
Abstract
Background
A subcutaneous (SC) formulation of infliximab biosimilar CT-P13 is approved in Europe for the treatment of adult patients with rheumatoid arthritis (RA). It may offer improved efficacy versus intravenous (IV) infliximab formulations.
Methods
A network meta-regression was conducted using individual patient data from two randomised trials in patients with RA, which compared CT-P13 SC with CT-P13 IV, and CT-P13 IV with reference infliximab IV. In this analysis, CT-P13 SC was compared with CT-P13 IV, reference infliximab IV and pooled data for both reference infliximab IV and CT-P13 IV. Outcomes included changes from baseline in 28-joint Disease Activity Score based on C-reactive protein (DAS28-CRP), Simplified Disease Activity Index (SDAI) and Clinical Disease Activity Index (CDAI), and rates of remission, low disease activity or clinically meaningful improvement in functional disability per Health Assessment Questionnaire–Disability Index (HAQ-DI).
Results
The two studies enrolled 949 patients with RA; pooled data for 840 and 751 patients were evaluable at weeks 30 and 54, respectively. For the CT-P13 SC versus pooled IV treatment arm comparison, differences in changes from baseline in DAS28-CRP (− 0.578; 95% confidence interval [CI] − 0.831, − 0.325; p < 0.0001), CDAI (− 3.502; 95% CI − 5.715, − 1.289; p = 0.002) and SDAI (− 4.031; 95% CI − 6.385, − 1.677; p = 0.0008) scores at 30 weeks were statistically significant in favour of CT-P13 SC. From weeks 30 to 54, the magnitude of the differences increased and remained statistically significant in favour of CT-P13 SC. Similar results were observed for the comparison of CT-P13 SC with CT-P13 IV and with reference infliximab IV. Statistically significant differences at week 30 favoured CT-P13 SC over the pooled IV treatment arms for the proportions of patients achieving EULAR-CRP good response, American College of Rheumatology (ACR) 50 and ACR70 responses, DAS28-CRP-defined remission, low disease activity (DAS28-CRP, CDAI and SDAI criteria) and clinically meaningful HAQ-DI improvement.
Conclusions
CT-P13 SC was associated with greater improvements in DAS28-CRP, CDAI and SDAI scores and higher rates of clinical response, low disease activity and clinically meaningful improvement in functional disability, compared with CT-P13 IV and reference infliximab IV.
Trial registration
EudraCT, 2016-002125-11, registered 1 July 2016; EudraCT 2010-018646-31, registered 23 June 2010.
Similar content being viewed by others


Background
Rheumatoid arthritis (RA) is a systemic inflammatory autoimmune disease that affects over 3.0 million people in Europe [1]. Symptoms include joint pain, swelling and stiffness, potentially leading to joint damage and irreversible disability [2,3,4,5]. Patients with RA have higher rates of disability than the general population [6], and many patients experience reduced work productivity and health-related quality of life (HRQoL) [7, 8].
Optimal use of medication is a key strategy for effective RA management [9]. The primary treatment target for patients with RA is sustained remission, with low disease activity as an alternative target, particularly for patients with long-standing disease [9, 10]. European League Against Rheumatism (EULAR) recommendations advocate the initiation of therapy with disease-modifying antirheumatic drugs (DMARDs) as soon as a diagnosis of RA is made [9]. The addition of a biologic DMARD (bDMARD) or targeted synthetic DMARD (tsDMARDs) is recommended when treatment targets are not achieved with the first conventional synthetic DMARD (csDMARD) and poor prognostic factors are present [9].
Tumour necrosis factor inhibitors (TNFis) are a class of bDMARD that are well tolerated and have been shown to effectively reduce disease activity and structural joint damage [9, 11]. Five TNFis are currently approved for the treatment of RA (infliximab, adalimumab, etanercept, golimumab and certolizumab pegol) [9]. Most of these are administered subcutaneously (SC); only reference infliximab intravenous (IV; Remicade®, Janssen Biologics BV) and biosimilars of infliximab are administered intravenously (although the feasibility of SC administration was reported as early as 2006 [12]). CT-P13 IV (Remsima® IV, Celltrion Healthcare Co., Ltd.), an infliximab biosimilar, received European Union (EU) authorisation for the treatment of adult patients with RA in 2013 [13] and US approval in 2016 [14]. Subsequently, CT-P13 SC (Remsima® SC, Celltrion Healthcare Co., Ltd), the only SC formulation of infliximab, received EU approval in 2019 [13, 15]. For maintenance therapy in patients with RA, the approved doses for CT-P13 IV and CT-P13 SC are 3 mg/kg every 8 weeks (Q8W) and 120 mg every 2 weeks (Q2W), respectively [13].
CT-P13 SC and CT-P13 IV were approved based on the results of the pivotal 3.5 (NCT03147248; EudraCT No. 2016-002125-11) and 3.1 (NCT01217086; EudraCT No. 2010-018646-31) trials, respectively. The CT-P13 3.5 trial was a randomised, multicentre, parallel-group, phase I/III study that enrolled 357 patients with active RA [16]. In this study, non-inferiority of CT-P13 SC to CT-P13 IV was demonstrated using assessment of the change from baseline in the 28-joint Disease Activity Score based on C-reactive protein (DAS28-CRP) at week 22, with a statistically significant treatment difference of 0.27 (95% confidence interval [CI] 0.02, 0.52) for the SC versus the IV treatment arm, although the 95% CI was higher than the predefined non-inferiority margin of − 0.06. Other efficacy outcomes were generally comparable between the SC and IV treatment arms up to week 22 and favoured the SC arm at week 30 [16]. The CT-P13 3.1 trial was a randomised, double-blind, parallel-group, phase III study of 606 patients with RA [17, 18]. In this study, therapeutic equivalence was established between CT-P13 IV and reference infliximab IV at week 30 [17], and similar efficacy and safety profiles were demonstrated up to week 54 [18].
To date, there have been no head-to-head comparisons of CT-P13 SC versus reference infliximab IV, and the comparison of CT-P13 SC versus CT-P13 IV was limited to 30 weeks as all patients randomised to CT-P13 IV in the 3.5 trial were switched to CT-P13 SC at week 30. However, the availability of patient-level data from the CT-P13 3.1 and 3.5 trials allows a mixed treatment comparison of CT-P13 SC versus infliximab IV at weeks 30 and 54. The objective of this analysis was to examine whether CT-P13 SC provides added clinical value compared with CT-P13 IV and reference infliximab IV using individual patient data (IPD) from the CT-P13 3.1 and 3.5 trials.
Methods
Data sources
Data were sourced from two randomised controlled trials (RCTs): the CT-P13 3.1 trial, which compared CT-P13 IV with reference infliximab IV [17, 18], and the CT-P13 3.5 trial, which compared CT-P13 IV with CT-P13 SC [16]. In both trials, patients received a similar dose of methotrexate at 12.5–25 mg/week (along with folic acid at a dose of ≥ 5 mg/week), which was maintained throughout the study [16, 17]. At baseline in the CT-P13 3.1 trial, the mean (standard deviation [SD]) methotrexate dose was 15.6 (3.1) and 15.6 (3.2) mg for patients in the CT-P13 IV and reference infliximab IV treatment arms, respectively [17]. In the CT-P13 3.5 trial, the mean (SD) methotrexate dose at baseline was 17.0 (4.0) and 17.4 (4.0) mg/week in the CT-P13 SC and CT-P13 IV treatment arms, respectively [16]. IPD, including patient characteristics and outcomes, were sourced from both trials.
The CT-P13 3.1 and 3.5 clinical trials both included a CT-P13 IV treatment arm, permitting indirect comparison of CT-P13 SC with either reference infliximab IV or with pooled data for reference infliximab IV and CT-P13 IV, at week 30.
Treatment comparisons
CT-P13 SC was compared to (1) CT-P13 IV (from studies 3.1 and 3.5), (2) reference infliximab IV (from study 3.1) and (3) pooled data including both reference infliximab IV and CT-P13 IV (from studies 3.1 and 3.5).
Efficacy outcomes of interest
Efficacy outcomes included in the present analysis were the change from baseline in DAS28-CRP [19]; change from baseline in Simplified Disease Activity Index (SDAI) [20]; change from baseline in Clinical Disease Activity Index (CDAI) [20]; remission based on SDAI and CDAI, Boolean remission (TJC ≤ 1, SJC ≤ 1, CRP ≤ 1 mg/dL and PGA ≤ 1 at any time point), and remission also defined as DAS-28-CRP (< 2.6) [21]; low disease activity based on DAS28-CRP (≤ 3.2), CDAI (≤ 10.0) and SDAI (≤ 11.0) [22]; EULAR response [23]; and American College of Rheumatology (ACR) response (ACR20, ACR50 and ACR70) [24, 25]. Additionally, functional disability was assessed based on the proportion of patients with a change in Health Assessment Questionnaire–Disability Index (HAQ-DI) that was equal to or greater than the minimal clinically important difference (MCID) of 0.22 [26, 27].
Feasibility assessment
The studies contributing data to the present analyses had similar eligibility criteria and the same target population. Definitions of all outcomes of interest were also the same. Baseline demographics (e.g. age, sex, body mass index) were similar between studies, although a higher proportion of Asian patients were enrolled in the 3.1 trial versus the 3.5 trial (Table 1). Baseline clinical characteristics were also similar in terms of objectively measured parameters, such as CRP, erythrocyte sedimentation rate (ESR), DAS28-CRP, 28-joint Disease Activity Score based on ESR (DAS28-ESR), Swollen 28-Joint Count (SJC) and Tender 28-Joint Count (TJC) (Table 1). Small differences were observed in patients’ and physicians’ global assessment of disease activity; these measures were numerically greater in the 3.5 trial, compared to the 3.1 trial (Table 1).
Statistical analyses
Baseline participant characteristics were presented using descriptive statistics: mean ± SD for continuous variables and percentages for categorical variables. All statistical analyses were conducted using SAS statistical software (version 9.3, SAS Institute, Cary, USA). Two-sided tests were used and p-values < 0.05 were considered statistically significant.
Data imputation at week 54
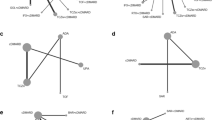
Because all participants randomised to CT-P13 IV in the 3.5 trial switched to CT-P13 SC at week 30, week 54 effects in the CT-P13 IV treatment arm were imputed using regression methods based on the effects observed in the CT-P13 IV treatment arm of the CT-P13 3.1 trial (Fig. 1). Linear regression models were fitted using IPD from the CT-P13 IV treatment arm of the CT-P13 3.1 trial. The dependent variables were the changes from baseline to week 54 in DAS28-CRP, CDAI and SDAI, respectively. The model covariates were the values of the modelled outcome at baseline and the change from baseline to week 30, as well as possible confounders selected from a list shown in Additional file 1, Table S1. The selection of potential confounders was performed in three steps: (1) the association of each variable listed in Table S1 with the change from baseline to week 54 in modelled outcome was tested; (2) if several variables were correlated (Pearson r ≥ 0.6 for continuous variables; see correlation coefficients in Additional file 1, Table S2), only the variable with the strongest association with the modelled outcomes was retained; and (3) all selected variables from step 2 were entered in the model and a backward selection procedure was applied to further reduce the list of variables.

Imputation of week-54 CT-P13 3.5 results based on CT-P13 regression model. EU, European Union; IV, intravenous; MTX, methotrexate; PD, pharmacodynamics; PK, pharmacokinetics
R2 was used to assess the quality of the models. R2 was 0.48 for the model predicting change from baseline in DAS28-CRP score and 0.61 for the models predicting the changes from baseline in CDAI and SDAI, demonstrating that the quality of models was acceptable. The models are presented in Additional file 1, Table S3.
The obtained regression models provided predictions of the mean changes from baseline to week 54 in DAS28-CRP, CDAI and SDAI and associated SDs. A multiple imputation method was used to account for the uncertainty around the predicted values [28]. For each patient, 10 values were generated randomly from the statistical distribution around the predicted scores, thus generating 10 datasets on which the meta-regression models were estimated. Imputation was not performed for binary outcomes because prediction of the outcome itself was surrounded with a large degree of uncertainty.
Meta-regression
A network meta-regression using IPD is recommended as the “gold standard” method to adjust for treatment effect modifiers when IPD are available for all considered studies [29, 30]; this method was implemented here in accordance with relevant methodological guidelines [29].
Two series of analyses were performed, using two definitions of the treatment variable: treatment variable with three levels (CT-P13 SC, CT-P13 IV, reference infliximab IV) and treatment variable with two levels (CT-P13 SC, infliximab IV [pooled data for CT-P13 IV and reference infliximab IV]).
Multivariate mixed models, with normal distribution and identity link function for continuous outcomes and binomial distribution and logit link function for binary outcomes, were fitted at weeks 30 and 54. Dependent variables were the outcomes of interest, as listed above. Model covariates were selected among variables listed in Additional file 1, Table S1. The same 3-step approach as described for the imputation model above was used for the models for DAS28-CRP, CDAI and SDAI change from baseline, EULAR good response (CRP criteria), ACR20, ACR50, ACR70, Boolean remission and HAQ-DI MCID (≥ 0.22) at week 30. For binary outcomes based on DAS28-CRP, CDAI and SDAI, the same covariates as in the model for corresponding continuous outcomes were used. For outcomes at week 54, the same covariates as for the corresponding outcomes at week 30 were used. In addition, a variable representing study 3.1 or 3.5 was entered in all regression models as a random effect.
Analysis outputs included treatment differences with associated 95% CIs for continuous outcomes, and odd ratios (OR) with associated 95% CIs for binary outcomes.
The treatment effect at week 54, for each continuous outcome (DAS28-CRP, CDAI and SDAI), was obtained as the mean of treatment effects estimated from the 10 simulated datasets, and the associated variance coefficient was calculated as the sum of the variance of estimated treatment effect within simulations and variance between simulations [28, 31]. p-value and 95% CI calculations considered a normal distribution of finally obtained coefficients.
Results
Mixed treatment comparison at week 30
Figure 2 presents pooled data for unadjusted estimates of outcomes at weeks 30 and 54, according to treatment. Table 2 presents pooled data for unadjusted response and remission rates at week 30, according to treatment.

Change from baseline in DAS28-CRP (a), CDAI (b) and SDAI (c) at weeks 30/54. CDAI, Clinical Disease Activity Index; CRP, C-reactive protein; DAS28-CRP, 28-joint Disease Activity Score based on C-reactive protein; IV, intravenous; SDAI, Simple Disease Activity Index; SC, subcutaneous; SD, standard deviation. *CT-P13 IV vs CT-P13 SC p < 0.05, CT-P13 SC vs reference infliximab IV p < 0.05 and CT-P13 IV vs reference infliximab IV p ≥ 0.05. p-values derive from the meta-regression efficacy analysis of week-30 and week-54 data (Tables 3 and 4)
A total of 840 patients were included in the meta-regression efficacy analysis of week 30 data (Table 3). For the comparison between CT-P13 SC and pooled IV treatment arms, the difference in the change from baseline in DAS28-CRP score was statistically significant (− 0.578; 95%: CI − 0.831, − 0.325; p < 0.0001), suggesting superior efficacy with CT-P13 SC. Differences in the changes from baseline in CDAI and SDAI scores were also statistically significant (− 3.502; 95% CI − 5.715, − 1.289; p = 0.002; and − 4.031; 95% CI − 6.385, − 1.677; p = 0.0008, respectively), suggesting superior efficacy with CT-P13 SC versus the pooled IV treatment arms. Similar results were obtained for the comparisons of CT-P13 SC versus CT-P13 IV, and CT-P13 SC versus reference infliximab IV (Table 3). The difference in DAS28-CRP (− 0.693) compared to reference infliximab was clinically meaningful [32].
Statistically significant differences favouring CT-P13 SC versus the pooled IV treatment arms were also observed for the majority of binary outcomes at week 30 (Table 3). The probability of achieving an ACR20 response, disease remission based on CDAI or SDAI, and Boolean remission were the only outcomes for which the effect of treatment was not statistically significantly different, although a numerical trend was observed in favour of CT-P13 SC. The odds of achieving a EULAR good response (CRP criteria) were > 2-fold higher for CT-P13 SC compared with all considered IV treatment arms. Similarly, the odds of achieving the outcome of low disease activity according to DAS28-CRP, CDAI and SDAI were also > 2-fold higher for CT-P13 SC versus all considered IV treatment arms. The ORs of patients achieving an ACR50 response ranged from 1.60 to 1.81 for the CT-P13 SC arm versus pooled IV treatment arms or reference infliximab IV; ORs of patients achieving ACR70 responses ranged from 1.72 to 2.18.
The proportion of patients achieving a clinically meaningful improvement in HAQ-DI at week 30 was statistically significantly higher with CT-P13 SC versus the pooled IV treatment arms (p = 0.03) (Table 3).
Mixed treatment comparison at week 54
Altogether, 751 patients were included in the meta-regression efficacy analysis of week-54 data, which included imputed values (Table 4). From week 30 to week 54, the magnitude of the difference in the SC arm versus pooled IV treatment arms increased and the treatment difference remained statistically significant at week 54 for changes in DAS28-CRP, CDAI and SDAI, with mean differences (95% CI) estimated at − 0.876 (− 1.119, − 0.633; p < 0.0001), − 6.484 (− 9.026, − 3.942; p < 0.0001) and − 7.302 (− 9.711, − 4.893, p < 0.0001), respectively.
Discussion
This study compared CT-P13 SC with infliximab IV using IPD network meta-regression techniques, allowing us to compare CT-P13 SC with infliximab IV when no head-to-head trial was available. Network meta-analysis (NMA), a broader analytical framework encompassing IPD network meta-analysis, is recommended by health technology assessment agencies and scientific societies, including the National Institute for Health and Care Excellence (NICE) and the International Society for Pharmacoeconomics and Outcomes Research (ISPOR) [29, 33]. NMA is considered an ideal approach as it is capable of synthesising reliable quantitative evidence about treatment effects. The validity of NMA, particularly IPD network meta-regression, relies on the comparability of studies. In our analysis, the populations represented in the two studies had characteristics that were generally similar and the same outcome measurement scales were used. The present analysis compared the efficacy of CT-P13 SC with established IV formulations of infliximab in adult RA patients co-treated with methotrexate. Outcomes data from the CT-P13 3.5 trial appeared to favour CT-P13 SC over CT-P13 IV at week 30. Furthermore, combined analysis of data from the CT-P13 3.1 and 3.5 trials showed a statistically significant difference between the CT-P13 SC and pooled IV treatment arms for the change from baseline in DAS28-CRP, CDAI and SDAI scores; the results favoured CT-P13 SC at both the week-30 and week-54 time points. Higher response rates (e.g. ACR50/70, EULAR good response [CRP criteria]), rates of low disease activity (DAS28-CRP, CDAI and SDAI criteria), and DAS28-CRP remission rates were also observed with CT-P13 SC compared with the pooled IV treatment arms, as evaluated at the week-30 time point. Similar results were observed for the CT-P13 SC versus reference infliximab IV comparison.
Differences in the pharmacokinetic profiles of the SC and IV formulations likely account for the improved treatment outcomes observed with CT-P13 SC compared with infliximab IV in the present analysis. CT-P13 SC is administered more frequently than infliximab IV (e.g. Q2W compared with Q8W, respectively) [13]. Consequently, compared with infliximab IV, CT-P13 SC achieves a more stable steady-state serum concentration and higher trough serum concentrations (Ctrough) [13, 17]. For example, the median Ctrough level of CT-P13 SC 120 mg in study 3.5 was 11.65 μg/mL [15]. Therefore, the Ctrough with Q2W dosing of CT-P13 SC 120 mg is approximately 11 times higher than the target concentration of 1 μg/mL, which is the therapeutic threshold for the treatment of RA and the approximate Ctrough achieved with Q8W dosing of infliximab IV [17, 34].
Trough serum levels of infliximab have been shown to correlate with the clinical response to infliximab treatment [35,36,37]. An analysis of data from the RISING study showed that in patients receiving reference infliximab IV Q8W, median trough serum infliximab concentrations were 3.0 (interquartile range 1.5–7.2), 1.1 (< 0.1–3.6) and < 0.1 (< 0.1–0.3) μg/mL for those achieving a EULAR good, moderate or no response, respectively, at week 54 [37]. The same study showed a significant association between clinical response and reduction in disease activity with higher trough serum infliximab levels (p < 0.001) [37]. Consistent with these findings, Wolbink and colleagues reported significantly lower trough serum infliximab levels in non-responders than responders and showed that low serum levels correlated with poor clinical improvement based on DAS28-CRP [35]. In further support of the correlation between low trough infliximab concentrations and poor clinical response, pharmacokinetic models developed for non-responsive patients in the ATTRACT trial predicted that shortening the dosing interval would have a better effect on maintaining higher trough serum levels of infliximab than increasing the dose, which in turn would increase treatment efficacy [36].
In summary, considering the totality of the evidence, it is plausible that improved efficacy of CT-P13 SC is due to achieving higher Ctrough levels through more frequent administration, compared with infliximab IV. Furthermore, there were no clinically meaningful differences between the safety profiles of CT-P13 SC and CT-P13 IV in study 3.5 [16], suggesting that higher trough serum concentrations observed with CT-P13 SC do not appear to be associated with an increased risk of adverse events.
Several limitations should be taken into consideration when interpreting the results of the present analysis. First, data were obtained from only two trials, and data from other infliximab RCTs were not included in the analysis. It was deemed that the integration of aggregate data from infliximab RCTs without CT-P13 SC would not add substantial value to this analysis, although the inclusion of more studies would arguably better account for variability in outcomes of infliximab IV between studies. Furthermore, the comparison of data from earlier infliximab trials with data from more recently conducted trials, such as study 3.1, may be inappropriate due to differences in disease duration and progression in the enrolled populations [17, 20, 38, 39]. A second limitation was the use of imputation for missing values (i.e. week-54 data), which relies on the assumption that the evolution of clinical scores after week 30 in patients who remained on CT-P13 IV in the 3.5 trial would be comparable to that observed in study 3.1. However, the uncertainty associated with extrapolation was fully accounted for in the reported 95% CIs, based on multiple imputation. Moreover, due to the high uncertainty associated with predicting binary outcomes at week 54, mixed treatment comparisons were reported as continuous variables only [33]. Thirdly, radiological outcomes were not assessed in the present meta-regression, as structural damage was not evaluated in the CT-P13 3.5 trial; however, other studies provide evidence that infliximab is effective in reducing radiological evidence of synovitis and erosions in the long term [40]. Finally, as the focus of the present analysis was the comparative efficacy of CT-P13 SC and infliximab IV, safety endpoints were not analysed. However, safety data from study 3.5 suggest that the safety profile of the CT-P13 SC is at least similar, or even favourable, compared with CT-P13 IV; for example, the proportions of patients experiencing treatment-emergent adverse events (TEAEs), serious TEAEs, TEAEs causing drug discontinuation, and infection were numerically lower in the SC arm, compared with the IV treatment arm, up to week 54 [16] (Additional file 1, Table S4). Finally, long-term data regarding CT-P13 SC safety and efficacy are limited and should be collected in future studies.
Conclusions
This meta-regression of IPD from two randomised trials showed that CT-P13 SC was associated with greater improvements in DAS28-CRP, CDAI and SDAI scores; higher proportions of patients achieving ACR and EULAR responses; low disease activity; and clinically meaningful improvements in functional disability, compared with CT-P13 IV and reference infliximab IV. Thus, CT-P13 SC may be a valuable alternative to infliximab IV.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACR:
-
American College of Rheumatology
- AE:
-
Adverse event
- anti-CCP:
-
Anti-cyclic citrullinated peptide
- bDMARD:
-
Biologic synthetic disease-modifying antirheumatic drug
- BMI:
-
Body mass index
- CDAI:
-
Clinical Disease Activity Index
- CI:
-
Confidence interval
- CRP:
-
C-reactive protein
- C trough :
-
Trough serum concentration
- DAS28-CRP:
-
28-Joint Disease Activity Score based on C-reactive protein
- DAS28-ESR:
-
28-Joint Disease Activity Score based on erythrocyte sedimentation rate
- DMARD:
-
Disease-modifying antirheumatic drug
- ESR:
-
Erythrocyte sedimentation rate
- EU:
-
European Union
- EULAR:
-
European League Against Rheumatism
- HAQ-DI:
-
Health Assessment Questionnaire–Disability Index
- HRQoL:
-
Health-related quality of life
- IFX:
-
Infliximab
- IPD:
-
Individual patient data
- ISPOR:
-
International Society for Pharmacoeconomics and Outcomes Research
- IV:
-
Intravenous
- MCID:
-
Minimal clinically important difference
- MTX:
-
Methotrexate
- NICE:
-
National Institute for Health and Care Excellence
- NMA:
-
Network meta-analysis
- OR:
-
Odds ratio
- PADA:
-
Patient Global Assessment of Disease Activity
- PAPA:
-
Patient Assessment of Pain
- PHDA:
-
Physician Assessment of Disease Activity
- PK:
-
Pharmacokinetic(s)
- Q#W:
-
Every # weeks
- RA:
-
Rheumatoid arthritis
- RCT:
-
Randomised controlled trial
- SC:
-
Subcutaneous
- SD:
-
Standard deviation
- SDAI:
-
Simplified Disease Activity Index
- SF-36:
-
36-Item Short-Form Health Survey
- SJC28:
-
Swollen 28-Joint Count
- TEAE:
-
Treatment-emergent adverse event
- TESAE:
-
Treatment-emergent serious adverse event
- TJC28:
-
Tender 28-Joint Count
- TNFi:
-
Tumour necrosis factor inhibitor
- tsDMARD:
-
Targeted synthetic disease-modifying antirheumatic drug
References
Safiri S, Kolahi AA, Hoy D, Smith E, Bettampadi D, Mansournia MA, et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: a systematic analysis of the Global Burden of Disease study 2017. Ann Rheum Dis. 2019;78(11):1463–71. https://doi.org/10.1136/annrheumdis-2019-215920.
Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Primers. 2018;4:18002.
Centers for Disease Control and Prevention. Rheumatoid arthritis (RA). Available from: https://www.cdc.gov/arthritis/basics/rheumatoid-arthritis.html. [cited 2021 January 29].
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38. https://doi.org/10.1016/S0140-6736(16)30173-8.
Sparks JA. Rheumatoid arthritis. Ann Intern Med. 2019;170(1):ITC1–ITC16. https://doi.org/10.7326/AITC201901010.
Sokka T, Krishnan E, Häkkinen A, Hannonen P. Functional disability in rheumatoid arthritis patients compared with a community population in Finland. Arthritis Rheum. 2003;48(1):59–63. https://doi.org/10.1002/art.10731.
West E, Jonsson SW. Health-related quality of life in rheumatoid arthritis in Northern Sweden: a comparison between patients with early RA, patients with medium-term disease and controls, using SF-36. Clin Rheumatol. 2005;24(2):117–22. https://doi.org/10.1007/s10067-004-0976-6.
Puolakka K, Kautiainen H, Mottonen T, Hannonen P, Hakala M, Korpela M, et al. Predictors of productivity loss in early rheumatoid arthritis: a 5 year follow up study. Ann Rheum Dis. 2005;64(1):130–3. https://doi.org/10.1136/ard.2003.019034.
Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2020;79(6):685–99. https://doi.org/10.1136/annrheumdis-2019-216655.
Smolen JS, Breedveld FC, Burmester GR, Bykerk V, Dougados M, Emery P, et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann Rheum Dis. 2016;75(1):3–15. https://doi.org/10.1136/annrheumdis-2015-207524.
Maini R, St Clair EW, Breedveld F, Furst D, Kalden J, Weisman M, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999;354(9194):1932–9.
Westhovens R, Houssiau F, Joly J, Everitt DE, Zhu Y, Sisco D, et al. A phase I study assessing the safety, clinical response, and pharmacokinetics of an experimental infliximab formulation for subcutaneous or intramuscular administration in patients with rheumatoid arthritis. J Rheumatol. 2006;33(5):847–53.
European Medicines Agency. Remsima summary of product characteristics. Available from: https://www.ema.europa.eu/en/documents/product-information/remsima-epar-product-information_en.pdf. [cited 2021 January 29].
US Food and Drug Administration. Highlights of prescribing information, inflectra (infliximab-dyyb). Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125544s008lbl.pdf. [cited 2021 January 29].
European Medicines Agency. Assessment report on extension(s) of marketing authorisation: Remsima. Available from: https://www.ema.europa.eu/en/documents/variation-report/remsima-h-c-2576-x-0062-epar-assessment-report-variation_en.pdf. [cited 2021 January 29].
Westhovens R, Wiland P, Zawadzki M, Ivanova D, Berrocal Kasay A, Chalouhi El-Khouri E et al. Efficacy, pharmacokinetics and safety of subcutaneous versus intravenous CT-P13 in rheumatoid arthritis: a randomized phase I/III trial. . Rheumatology (Oxford). 2020;doi: https://doi.org/10.1093/rheumatology/keaa580 [epub ahead of print].
Yoo DH, Hrycaj P, Miranda P, Ramiterre E, Piotrowski M, Shevchuk S, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72(10):1613–20. https://doi.org/10.1136/annrheumdis-2012-203090.
Yoo DH, Racewicz A, Brzezicki J, Yatsyshyn R, Arteaga ET, Baranauskaite A, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2016;18(1):82. https://doi.org/10.1186/s13075-016-0981-6.
Prevoo ML, Van ‘t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38(1):44–8. https://doi.org/10.1002/art.1780380107.
Aletaha D, Smolen J. The Simplified Disease Activity Index (SDAI) and the Clinical Disease Activity Index (CDAI): a review of their usefulness and validity in rheumatoid arthritis. Clin Exp Rheumatol. 2005;23(5 Suppl 39):S100–8.
Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, et al. American College of Rheumatology/European League Against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Arthritis Rheum. 2011;63(3):573–86. https://doi.org/10.1002/art.30129.
Anderson J, Caplan L, Yazdany J, Robbins ML, Neogi T, Michaud K, et al. Rheumatoid arthritis disease activity measures: American College of Rheumatology recommendations for use in clinical practice. Arthritis Care Res (Hoboken). 2012;64(5):640–7. https://doi.org/10.1002/acr.21649.
Fransen J, van Riel PL. The Disease Activity Score and the EULAR response criteria. Rheum Dis Clin N Am. 2009;35(4):745–57, vii-viii. https://doi.org/10.1016/j.rdc.2009.10.001.
Felson DT, Anderson JJ, Boers M, Bombardier C, Chernoff M, Fried B, et al. The American College of Rheumatology preliminary core set of disease activity measures for rheumatoid arthritis clinical trials. The Committee on Outcome Measures in Rheumatoid Arthritis Clinical Trials. Arthritis Rheum. 1993;36(6):729–40. https://doi.org/10.1002/art.1780360601.
Felson DT, Anderson JJ, Lange ML, Wells G, LaValley MP. Should improvement in rheumatoid arthritis clinical trials be defined as fifty percent or seventy percent improvement in core set measures, rather than twenty percent? Arthritis Rheum. 1998;41(9):1564–70. https://doi.org/10.1002/1529-0131(199809)41:9<1564::AID-ART6>3.0.CO;2-M.
Behrens F, Koehm M, Schwaneck EC, Schmalzing M, Gnann H, Greger G, et al. Use of a “critical difference” statistical criterion improves the predictive utility of the Health Assessment Questionnaire-Disability Index score in patients with rheumatoid arthritis. BMC Rheumatol. 2019;3(1):51. https://doi.org/10.1186/s41927-019-0095-2.
Kosinski M, Zhao SZ, Dedhiya S, Osterhaus JT, Ware JE Jr. Determining minimally important changes in generic and disease-specific health-related quality of life questionnaires in clinical trials of rheumatoid arthritis. Arthritis Rheum. 2000;43(7):1478–87. https://doi.org/10.1002/1529-0131(200007)43:7<1478::AID-ANR10>3.0.CO;2-M.
Harel O, Zhou X-H. Multiple imputation: review of theory, implementation and software. Stat Med. 2007;26(16):3057–77. https://doi.org/10.1002/sim.2787.
Phillippo D, Ades AE, Dias S, Palmer S, K.R. A, Welton NJ. NICE DSU technical support document 18: Methods for population-adjusted indirect comparisons in submissions to NICE. Available from: http://nicedsu.org.uk/technical-support-documents/population-adjusted-indirect-comparisons-maic-and-stc/. [cited 2021 January 29].
Phillippo DM, Dias S, Ades AE, Belger M, Brnabic A, Schacht A, et al. Multilevel network meta-regression for population-adjusted treatment comparisons. J R Stat Soc A Stat. 2020;183(3):1189–210. https://doi.org/10.1111/rssa.12579.
Rubin DB. Multiple imputation for nonresponse in surveys. New York: John Wiley & Sons Inc.; 1987. https://doi.org/10.1002/9780470316696.
Fransen J, van Riel PL. The Disease Activity Score and the EULAR response criteria. Clin Exp Rheumatol. 2005;23(5 Suppl 39):S93–9.
Hoaglin DC, Hawkins N, Jansen JP, Scott DA, Itzler R, Cappelleri JC, et al. Conducting indirect-treatment-comparison and network-meta-analysis studies: report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: part 2. Value Health. 2011;14(4):429–37. https://doi.org/10.1016/j.jval.2011.01.011.
Rahman MU, Strusberg I, Geusens P, Berman A, Yocum D, Baker D, et al. Double-blinded infliximab dose escalation in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66(9):1233–8. https://doi.org/10.1136/ard.2006.065995.
Wolbink GJ, Voskuyl AE, Lems WF, de Groot E, Nurmohamed MT, Tak PP, et al. Relationship between serum trough infliximab levels, pretreatment C reactive protein levels, and clinical response to infliximab treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64(5):704–7. https://doi.org/10.1136/ard.2004.030452.
St Clair EW, Wagner CL, Fasanmade AA, Wang B, Schaible T, Kavanaugh A, et al. The relationship of serum infliximab concentrations to clinical improvement in rheumatoid arthritis: results from ATTRACT, a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46(6):1451–9. https://doi.org/10.1002/art.10302.
Takeuchi T, Miyasaka N, Inoue K, Abe T, Koike T. Impact of trough serum level on radiographic and clinical response to infliximab plus methotrexate in patients with rheumatoid arthritis: results from the RISING study. Mod Rheumatol. 2009;19(5):478–87. https://doi.org/10.3109/s10165-009-0195-8.
Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343(22):1594–602. https://doi.org/10.1056/NEJM200011303432202.
Aletaha D, Ward MM. Duration of rheumatoid arthritis influences the degree of functional improvement in clinical trials. Ann Rheum Dis. 2006;65(2):227–33. https://doi.org/10.1136/ard.2005.038513.
Quinn MA, Conaghan PG, O'Connor PJ, Karim Z, Greenstein A, Brown A, et al. Very early treatment with infliximab in addition to methotrexate in early, poor-prognosis rheumatoid arthritis reduces magnetic resonance imaging evidence of synovitis and damage, with sustained benefit after infliximab withdrawal: results from a twelve-month randomized, double-blind, placebo-controlled trial. Arthritis Rheumatism. 2005;52(1):27–35.
Acknowledgements
Statistical analysis and the development of a draft outline and subsequent drafts in consultation with the authors was provided by Samuel Aballéa at Creativ-Ceutical (Paris, France) and Aix-Marseille University (Marseille, France) and Renata Majewska at Creativ-Ceutical (Paris, France), and funded by Celltrion Healthcare (Incheon, Republic of Korea). Editorial support was provided by Beatrice Tyrrell, DPhil, at Aspire Scientific Limited (Bollington, UK), and funded by Celltrion Healthcare (Incheon, Republic of Korea).
Funding
The analysis was sponsored by Celltrion Healthcare (Incheon, Republic of Korea).
Author information
Authors and Affiliations
Contributions
BC, YA, RA, RC, PD, FI, MTN, SJL and DHY contributed to data analysis or interpretation. MT, TSK and JN contributed to study design, data collection and data analysis or interpretation. GP contributed to data collection and data analysis or interpretation. All authors reviewed and critically revised the manuscript, approved the final draft and are accountable for the accuracy and integrity of the article.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
BC received honoraria from AbbVie, Bristol-Myers Squibb, Gilead, Janssen, Lilly, Merck, Novartis, Pfizer, Roche-Chugai, Sanofi and UCB; and research grants from Novartis, Pfizer and Roche. YA declares no competing interests. RA received honoraria from AbbVie, Bristol-Myers Squibb, Celltrion, Gilead, Janssen, Lilly, Merck, Novartis, Pfizer, Roche-Chugai and UCB; and research grants from Novartis, Pfizer and Roche. RC received a speaker’s fee and a consultation grant from AbbVie, BMS, Celltrion, Fresenius-Kabi, Gilead-Galapagos, Lilly, MSD, Pfizer, Roche, Samsung-Bioepis, Sanofi and UCB. PD received speaker’s fees from AbbVie, Bristol-Myers Squibb, Galapagos, Lilly and Sanofi. FI received a speaker’s fee and consultation grant from AbbVie, Actelion, BMS, Biogen, Lilly, MSD, Pfizer, Roche, Sanofi and UCB. MTN received consulting fees from AbbVie, Celgene, Celltrion, Eli Lilly, Janssen and Sanofi; speakers fees from AbbVie, Bristol-Myers Squibb, Eli Lilly, Roche and Sanofi; and research funding from AbbVie, Bristol-Myers Squibb, Celgene, Eli Lilly, Janssen, MSD, Mundipharma, Novartis, Pfizer, Roche and Sanofi. MT is a consultant at Creativ-Ceutical, an international consulting firm providing services to public and private organisations. Creativ-Ceutical provides services for most pharmaceutical industry companies including Celltrion. SJL is a full-time employee of Celltrion Inc. TSK is a full-time employee of Celltrion Healthcare. JN is a full-time employee of Celltrion Healthcare. GP is a full-time employee of Celltrion Inc. DHY is on the speaker’s bureau for Celltrion and Celltrion Healthcare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
List of variables used as potential covariates. Table S2. Pearson correlation between baseline parameters. Table S3. Prediction models in Remsima IV treatment arm (Study 3.1). Table S4. Comparison of safety outcomes in Studies 3.5 and 3.1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Combe, B., Allanore, Y., Alten, R. et al. Comparative efficacy of subcutaneous (CT-P13) and intravenous infliximab in adult patients with rheumatoid arthritis: a network meta-regression of individual patient data from two randomised trials. Arthritis Res Ther 23, 119 (2021). https://doi.org/10.1186/s13075-021-02487-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-021-02487-x