV (Table 1). Among these, HuNoV GI was detected in 244 specimens (around 5% of all specimens) and HuNoV GII in 2437 (around 53%). Detailed data on the seasonal variations, detected genotypes, and the outbreak site are shown in Tables 2 and 3 and Fig. 1. First, during the 2012–2015 seasons, GII.4 was detected in many cases in all patient sites, including outbreaks at childcare (0–6 years old), educational facilities (6–15 years old), and elderly nursing homes, and cases involving food poisoning. The GII.2 was the main genotype detected in outbreaks at childcare and educational facilities in the 2016/2017 season. GII.6 was mainly detected in the 2013/2014 season in outbreaks at childcare and educational facilities. GII.17 was suddenly detected in outbreaks involving food poisoning from the 2014/2015 season. GII.4 reemerged and caused outbreaks at childcare and educational facilities during the 2017/2018 season. In addition, GII.2, GII.4, and GII.6 were detected during September–March, whereas GII.17 was detected during January–April (Fig. 1). Finally, the GI virus was mainly detected in the 2014/2015 season from outbreaks at childcare and educational facilities and from cases involving food poisoning (Table 2). Moreover, unlike GII virus, GI virus was sporadically detected throughout the seasons (Fig. 1). These results suggest that various types of HuNoV were associated with the outbreaks of gastroenteritis in Ibaraki Prefecture.

Relationship between the occurrence of outbreaks for each genotype of NoV and the site of infection. The number of outbreaks of a GI, b GII.2, c GII.4, d GII.6, and e GII.17 is shown as a line graph. Blue indicates cases at childcare and educational facilities, red indicates cases involving food poisoning, orange indicates cases at elderly nursing homes, and green indicates others. The vertical axis shows the number of outbreaks, and the horizontal axis shows the season and month of appearance
Relationships among age, viral load, and HuNoV genotype in the fecal specimens
In the present study, we analyzed the relationships among age, viral load, and HuNoV genotype in the fecal specimens. The ages of the patients were significantly lower in whom GII.2 and GII.6 were detected than in whom GI, GII.4, and GII.17 were detected. Moreover, the age of patients in whom GII.6 was detected was significantly lower than for GII.2 (Table 4). We also analyzed the viral loads for some genotypes, including GI, GII.2, GII.4, GII.6, and GII.17, using real-time (RT)-PCR in the fecal specimens (Table 5). The HuNoV genome copy numbers of GII.2 were significantly higher than that of GI, GII.4, GII.6, and GII.17. These results suggested that the patients with GII.2 excreted more viruses than those infected with viruses of other genotypes.
Phylogeny of the detected HuNoV viruses
We performed a phylogenetic analysis based on the VP1 gene sequences of GI and genotypes of GII; GII.2, GII.4, GII.6, and GII.17 using the maximum likelihood (ML) method (Fig. 2a–e). First, 7 genotypes of GI virus such as GI.2, 3, 4, 5, 6, 7, and 9 were detected in this study (Fig. 2a). The tree of the genotype GII.2 formed three major clusters (Fig. 2b). GII.2 strains belonging to Clusters 1 and 2 were the main ones detected during the last two seasons. In the present tree, GII.2 virus detected in the 2016/17 season were classified into the Clusters 1 and 2, whereas the GII.2 virus mainly detected in the 2017/18 season were classified into Cluster 2. The GII.4 strains formed many clusters, although almost all of them were classified into the Sydney 2012 type (Fig. 2c). Among them, the GII.4 virus detected during the 2012–2015 seasons were genetically identical with a GII.4 prototype strain (accession no. JX459908), whereas the virus detected in some cases during 2015–2018 seasons were genetically identical with another prototype GII.4 (accession no. LC160215). GII.6 strains formed three clear clusters. Among these, the strains detected in the 2013/2014 season belonged to Cluster 1, whereas the strains detected in the 2012/2013 season mainly belonged to Cluster 2 (Fig. 2d). Most GII.17 strains belonged to Cluster 1 (Kawasaki308 type), whereas some strains belonged to Cluster 2 (Kawasaki323 type) (Fig. 2e). In the present cases, we detected genotype GII.17 in some cases during the 2014–2018 seasons, and these strains were genetically identical with a prototype of GII.17 virus (Kawasaki 308 strain). These results suggest that various genotypes of GII viruses have been associated with a range of outbreaks in Ibaraki Prefecture.





Gene phylogenetic tree was created by the ML method using 295 or 298 nt (GI) and 282 nt (GII) from the 5′-end of VP1. a Gene phylogenetic tree of GI. Nucleotide substitution model was GTR + Gamma. b Gene phylogenetic tree of GII.2. Nucleotide substitution model was K80 + Gamma. c Gene phylogenetic tree of GII.4. Nucleotide substitution model was K80 + Gamma. d Gene phylogenetic tree of GII.6. Nucleotide substitution model was K80 + Gamma. e Gene phylogenetic tree of GII.17. Nucleotide substitution model was GTR + Invariant. The strains detected in this study were shown as a black circle. Strains detected in multiple cases are shown in bold. The site of the outbreaks and their number are described at the end of the strain name. Cases at childcare and educational facilities (C) are colored blue, cases involving food poisoning (F) are colored red, those at elderly nursing homes (E) are colored orange, and others (O) are colored green
Pairwise distance of the strains
To analyze the genetic divergence of the present strains, we calculated the pairwise distances of the GI, GII.2, GII.4, GII.6, and GII.17 strains (Fig. 3a–e). First, the pairwise distance value among the GI strains was 0.18 ± 0.07 (mean ± standard deviation [SD]), and the intra-genotypic pairwise distance value was 0.067 ± 0.061 (mean ± SD). The intra-genotypic pairwise distance value of the present GII.2 was 0.020 ± 0.012 (mean ± SD), whereas that of the GII.4, GII6, and GII.17 values were 0.021 ± 0.010 (mean ± SD), 0.046 ± 0.036 (mean ± SD), and 0.017 ± 0.015 (mean ± SD), respectively. Overall, these genetic distances were relatively short within the same cluster. Thus, the results suggest that the strains analyzed here had not undergone wide genetic divergence.

Distributions of the pairwise distance values of partial VP1 gene of NoV detected in Ibaraki Prefecture. a GI was analyzed for a total of 42 strains. b GII.2 was analyzed for a total of 48 strains. c GII.4 was analyzed for a total of 92 strains. d GII.6 was analyzed for a total of 22 strains. e GII.17 was analyzed for a total of 23 strains. The vertical axis shows the number of sequence pairs, and the horizontal axis shows the pairwise distance. Blue indicates different clusters, and gray indicates the same cluster
Discussion
In this study, we performed a molecular epidemiological study of HuNoV infection in Ibaraki Prefecture, Japan, during the 2012–2018 seasons. The main findings were as follows: (i) various HuNoV genotypes including GII.2, GII.4, GII.6, and GII.17 were associated with the outbreaks of gastroenteritis in Ibaraki Prefecture; (ii) the GII.2-infected subjects showed a higher viral load in fecal specimens than those infected with viruses of other genotypes; and (iii) the detected strains had relatively low genetic divergence.
It has been reported that, although other GII genotypes were previously prevalent, GII.4 Den Haag 2006b suddenly emerged and caused pandemics in the 2006/2007 season [18, 19]. Moreover, variants (Den Haag 2006b type, New Orleans 2009 type, and Sydney 2012 type) of the GII.4 caused many outbreaks up to the 2013/2014 season [8]. However, after the 2014/2015 season, not only was GII.4 prevalent but also other genotypes, such as GII.2 and GII.17, were associated with outbreaks [13, 20]. A possible reason for the alterations of the prevalent GII genotypes; is that acquired herd immunities due to large outbreaks may affect human population [6, 21, 22]. Overall, the identified trends regarding the prevalent genotypes in Ibaraki Prefecture are compatible with those in other reports [13, 17, 23,24,25,26,27].
Previous studies have demonstrated that the GII virus was frequently detected compared with the GI virus from the NoV infection [28,29,30,31], which was consistent with our results. In contrast, both the GI and GII virus genomes were detected in environmental water at equivalent frequencies using real-time RT-PCR [32, 33] possibly due to the difference in stability between the GI and GII capsid proteins [34]. Indeed, Pogan et al. [34] showed that, unlike the GII.17 virus, the GI.1 virus may not be stable at high pH (over pH 8) using virus-like particles; however, this study did not examine the infectivity. We speculate that the stabilities of the virus particles between GI and GII viruses reflect the infectivity of these viruses to humans.
The phylogenetic tree of GII.4 created here showed that almost all detected strains were of the Sydney 2012 type, although these strains formed many small clusters in the tree (Fig. 3c). Previous reports have suggested that GII.4 suddenly emerged and caused pandemics of gastroenteritis in the 2006/2007 season (Den Haag 2006b type) and that some GII.4 variants such as Osaka 2007, Apeldoorn 2007, New Orleans 2009, and Sydney 2012 were subsequently generated [4, 35,36,37]. Among these, Sydney 2012 type caused as many pandemics of gastroenteritis as Den Haag 2006b type [4, 35]. The results suggested that the GII.4 Sydney variant was also associated with gastroenteritis outbreaks at childcare and educational facilities, in cases of food poisoning, and at elderly nursing homes. This finding is compatible with the previous reports [36, 38], which suggested that GII.4 was the most dominant type during the 2006–2014 seasons, whereas a small number of GII.2 were detected in this period [20, 39, 40]. However, GII.2 was the most prevalent type in the 2016/2017 season in various countries, including Germany, France, USA, China, and Japan [16, 17, 25, 26]. In Ibaraki Prefecture, GII.2 was also detected from many outbreaks in the 2016/2017 season associated with childcare and educational facility. In the phylogenetic tree created here, distinct clusters were formed by the GII.2 strains detected in the 2016/2017 season and those from other seasons [20, 40]. It is suggested that the GII.2 strains detected in the 2016/2017 season were recombinant, which is compatible with the findings from very recent studies [16]. Although we did not examine the polymerase type of the present GII.2 strains, such recombination may have been associated with the prevalence of GII.2 in Ibaraki Prefecture. Next, GII.17 was detected from the 2013/2014 season onwards, which was associated with many food poisoning outbreaks in Ibaraki (Table 3). Moreover, the periods of greatest prevalence differed between GII.17 and other genotypes such as GII.2, GII.4, and GII.6 (Fig. 1). The reason for this is not understood, but this finding is also compatible with previous reports [41, 42].
Next, we examined the viral loads among infections with viruses of various genotypes including GII.2, GII.4, GII.6, and GII.17. The results showed that the viral loads of GII.2 were higher than for the other genotypes and the age of patients infected with HuNoV GII.6 was lower than for the other genotypes. Previous reports suggested that the HuNoV viral loads in feces are associated with the age and immunity status of the hosts, although the reasons for this are not known [43, 44]. Although there are few previous reports describing the viral load of HuNoV, the propagation rate of GII.2 may not be higher than that of other genotypes [45, 46]. A possible reason for this is that we did not examine the differences in propagation among the genotypes and did not take into account the number of days since the patient had developed symptoms in this study. Moreover, the methods used in this study possibly cannot be used to analyze samples containing low numbers of the NoV genomes (approximately > 10 copies/experiment). However, if samples contain large numbers of genomes of different NoV genogroups, we may be able to analyze genotypes of plural NoV genogroup in each sample. In this study, we could analyze plural NoV genogroups in 23 samples of 19 cases. Currently, Next Generation Sequencing is expensive; therefore, in this study, we used conventional methods to analyze the samples as previously described [47]. Thus, further studies may be needed to clarify the epidemiology of HuNoV.
Conclusions
We showed in this study that many HuNoV genotypes, including GII.2, GII.4, GII.6, and GII.17, were associated with various types of outbreak sites (at childcare and educational facilities, in cases of food poisoning, and at elderly nursing homes) in this study. These genotypes emerged in recent years, and they exhibited distinct patterns of prevalence. Moreover, differences in the outbreak sites and viral load of patients were identified among the genotypes. To better understand the molecular epidemiology of HuNoV infection, ongoing molecular epidemiological studies may be needed.
Methods
Sample collection
Fecal specimens and patient information were collected for the following two types of outbreak cases among the surveillance system in Ibaraki Prefecture in Japan: (1) group cases that suspected outbreaks of human-to-human infectious disease and (2) group cases that suspected outbreaks of foodborne infectious disease. Public health centers collected information and specimens from the patients in both cases. In this study, we targeted population outbreak patients; therefore, whether the patients were administered to hospitals remains unknown. Patients without data on sex and age were omitted, and a total of 4588 specimens were collected by surveillance in the six seasons from September 2012 to August 2018 (Table 1).
Epidemiological data analyses
For each genotype, we compared and considered the epidemiological data of specimens positive for HuNoV GII (season, age group, viral load, and site of infection). Infection cases were classified into the following four groups regarding the site of infection as well as the age of the patients: (1) kindergarten, nursery school, and primary school (childcare and educational facilities: C), (2) suspected food poisoning (F), (3) elderly nursing homes (E), and (4) others (O).
In this study, food poisoning was defined as the outbreaks of the gastroenteritis due to foods served for commercial purposes from the food provision facility.
Detecting norovirus GII, sequencing, and genotyping
Fecal specimens were adjusted to 10 wt% with phosphate-buffered saline and centrifuged at 10,000×g for 10 min at 4 °C. The nucleic acids were extracted from the supernatant using QIAamp Viral RNA Mini Kit (Qiagen). Subsequently, complementary DNA (cDNA) was prepared by reverse transcription using PrimeScript™ RT Reagent Kit (Perfect Real Time) (Takara Bio). It was then used for quantitative polymerase chain reaction (q-PCR), which was performed using the TaqMan probe PCR system as described previously [48].
All RNA for which HuNoV GI and GII were determined to be positive by q-PCR was amplified using the PrimeScript™ II High Fidelity One Step RT-PCR Kit (Takara Bio) with G1SKF/G1SKR and G2SKF/G2SKR primers, respectively [47]. The nucleic acid sequence of the PCR product was determined by direct sequencing using the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). The resulting sequence was genotyped using the Norovirus Genotyping Tool [49]. If the genotypes were the same among samples collected in the same case, one sequence was selected, and a dataset of the gene sequence was prepared.
Calculation of pairwise distance
We analyzed pairwise distances to assess the genetic distances between human GII strains detected in Ibaraki Prefecture. Among the viral genes, 100% matched strains were omitted and pairwise distance values were calculated using MEGA 6 [50].
Phylogenetic tree analysis
The obtained gene sequence was compiled for each genotype, and a dataset was obtained by adding standard strains. We revealed the nucleotide substitution model with KAKUSAN 4 [51] and performed a phylogenetic tree analysis using the maximum likelihood method with MEGA 6 [50]. The strains detected in this study are indicated as a black circle. When 100% homologous sequence strains were detected in the same season, only one strain was retained and indicated in bold; the other sequence(s) was omitted from the dataset. The sites regarding the outbreaks and their number are described at the end of the strain name. In addition, cases involving childcare and educational facilities (C) are colored blue, those involving cases of food poisoning (F) are colored red, those at elderly nursing homes (E) are colored orange, and others (O) are colored green.
Statistical analysis
Statistical analysis was performed using EZR software [52]. After conducting the Kruskal–Wallis test as a statistical analysis on the age and viral load distribution of patients in each genotype, Holm’s multiple comparison test was performed.
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its Additional files).
Abbreviations
- HuNoV:
-
human norovirus
- GI:
-
genogroup I
- GII:
-
genogroup II
- PCR:
-
polymerase chain reaction
- cDNA:
-
complementary DNA
- q-PCR:
-
real-time polymerase chain reaction
- ML:
-
maximum likelihood
References
Green KY. Caliciviridae: the noroviruses, Ch. 20. In: Knipe DM, Howley PM, editors. Fields virology, vol. 1. 6th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins Press; 2013. p. 582–608.
Kobayashi M, Matsushima Y, Motoya T, Sakon N, Shigemoto N, Okamoto-Nakagawa R, Nishimura K, Yamashita Y, Kuroda M, Saruki N, Ryo A, Saraya T, Morita Y, Shirabe K, Ishikawa M, Takahashi T, Shinomiya H, Okabe N, Nagasawa K, Suzuki Y, Katayama K, Kimura H. Molecular evolution of the capsid gene in human norovirus genogroup II. Sci Rep. 2016;6:29400. https://doi.org/10.1038/srep29400.
Kobayashi M, Yoshizumi S, Kogawa S, Takahashi T, Ueki Y, Shinohara M, Mizukoshi F, Tsukagoshi H, Sasaki Y, Suzuki R, Shimizu H, Iwakiri A, Okabe N, Shirabe K, Shinomiya H, Kozawa K, Kusunoki H, Ryo A, Kuroda M, Katayama K, Kimura H. Molecular evolution of the capsid gene in norovirus genogroup I. Sci Rep. 2015;5:13806. https://doi.org/10.1038/srep13806.
Vinjé J. Advances in laboratory methods for detection and typing of norovirus. J Clin Microbiol. 2015;53:373–81. https://doi.org/10.1128/JCM.01535-14.
Kumazaki M, Usuku S. Genetic analysis of norovirus GII.4 variant strains detected in outbreaks of gastroenteritis in Yokohama, Japan, from the 2006–2007 to the 2013–2014 Seasons. PLoS ONE. 2015;10:e0142568. https://doi.org/10.1371/journal.pone.0142568.
Sakon N, Yamazaki K, Nakata K, Kanbayashi D, Yoda T, Mantani M, Kase T, Takahashi K, Komano J. Impact of genotype-specific herd immunity on the circulatory dynamism of norovirus: a 10-year longitudinal study of viral acute gastroenteritis. J Infect Dis. 2016;211:879–88.
Xue C, Pan L, Zhu W, Wang Y, Fu H, Cui C, Lu L, Qiao S, Xu B. Molecular epidemiology of genogroup II norovirus infections in acute gastroenteritis patients during 2014–2016 in Pudong New Area, Shanghai, China. Gut Pathog. 2018;10:7. https://doi.org/10.1186/s13099-018-0233-1.
Qiao N, Wang XY, Liu L. Temporal evolutionary dynamics of norovirus GII.4 variants in China between 2004 and 2015. PLoS ONE. 2016;11:e0163166. https://doi.org/10.1371/journal.pone.0163166.
Romero C, Tinoco YO, Loli S, Razuri H, Soto G, Silva M, Galvan P, Kambhampati A, Parashar UD, Kasper MR, Bausch DG, Simons MP, Lopman B. Incidence of norovirus-associated diarrhea and vomiting disease among children and adults in a community cohort in the Peruvian Amazon Basin. Clin Infect Dis. 2017;65:833–9. https://doi.org/10.1093/cid/cix423.
Bruggink LD, Moselen JM, Marshall JA. The molecular epidemiology of norovirus outbreaks in Victoria, 2014 to 2015. Commun Dis Intell Q Rep. 2017;41:E21–32.
Cannon JL, Barclay L, Collins NR, Wikswo ME, Castro CJ, Magaña LC, Gregoricus N, Marine RL, Chhabra P, Vinjé J. Genetic and epidemiologic trends of norovirus outbreaks in the United States from 2013 to 2016 demonstrated emergence of novel GII.4 recombinant viruses. J Clin Microbiol. 2017;55:2208–21. https://doi.org/10.1128/jcm.00455-17.
Suzuki Y, Doan YH, Kimura H, Shinomiya H, Shirabe K, Katayama K. Predicting genotype compositions in norovirus seasons in Japan. Microbiol Immunol. 2016;60:418–26. https://doi.org/10.1111/1348-0421.12384.
Matsushima Y, Ishikawa M, Shimizu T, Komane A, Kasuo S, Shinohara M, Nagasawa K, Kimura H, Ryo A, Okabe N, Haga K, Doan YH, Katayama K, Shimizu H. Genetic analyses of GII.17 norovirus strains in diarrheal disease outbreaks from December 2014 to March 2015 in Japan reveal a novel polymerase sequence and amino acid substitutions in the capsid region. Euro Surveill. 2015;20:21173.
Sakon N, Sadamasu K, Shinkai T, Hamajima Y, Yoshitomi H, Matsushima Y, Takada R, Terasoma F, Nakamura A, Komano J, Nagasawa K, Shimizu H, Katayama K, Kimura H. Foodborne outbreaks caused by human norovirus GII.P17-GII.17-contaminated Nori, Japan, 2017. Emerg Infect Dis. 2018;24:920–3. https://doi.org/10.3201/eid2405.171733.
Ao Y, Cong X, Jin M, Sun X, Wei X, Wang J, Zhang Q, Song J, Yu J, Cui J, Qi J, Tan M, Duan Z. Genetic analysis of re-emerging GII.P16-GII.2 noroviruses in 2016–2017 in China. J Infect Dis. 2018. https://doi.org/10.1093/infdis/jiy182.
Nagasawa K, Matsushima Y, Motoya T, Mizukoshi F, Ueki Y, Sakon N, Murakami K, Shimizu T, Okabe N, Nagata N, Shirabe K, Shinomiya H, Suzuki W, Kuroda M, Sekizuka T, Ryo A, Fujita K, Oishi K, Katayama K, Kimura H. Phylogeny and immunoreactivity of norovirus GII.P16-GII.2, Japan, Winter 2016–17. Emerg Infect Dis. 2018;24:144–8.
Niendorf S, Jacobsen S, Faber M, Eis-Hübinger AM, Hofmann J, Zimmermann O, Höhne M, Bock CT. Steep rise in norovirus cases and emergence of a new recombinant strain GII.P16-GII.2, Germany, winter 2016. Euro Surveill. 2016;22:30447. https://doi.org/10.2807/1560-7917.es.2017.22.4.30447.
Siebenga JJ, Vennema H, Zheng DP, Vinjé J, Lee BE, Pang XL, Ho EC, Lim W, Choudekar A, Broor S, Halperin T, Rasool NB, Hewitt J, Greening GE, Jin M, Duan ZJ, Lucero Y, O’Ryan M, Hoehne M, Schreier E, Ratcliff RM, White PA, Iritani N, Reuter G, Koopmans M. Norovirus illness is a global problem: emergence and spread of norovirus GII.4 variants, 2001–2007. J Infect Dis. 2009;200:802–12. https://doi.org/10.1086/605127.
Zhirakovskaia EV, Tikunov AY, Bodnev SA, Klemesheva VV, Netesov SV, Tikunova NV. Molecular epidemiology of noroviruses associated with sporadic gastroenteritis in children in Novosibirsk, Russia, 2003–2012. J Med Virol. 2015;87:740–53. https://doi.org/10.1002/jmv.24068.
Nagasawa K, Matsushima Y, Motoya T, Mizukoshi F, Ueki Y, Sakon N, Murakami K, Shimizu T, Okabe N, Nagata N, Shirabe K, Shinomiya H, Suzuki W, Kuroda M, Sekizuka T, Suzuki Y, Ryo A, Fujita K, Oishi K, Katayama K, Kimura H. Genetic analysis of human norovirus strains in Japan in 2016–2017. Front Microbiol. 2018;9:1. https://doi.org/10.3389/fmicb.2018.00001.
Eden JS, Hewitt J, Lim KL, Boni MF, Merif J, Greening G, Ratcliff RM, Holmes EC, Tanaka MM, Rawlinson WD, White PA. The emergence and evolution of the novel epidemic norovirus GII.4 variant Sydney 2012. Virology. 2014;450–451:106–13. https://doi.org/10.1016/j.virol.2013.12.005.
Debbink K, Lindesmith LC, Donaldson EF, Baric RS. Norovirus immunity and the great escape. PLoS Pathog. 2012;8:e1002921. https://doi.org/10.1371/journal.ppat.1002921.
Allen DJ, Adams NL, Aladin F, Harris JP, Brown DW. Emergence of the GII-4 norovirus Sydney 2012 strain in England, winter 2012–2013. PLoS ONE. 2014;9:e88978. https://doi.org/10.1371/journal.pone.0088978.
van Beek J, Ambert-Balay K, Botteldoorn N, Eden JS, Fonager J, Hewitt J, Iritani N, Kroneman A, Vennema H, Vinjé J, White PA, Koopmans M, NoroNet. Indications for worldwide increased norovirus activity associated with emergence of a new variant of genotype II.4, late 2012. Euro Surveill. 2013;18:8–9.
Bidalot M, Théry L, Kaplon J, De Rougemont A, Ambert-Balay K. Emergence of new recombinant noroviruses GII.p16-GII.4 and GII.p16-GII.2, France, winter 2016 to 2017. Euro Surveill. 2017;22:30508. https://doi.org/10.2807/1560-7917.es.2017.22.15.30508.
Kwok K, Niendorf S, Lee N, Hung TN, Chan LY, Jacobsen S, Nelson EAS, Leung TF, Lai RWM, Chan PKS, Chan MCW. Increased detection of emergent recombinant norovirus GII.P16-GII.2 strains in young adults, Hong Kong, China, 2016–2017. Emerg Infect Dis. 2017;23:1852–5. https://doi.org/10.3201/eid2311.170561.
Luo LF, Qiao K, Wang XG, Ding KY, Su HL, Li CZ, Yan HJ. Acute gastroenteritis outbreak caused by a GII.6 norovirus. World J Gastroenterol. 2015;21:5295–302. https://doi.org/10.3748/wjg.v21.i17.5295.
Hoa Tran TN, Trainor E, Nakagomi T, Cunliffe NA, Nakagomi O. Molecular epidemiology of noroviruses associated with acute sporadic gastroenteritis in children: global distribution of genogroups, genotypes and GII.4 variants. J Clin Virol. 2013;56:185–93. https://doi.org/10.1016/j.jcv.2012.11.011.
Bruggink LD, Oluwatoyin O, Sameer R, Witlox KJ, Marshall JA. Molecular and epidemiological features of gastroenteritis outbreaks involving genogroup I norovirus in Victoria, Australia, 2002–2010. J Med Virol. 2012;84:1437–48. https://doi.org/10.1002/jmv.23342.
Nakamura N, Kobayashi S, Minagawa H, Matsushita T, Sugiura W, Iwatani Y. Molecular epidemiology of enteric viruses in patients with acute gastroenteritis in Aichi prefecture, Japan, 2008/09–2013/14. J Med Virol. 2016;88:1180–6. https://doi.org/10.1002/jmv.24445.
Hasing ME, Lee BE, Qiu Y, Xia M, Pabbaraju K, Wong A, Tipples G, Jiang X, Pang XL. Changes in norovirus genotype diversity in gastroenteritis outbreaks in Alberta, Canada: 2012–2018. BMC Infect Dis. 2019;19:177. https://doi.org/10.1186/s12879-019-3792-y.
Haramoto E, Katayama H, Oguma K, Ohgaki S. Application of cation-coated filter method to detection of noroviruses, enteroviruses, adenoviruses, and torque teno viruses in the Tamagawa River in Japan. Appl Environ Microbiol. 2005;71:2403–11.
Haramoto E, Kitajima M, Kishida N, Katayama H, Asami M, Akiba M. Occurrence of viruses and protozoa in drinking water sources of Japan and their relationship to indicator microorganisms. Food Environ Virol. 2012;4:93–101. https://doi.org/10.1007/s12560-012-9082-0.
Pogan R, Schneider C, Reimer R, Hansman G, Uetrecht C. Norovirus-like VP1 particles exhibit isolate dependent stability profiles. J Phys: Condens Matter. 2018;30:064006. https://doi.org/10.1088/1361-648X/aaa43b.
White PA. Evolution of norovirus. Clin Microbiol Infect. 2014;20:741–5. https://doi.org/10.1111/1469-0691.12746.
Vega E, Barclay L, Gregoricus N, Shirley SH, Lee D, Vinjé J. Genotypic and epidemiologic trends of norovirus outbreaks in the United States, 2009 to 2013. J Clin Microbiol. 2014;52:147–55. https://doi.org/10.1128/JCM.02680-13.
Motoya T, Nagasawa K, Matsushima Y, Nagata N, Ryo A, Sekizuka T, Yamashita A, Kuroda M, Morita Y, Suzuki Y, Sasaki N, Katayama K, Kimura H. Molecular evolution of the VP1 gene in human norovirus GII.4 variants in 1974–2015. Front Microbiol. 2017;8:2399. https://doi.org/10.3389/fmicb.2017.02399.
Parra GI, Squires RB, Karangwa CK, Johnson JA, Lepore CJ, Sosnovtsev SV, Green KY. Static and evolving norovirus genotypes: implications for epidemiology and immunity. PLoS Pathog. 2017;13:e1006136. https://doi.org/10.1371/journal.ppat.1006136.
Kumazaki M, Usuku S. Norovirus genotype distribution in outbreaks of acute gastroenteritis among children and older people: an 8-year study. BMC Infect Dis. 2016;16:643.
Mizukoshi F, Nagasawa K, Doan YH, Haga K, Yoshizumi S, Ueki Y, Shinohara M, Ishikawa M, Sakon N, Shigemoto N, Okamoto-Nakagawa R, Ochi A, Murakami K, Ryo A, Suzuki Y, Katayama K, Kimura H. Molecular evolution of the RNA-dependent RNA polymerase and capsid genes of human norovirus genotype GII.2 in Japan during 2004–2015. Front Microbiol. 2017;8:705. https://doi.org/10.3389/fmicb.2017.00705.
Gao Z, Liu B, Huo D, Yan H, Jia L, Du Y, Qian H, Yang Y, Wang X, Li J, Wang Q. Increased norovirus activity was associated with a novel norovirus GII.17 variant in Beijing, China during winter 2014–2015. BMC Infect Dis. 2015;15:574. https://doi.org/10.1186/s12879-015-1315-z.
Zhang P, Chen L, Fu Y, Ji L, Wu X, Xu D, Han J. Clinical and molecular analyses of norovirus-associated sporadic acute gastroenteritis: the emergence of GII.17 over GII.4, Huzhou, China, 2016. BMC Infect Dis. 2016;16:717. https://doi.org/10.1186/s12879-016-2033-x.
Chan MC, Leung TF, Kwok AK, Lee N, Chan PK. Characteristics of patients infected with norovirus GII.4 Sydney, Hong Kong, China. Emerg Infect Dis. 2012;2014(20):658–61. https://doi.org/10.3201/eid2004.13457.
Portes SAR, Carvalho-Costa FA, Rocha MS, Fumian TM, Maranhão AG, de Assis RM, Xavier MDPTP, Rocha MS, Miagostovich MP, Leite JPG, Volotão EM. Enteric viruses in HIV-1 seropositive and HIV-1 seronegative children with diarrheal diseases in Brazil. PLoS ONE. 2017;12:e0183196. https://doi.org/10.1371/journal.pone.0183196.
Costantini V, Morantz EK, Browne H, Ettayebi K, Zeng XL, Atmar RL, Estes MK, Vinjé J. Human norovirus replication in human intestinal enteroids as model to evaluate virus inactivation. Emerg Infect Dis. 2018;24:1453–64. https://doi.org/10.3201/eid2408.180126.
Kirby AE, Streby A, Moe CL. Vomiting as a symptom and transmission risk in norovirus illness: evidence from human challenge studies. PLoS ONE. 2016;11:e0143759. https://doi.org/10.1371/journal.pone.0143759.
Kojima S, Kageyama T, Fukushi S, Hoshino FB, Shinohara M, Uchida K, Natori K, Takeda N, Katayama K. Genogroup-specific PCR primers for detection of Norwalk-like viruses. J Virol Methods. 2002;100:107–14.
Kageyama T, Shinohara M, Uchida K, Fukushi S, Hoshino FB, Kojima S, Takai R, Oka T, Takeda N, Katayama K. Coexistence of multiple genotypes, including newly identified genotypes, in outbreaks of gastroenteritis due to Norovirus in Japan. Clin Microbiol. 2004;42:2988–95.
Kroneman A, Vennema H, Deforche K, Avoort HV, Peñaranda S, Oberste MS, Vinjé J, Koopmans M. An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol. 2011;51:121–5. https://doi.org/10.1016/j.jcv.2011.03.006.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Tanabe AS. Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol Ecol Resour. 2011;11:914–21.
Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48:452–8.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
TM was responsible for all sample preparation and testing and performed data collection and manuscript preparation. MU, AS, KG, and ID provided valuable assistance in sample preparation. SF, NN, and YI edited the manuscript and obtained necessary funding. KO, JA, YM, and TI drafted and revised the manuscript. AR, NS, and KK provided significant input into decisions regarding manuscript content, editing, and manuscript preparation. HK designed and supervised the overall study, as well as conceptualized, drafted, and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by Ibaraki Prefecture Epidemiological Research Joint Ethics Review Committee, Ibaraki, Japan (Approval Number: H30-1).
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Motoya, T., Umezawa, M., Saito, A. et al. Variation of human norovirus GII genotypes detected in Ibaraki, Japan, during 2012–2018. Gut Pathog 11, 26 (2019). https://doi.org/10.1186/s13099-019-0303-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13099-019-0303-z