
Abstract
Background
Osteoarthritis (OA) is the most prevalent form of arthritis and the major cause of disability and overall diminution of quality of life in the elderly population. Currently there is no cure for OA, partly due to the large gaps in our understanding of its underlying molecular and cellular mechanisms. Macrophage migration inhibitory factor (MIF) is a procytokine that mediates pleiotropic inflammatory effects in inflammatory diseases such as rheumatoid arthritis (RA) and ankylosing spondylitis (AS). However, data on the role of MIF in OA is limited with conflicting results. We undertook this study to investigate the role of MIF in OA by examining MIF genotype, mRNA expression, and protein levels in the Newfoundland Osteoarthritis Study.
Methods
One hundred nineteen end-stage knee/hip OA patients, 16 RA patients, and 113 healthy controls were included in the study. Two polymorphisms in the MIF gene, rs755622, and -794 CATT5-8, were genotyped using polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) and PCR followed by automated capillary electrophoresis, respectively. MIF mRNA levels in articular cartilage and subchondral bone were measured by quantitative polymerase chain reaction. Plasma concentrations of MIF, tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β) were measured by enzyme-linked immunosorbent assay.
Results
rs755622 and -794 CATT5-8 genotypes were not associated with MIF mRNA or protein levels or OA (all p ≥ 0.19). MIF mRNA level in cartilage was lower in OA patients than in controls (p = 0.028) and RA patients (p = 0.004), while the levels in bone were comparable between OA patients and controls (p = 0.165). MIF protein level in plasma was lower in OA patients than in controls (p = 3.01 × 10−10), while the levels of TNF-α, IL-6 and IL-1β in plasma were all significantly higher in OA patients than in controls (all p ≤ 0.0007). Multivariable logistic regression showed lower MIF and higher IL-1β protein levels in plasma were independently associated with OA (OR per SD increase = 0.10 and 8.08; 95% CI = 0.04–0.19 and 4.42–16.82, respectively), but TNF-α and IL-6 became non-significant.
Conclusions
Reduced MIF mRNA and protein expression in OA patients suggested MIF might have a protective role in OA and could serve as a biomarker to differentiate OA from other joint disorders.
Similar content being viewed by others


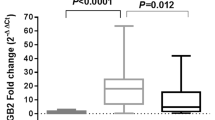
Background
Osteoarthritis (OA), the most common form of arthritis, is characterized by the loss of articular cartilage due to an imbalance between extracellular matrix destruction and repair, accompanied with osteophyte formation at joint margins and changes in other joint tissues [1]. OA causes pain and functional limitations in affected joints and is the major cause of disability and overall diminution of quality of life in the elderly population [2]. At present, there is no cure for it yet, partly because of the large gaps in our understanding of the underlying molecular and cellular mechanisms of OA.
Risk factors such as older age, female sex, obesity, and joint injury have been associated with OA [3]. Chondrocyte, the only cellular component in articular cartilage, synthesizes cartilage matrix proteins such as collagens and proteoglycans and maintains the structural and functional integrity of the matrix. Chondrocyte plays a critical role in the pathogenesis of OA in responding to excess or abnormal mechanical loading and biological stimuli such as dysregulated cytokine activities [4]. In OA, excess pro-inflammatory cytokines are produced by joint tissue cells including chondrocytes and released into synovial fluid, and then act on the resident cells in an autocrine-paracrine manner [5]. In response to the elevated level of pro-inflammatory cytokines, the expression of proteinases such as metalloproteinases (MMPs) and aggrecanases is upregulated, and compensatory synthesis pathways are downregulated in chondrocytes, which leads to degradation of cartilage matrix and deterioration in the structural and functional properties of cartilage [4]. The cartilage breakdown products released into the synovial fluid promote synovial inflammation, which produces more pro-inflammatory cytokines and proteases and creates a vicious cycle of more cartilage being degraded and subsequently provoking more inflammation [6].
Macrophage migration inhibitory factor (MIF) is a glucocorticoid (GC)-induced mediator that plays a central role in promoting inflammation by overriding the anti-inflammatory action of GCs [7]. Because of this, numerous studies have been conducted to investigate the role of MIF in diseases, particularly autoimmune disorders such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) where GC therapy is required [8]. Studies have found that MIF is overexpressed in serum, synovial fluid and cultured fibroblast-like synoviocytes (FLS) from RA patients compared with those from controls [9], indicating a significant role of MIF in RA. Similarly, MIF is also implicated in the pathogenesis of SLE [10]. Chronic inflammation has been implicated in OA and cytokines such as tumor necrosis factor alpha (TNF-α), interlukin-6 (IL-6), and interlukin-1β (IL-1β) have been reported to be associated with OA [11,12,13]. However, data on the role of MIF in OA is limited with conflicting results [14,15,16,17,18,19]. Therefore, we undertook this study to investigate the potential role of MIF in OA by systematically examining MIF genotypes, mRNA and protein expressions in a well-established OA cohort—the Newfoundland Osteoarthritis Study (NFOAS) [20].
Methods
Study participants
Study participants were derived from the NFOAS, which was initiated in 2011 to study the genetic, biochemical and epigenetic markers of OA [20]. OA and RA patients were all at the end-stage of the disease and underwent total joint replacement (TJR) surgery in St. John’s, Newfoundland and Labrador (NL), Canada, between 2011 and 2017. Controls were from the same population and had no self-reported arthritis at all joints. Diagnosis of OA and RA was based on American College of Rheumatology (ACR) and 2010 ACR/European League Against Rheumatism (EULAR) clinical diagnostic criteria [21,22,23] and was confirmed by pathological report after TJR surgery. The study was approved by the Health Research Ethics Authority of Newfoundland and Labrador (HREB # 2011.311) and written consent was obtained from all participants.
Demographic and joint pain data collection
Date of birth and sex data were collected with a general health questionnaire, and age at the time of TJR surgery was then calculated. Weight and height data were retrieved from Eastern Health Meditech Health Care Information System, and body mass index (BMI) was calculated as weight in kg/squared height in meters. Joint pain of OA and RA patients was assessed using the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) Likert version 3.0. WOMAC pain subscale scores 0–20, with 0 representing no pain and 20 representing the most severe pain.
Plasma separation and blood DNA extraction
Whole blood samples were collected into Vacutainer EDTA tubes following at least 8 h of fasting. Plasma was separated by centrifugation at 2000 RPM for 10 min and stored at − 80 °C until analysis. Blood DNA was extracted by modified salting-out method [24]. Briefly, for 8 ml of whole blood, after plasma was removed, 25 ml of red cell lysis buffer (140 mM NH4Cl, 17 mM Tris-HCl, pH 7.65) was added and the sample was then incubated at 37 °C in water bath for 10 min. After centrifugation at 2500 RPM for 5 min and removal of supernatant, another 15 ml of red cell lysis buffer was added, followed by incubation and centrifugation. The pellet was then mixed with 3 ml of nuclei lysis buffer (400 mM NaCl, 2 mM EDTA, 10 mM Tris-HCl, pH 8.0) by vortex. Two hundred microliters of 10% SDS and 500 μl of pronase E solution (3 mg/ml pronase E, 1% SDS, 2 mM EDTA) were added and sample was incubated at 37 °C in water bath till the pellet was digested completely. One milliliter of saturated NaCl (6 M) was added and the sample was vigorously mixed for 15 s and then centrifuged at 2800 RPM for 18 min. The supernatant was carefully transferred into a new tube, and two volumes of absolute ethanol was added. The tube was inverted for a few times and precipitated DNA was fished out with a Pasteur pipette and washed with 70% ethanol. DNA pellet was dried at room temperature, dissolved in 500 μl of TE buffer (1 mM EDTA, 10 mM Tris-HCl, pH 8.0), and stored at 4 °C.
MIF genotyping for single nucleotide polymorphism (SNP) -173 G/C and microsatellite -794 CATT5-8
A functional SNP in the promoter region of MIF, -173 G/C (rs755622), was genotyped by polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) method. The 366-bp polymorphic fragment was amplified with forward primer 5′-ACTAAGAAAGACCCGAGGC-3′ and reverse primer 5′-GGGGCACGTTGGTGTTTAC-3′ in a 20 μl reaction containing 100 ng genomic DNA, 0.1 μM forward and reverse primer each, 0.2 mM dNTPs, 1 mM MgCl2, 2 μl 10× PCR buffer, and 0.4 unit of Platinum Taq DNA polymerase (Invitrogen, Waltham, MA, USA, Cat # 10966034). Cycling conditions were as follows: initial activation at 95 °C for 10 min, followed by 95 °C for 1 min, 58 °C for 45 s, 72 °C for 45 s, repeated in 35 cycles, and a final extension at 72 °C for 7 min. Five microliters of PCR product was digested with 5 units of Alu I restriction endonuclease (Invitrogen, Waltham, MA, USA, Cat # 45200029) in a 20 μl reaction at 37 °C overnight. The digestion product was resolved on a 2% agarose gel, stained with SYBRTM Safe, and visualized using ultraviolet (UV) gel imaging system. The C allele that contains two Alu I cleavage sites yielded 205-, 98-, and 63-bp bands, while the G allele that contains one Alu I cleavage site yielded 268- and 98-bp bands.
Microsatellite -794 CATT5-8, also located in the promoter region of MIF, was genotyped by PCR followed by automated capillary electrophoresis. The forward primer was 5′-CTATGTCATGGCTTATCTTC-3′ labeled with 6-carboxyfluorescein (6-FAM) at 5′-end, and the reverse primer was 5′-TCCACTAATGGTAAACTCGG-3′. PCR reaction and cycling conditions were as above with 200 ng genomic DNA, 1.25 mM MgCl2, and annealing temperature 55 °C. PCR product was 1:10 diluted with ddH2O and sequenced on ABI 3730 DNA Analyzer (Applied Biosystems, Waltham, MA, USA). The CATT5-8 alleles were identified using Peak Scanner software V 1.0 (Applied Biosystems, Waltham, MA, USA).
RNA extraction and MIF gene expression measurement
Articular cartilage and subchondral bone tissues of OA patients and articular cartilage tissue of RA patients were retained from either tibial plateaus or femoral heads during TJR surgeries; those of controls were collected from femoral heads during hemiarthroplasty for hip fracture patients. Samples were flash-frozen and stored in liquid nitrogen until the experiment. RNA was extracted from the cartilage and bone samples as described previously [25]. mRNA levels of the MIF gene were measured by quantitative PCR (qPCR) method. Complementary DNA (cDNA) synthesis, qPCR, and relative quantification (RQ) of MIF gene expression were performed as described previously [25]. Table 1 presents the primer sequences, the sizes of PCR products, as well as primer efficiencies.
Enzyme-linked immunosorbent assay (ELISA) of MIF, TNF-α, IL-6, and IL-1β
Plasma levels of MIF, TNF-α, IL-6, and IL-1β were measured in duplicates using sandwich enzyme-linked immunosorbent assay (ELISA) kits (Human MIF DuoSet ELISA, DY289; DuoSet ELISA Ancillary Reagent Kit 2, DY008; Human TNF-alpha Quantikine HS ELISA, SSTA00E; Human IL-6 Quantikine HS ELISA Kit, SS600C; Human IL-1 beta Quantikine HS ELISA Kit, SSLB00D, R&D systems, Minneapolis, MN, USA). All assays were performed according to the manufacturer’s instructions. The intra-assay CV% of MIF, TNF-α, IL-6, and IL-1β were ≤ 2.5%, 2.2%, 4.7%, and 4.4%, respectively, and inter-assay CV% were ≤ 6.7%, 10.8%, and 10.7% for TNF-α, IL-6, and IL-1β, respectively.
Statistical analysis
Normality of distribution was tested with the Shapiro-Wilk test. Age, BMI, and WOMAC pain scores, which were not normally distributed, were compared with the Mann-Whitney U test. Sex distribution was tested with chi-squared test. Hardy–Weinberg equilibrium was assessed by Fisher’s exact test. Association of MIF genotypes with OA was tested by chi-squared test of the distribution of genotypes containing risk alleles, -173 C allele in rs755622 and 7 or above CATT-repeats in both alleles in -794 CATT5-8 [26]. MIF mRNA levels were natural log transformed and compared with independent sample Student’s t test. Plasma cytokine levels were natural log transformed and compared with the Mann-Whitney U test due to their non-normal distribution. Linear regression was used to test associations between MIF genotypes and mRNA and protein levels. Spearman correlation coefficients were calculated to evaluate the relationships between the levels of MIF and other pro-inflammatory cytokines in plasma. Logistic regression was utilized for adjustment for potential confounding factors. Natural log-transformed cytokine concentrations were standardized using Z score for the logistic regression. Significant level was defined at α = 0.05. All analyses were performed in R Studio with R version 3.6.3. Visualizations of the results were done with ggplot2 R package [27].
Results
A total of 248 study participants were included in the study, of which 119 were OA patients, 16 were RA patients, and 113 were controls. OA patients were significantly older than controls, had a higher BMI, and less females than controls and RA patients (all p < 0.04, Table 2). OA patients were also older than RA patients, but the difference was not statistically significant (p > 0.05, Table 2). There was no difference in age, BMI, or percentage of females between controls and RA patients (all p ≥ 0.13, Table 2).
MIF genotypes and OA
Two polymorphisms in the promoter region of MIF were genotyped for OA patients and controls. For rs755622, the overall frequency of GG genotype was 76%, and that of CC was 2%. The distribution of the genotypes showed no deviation from the Hardy-Weinberg equilibrium (p = 0.56). Minor allele frequency (MAF) was 13%, which was lower than reported in other Caucasian populations [28]. Frequency of genotypes containing C allele (GC + CC) was 20% in control group and 28% in OA group, but the difference was not statistically significant (p = 0.19). When examining hip (n = 64) and knee (n = 55) separately, GC + CC frequency was 28% in hip OA and 27% in knee OA patients, and it was not associated with OA in either hip or knee joint (p = 0.24 and 0.31 for hip and knee OA, respectively).
Six genotypes of -794 CATT5-8 were identified, 6/6, 6/7, 6/8, 7/7, 7/8, and 8/8. The frequencies were 8%, 33%, 3%, 41%, 15%, and 1%, respectively, which were also in accordance with the Hardy–Weinberg equilibrium (p = 0.35). Allele frequencies were 26%, 65%, and 10% for CATT6, CATT7, and CATT8. Compared to what was reported in other Caucasian populations [29], CATT5 allele was absent in our cohort, CATT6 had a lower frequency, while CATT7 and CATT8 had much higher frequencies. Frequency of genotypes containing 7 or above CATT-repeats in both alleles (7/7+7/8+8/8) was 56% in control group, 57% in OA group, 63% in hip OA, and 51% in knee OA. No significant association between -794 CATT5-8 and OA was observed (p = 0.83, 0.38 and 0.55 for OA, hip OA and knee OA, respectively). Genotype frequencies of rs755622 and -794 CATT5-8 in control and OA groups were presented in Table 3.
MIF mRNA levels in joint cartilage/bone and OA
MIF mRNA levels were assessed in a total of 59 articular cartilage and 43 subchondral bone samples including 13 control, 30 OA and 16 RA articular cartilage samples, and 6 control and 37 OA subchondral bone samples. The RA samples were included so that we could compare the difference between OA and an autoimmune disease that also involves chronic inflammation. In cartilage, natural log-transformed MIF mRNA level was significantly lower in OA patients than in controls (mean ± SD, logRQ = − 0.47 ± 0.32 vs. − 0.21 ± 0.35; p = 0.028) and RA patients (mean ± SD, logRQ = − 0.18 ± 0.28; p = 0.004). The levels were not significantly different between RA patients and controls (p = 0.747). When hip and knee joints were examined separately, the significances remained between hip OA patients (n = 21) and controls (mean ± SD, logRQ = − 0.49 ± 0.30 vs. - 0.21 ± 0.35; p = 0.02) and hip RA patients (n = 4; mean ± SD, logRQ = − 0.08 ± 0.18; p = 0.016). Knee OA patients (n = 9) also had a lower level of MIF mRNA than controls and knee RA patients (n = 12) (mean ± SD, logRQ = − 0.40 ± 0.38, − 0.21 ± 0.35, and − 0.21 ± 0.30, respectively), but the differences were not statistically significant (p ≥ 0.2). Natural log-transformed MIF mRNA levels were not significantly different between OA patients and controls in subchondral bone samples (mean ± SD, logRQ = 0.11 ± 0.47 vs. 0.41 ± 0.56; p = 0.165), which were all from hip joints (Fig. 1). There was no correlation between natural log-transformed MIF mRNA level and the two polymorphisms in MIF promoter region (p = 0.97 for rs755622 and p = 0.93 for -794 CATT5-8).

Boxplots for natural log-transformed MIF mRNA levels in control, OA, and RA groups. a Natural log-transformed MIF mRNA levels in control, OA, and RA groups in cartilage. b Natural log-transformed MIF mRNA levels in control and OA groups in subchondral bone. c Natural log-transformed MIF mRNA levels in control, hip OA, hip RA, knee OA, and knee RA groups in cartilage. MIF, macrophage migration inhibitory factor; OA, osteoarthritis; RA, rheumatoid arthritis; RQ, relative quantification; p values were obtained from independent sample Student’s t test
Association between plasma MIF levels and OA
Plasma MIF concentrations were measured for all 119 OA patients and 109 controls. The raw MIF concentration was natural log-transformed for normality because its distribution was highly skewed. The average natural log-transformed MIF level in OA patients was significantly lower than that in controls (mean ± SD, 1.43 ± 0.46 vs. 1.82 ± 0.40 log ng/ml; p = 3.01 × 10−10, Fig. 2). The significances remained when hip and knee OA were compared with control group separately (mean ± SD, 1.46 ± 0.46 and 1.40 ± 0.45 log ng/ml in hip and knee OA, respectively; p = 1.05 × 10−6 and 3.37 × 10−8, respectively). Natural log-transformed plasma MIF level was not correlated with MIF genotypes (p = 0.31 for rs755622 and p = 0.62 for -794 CATT5-8) or natural log-transformed MIF mRNA level in cartilage (p = 0.21).

Boxplots for natural log-transformed plasma cytokine levels. MIF, macrophage migration inhibitory factor; TNF-a, tumor necrosis factor alpha; IL-6, interlukin-6; IL-1β, interlukin-1β; OA, osteoarthritis. Log concentration for MIF: log ng/ml; log concentration for TNF-a, IL-6, and IL-1β: log pg/ml; p values were obtained from Mann-Whitney U test
Relationships between joint pain intensity and MIF levels and OA
Among the 30 OA and 16 RA patients with cartilage MIF mRNA level data, 29 OA and 11 RA patients had WOMAC pain scores available, ranging from 2 to 20. There was no correlation between cartilage MIF mRNA levels and WOMAC pain scores (p = 0.72). Among the 119 OA patients with plasma MIF concentration data, 111 had WOMAC pain scores available, also ranging from 2 to 20. There was no correlation between plasma MIF concentrations and WOMAC pain scores (p = 0.26). WOMAC pain scores were not significantly different between OA and RA patients (13.98 ± 4.55 vs. 15.36 ± 2.94, p = 0.44).
Relationships between OA, TNF-α, IL-6, IL-1β, and MIF
Given that MIF promotes the production of cytokines [7], we measured the levels of three inflammatory factors, namely TNF-α, IL-6, IL-1β in the same plasma samples. We found that the natural log-transformed concentrations of the three cytokines in OA patients were all significantly higher than those in controls (all p ≤ 0.0007) (Fig. 2). MIF was significantly correlated with IL-1β (rho = 0.18, p = 0.01) but not TNF-α or IL-6 (p > 0.2), and TNF-α, IL-6, and IL-1β were correlated with each other (p ≤ 0.01).
In the multivariable analysis with logistic regression including all four cytokines and age, sex, and BMI, we found that lower MIF and higher IL-1β natural log-transformed concentrations in plasma were independently associated with OA (OR per SD increase = 0.10 (95% CI = 0.04–0.19) and 8.08 (95% CI 4.42–16.82), respectively), while TNF-α and IL-6 became non-significant.
Discussion
We investigated the role of MIF in OA by systematically examining the association between OA and MIF genotypes, mRNA expression, and protein production and found that MIF might have a protective role in OA.
rs755622 and -794 CATT5-8 are two polymorphisms located in the promoter region of the MIF gene, and studies on their association with MIF expression have yielded conflicting results. Radstake TR et al. reported that rs755622 C and -794 CATT7 alleles were associated with higher MIF level in circulation in the Netherlands population [26], while Baños-Hernández CJ et al. and Cruz-Mosso et al. found that there was no association between these alleles and blood MIF mRNA level or serum MIF level in the southern Mexican population [30, 31]. In consistent with Baños-Hernández and Cruz-Mosso’s findings, we did not observe any association between these variants and MIF expression either at mRNA level in cartilage or protein level in plasma, suggesting the function of these two variants may be population specific. The MIF allelic structure was reported to have significant population stratification [32]. Zhong X et al. compared allele frequencies of -794 CATT5-8 in five different populations including Zambia, North African, Korean, Caucasian, and African American and found they were highly variable between populations, likely due to relatedness between populations and selection pressure [32]. Similarly, the allele frequencies of rs755622 between Caucasian, African, and African American also showed significant variation [29, 32]. Our study cohort had a lower MAF of rs755622, absence of CATT5 allele, lower frequency of CATT6 allele and higher frequencies of CATT7-8 alleles for -794 CATT5-8 compared to what was reported in other Caucasian populations [28, 29], supporting this and suggesting different regulatory mechanisms for MIF expression in the NL population which has an unique genetic structure as a genetic isolate [33].
These two polymorphisms have been associated with the susceptibility or severity of different diseases. rs755622 C and -794 CATT7 alleles were found to be associated with increased risk of RA [34], systemic sclerosis (SSc) [30], and SLE [31], and RA patients with rs755622 C allele and homozygotes for -794 CATT7 allele displayed higher levels of joint damage [26]. The current study was the first study to examine these variants in OA, and in contrast to what was reported in autoimmune diseases such as RA and SLE, we found neither of these variants was associated with OA.
However, while MIF mRNA level in cartilage was not correlated with MIF protein level in plasma, we found that MIF mRNA level in articular cartilage was significantly lower in OA patients than in controls and RA patients, and MIF protein level in plasma was also significantly lower in OA patients than in controls. While there was no data reported on MIF mRNA expression in human cartilage, several studies reported MIF protein levels in synovial fluid samples. Most of these studies did not have healthy controls but compared between OA and other autoimmune disease such as ankylosing spondylitis (AS) [14] and RA [15]. Consistent with our findings, they reported a significant lower level of MIF protein in OA patients than in AS and RA patients [14, 15], suggesting a different role of MIF between OA and autoimmune diseases. However, we found that there was no difference in MIF mRNA level in cartilage between RA patients and controls. This is in contrast to what was found in synovial fluid [35], suggesting that cartilage may not be a major contributor of MIF production in RA; rather, MIF may be released by other joint tissues such as synovium into synovial fluid and cause cartilage degradation. Further studies are needed to confirm this.
Previous reports on blood MIF level in OA were conflicting. While Liu M & Hu C found that serum level of MIF was significantly higher in radiographic OA patients than in controls and was associated with radiographic severity of the disease [18], Zhang PL et al. reported the level was similar between OA and healthy controls [17]. The discrepancy between these reports and our findings is unclear but could be partially explained by the difference in study populations. In addition to the difference in disease stage, these previous studies were from East Asia whereas our study population was Caucasian. Further, Zhang PL et al. [17] showed MIF levels in synovial fluid but not in serum were associated with the severity of self-reported pain in OA patients. We found no association between the WOMAC pain severity score and MIF protein level in plasma, in agreement with their findings that compared to that in the circulating system, the alteration of MIF concentration on a local level might have a closer relationship with the pain severity of the joints. We also found no association between the WOMAC pain severity score and MIF mRNA level in cartilage, suggesting a similar scenario to that in RA, that cartilage may not be the main source of MIF in synovial fluid in OA. A MIF knock-out mice study [16] showed that the deletion of MIF reduced OA severity in aged mice but not in surgical induced OA model, suggesting the role of MIF may be different in different subtypes of OA.
To the best of our knowledge, the current study was the first to have systematically examined the role of MIF in OA and documented evidence that MIF might have a protective role in OA. MIF is an intriguing cytokine which has multiple, sometimes opposite functions depending on the cellular source, the time of release, and the type of disease [14, 36]. When binding to CXCR2 or CXCR4, MIF promotes inflammation [37]; when binding to CD74, it appears to have beneficial effects [38]. MIF can also fine-tune inflammatory microenvironment to maintain homeostasis or a stable state of inflammation [39, 40] and exert organ- and tissue-protective effects during diseases [41, 42]. The beneficial effects of MIF have been reported in diseases such as community-acquired pneumonia [43], experimental liver fibrosis [44], and ischemic heart [38]. While how MIF exerts a beneficial effect in OA remains to be examined, in 3D culture, with MIF added to the medium, the expression of type II collagen (COL2) and aggrecan in chondrocytes was significantly enhanced while type X collagen (COL10) level was unchanged [45]. In mice transplanted with tissue-engineered cartilage from Mif +/+ and Mif −/− chondrocytes, Mif +/+ construct also had higher COL2 expression level but unchanged COL10 level [45]. These results suggested MIF secreted from chondrocytes could promote extracellular matrix (ECM) formation in an autocrine manner without inducing chondrocyte hypertrophy.
In OA joints, cartilage damage results in the production of damage-associated molecular patterns (DAMPs). DAMPs signal through pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) on chondrocytes, synovial macrophages, and FLS to induce production of inflammatory mediators through activation of innate immune system [46]. Cytokines including TNF-α, IL-1β, and IL-6 have been reported to be increased in OA patients [11, 47]. Consistent with these previous reports, we found that the concentrations of all three cytokines in plasma were significantly increased in OA patients compared to controls in the current study. The increased expression of these cytokines can augment production of chondrocyte-mediated catabolic proteinases and inhibit anabolic processes, and thus promote further ECM breakdown, perpetuating the cycle of inflammation and cartilage degradation [4]. MIF is thought to be an upstream regulator in innate immunity, mediates macrophage activation and retention, upregulates the expression of TLR4, and directly or indirectly promotes the production or expression of a large panel of pro-inflammatory molecules including cytokines such as TNF-α, IL-1β, and IL-6 [7, 48]. However, our data showed that MIF was not correlated with TNF-α or IL-6 and only had a moderate correlation with IL-1β. Further, the multivariable analysis showed that both MIF and IL-1β were independently and significantly associated with OA but not TNF-α or IL-6, suggesting that the increased levels of TNF-α, IL-1β, and IL-6 in OA were regulated by different mechanisms rather than MIF.
There are some limitations in the study. Hip and knee OA may have different etiologies. Our results showed that there was no difference in MIF protein levels in plasma between knee and hip OA patients, suggesting that MIF plays a similar role in knee and hip OA. However, when looking at MIF mRNA expression in cartilage in knee and hip OA separately, the significance was seen only in hip OA. There was a trend association for knee OA but statistically it was not significant, most likely due to small sample size. Secondly, due to the nature of the cross-sectional analyses, we were not able to assess the causal effect of MIF in OA. Longitudinal or Mendelian randomization studies are required to confirm this. Lastly, all study participants were from genetically homogenous NL population. Given that the allelic structure of MIF gene polymorphisms is strongly affected by the population studied, the generalizability of our finding to other populations may be limited.
Conclusions
Despite the MIF mRNA expression in cartilage was not correlated with the plasma MIF protein levels, we observed that both of them were reduced in OA patients, suggesting a protective role of MIF in OA which was different from what was reported in autoimmune disorders such as RA, AS, and SLE. Plasma MIF level could serve as a potential biomarker to differentiate OA from other joint diseases such as RA and AS.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 6-FAM:
-
6-Carboxyfluorescein
- ACR:
-
American College of Rheumatology
- AS:
-
Ankylosing spondylitis
- BMI:
-
Body mass index
- cDNA:
-
Complementary DNA
- COL10:
-
Type X collagen
- COL2:
-
Type II collagen
- DAMP:
-
Damage-associated molecular pattern
- ECM:
-
Extracellular matrix
- ELISA:
-
Enzyme-linked immunosorbent assay
- EULAR:
-
European League Against Rheumatism
- FLS:
-
Fibroblast-like synoviocytes
- GC:
-
Glucocorticoid
- IL-1β :
-
Interleukin-1 beta
- IL-6:
-
Interleukin-6
- MAF:
-
Minor allele frequency
- MIF:
-
Macrophage migration inhibitory factor
- MMP:
-
Metalloproteinase
- NFOAS:
-
Newfoundland Osteoarthritis Study
- OA:
-
Osteoarthritis
- PCR-RFLP:
-
Polymerase chain reaction–restriction fragment length polymorphism
- PRR:
-
Pattern recognition receptor
- qPCR:
-
Quantitative PCR
- RA:
-
Rheumatoid arthritis
- RQ:
-
Relative quantification
- SD:
-
Standard deviation
- SLE:
-
Systemic lupus erythematosus
- SNP:
-
Single nucleotide polymorphism
- SSc:
-
Systemic sclerosis
- TJR:
-
Total joint replacement
- TLR:
-
Toll-like receptor
- TNF-α:
-
Tumor necrosis factor-alpha
- UV:
-
Ultraviolet
- WOMAC:
-
Western Ontario and McMaster Universities Osteoarthritis Index
References
Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697–707.
Guccione AA, Felson DT, Anderson JJ, Anthony JM, Zhang Y, Wilson PW, Kelly-Hayes M, Wolf PA, Kreger BE, Kannel WB. The effects of specific medical conditions on the functional limitations of elders in the Framingham Study. Am J Public Health. 1994;84(3):351–8.
Zhang Y, Jordan JM. Epidemiology of osteoarthritis. Clin Geriatr Med. 2010;26(3):355–69.
Goldring SR, Goldring MB. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res. 2004;427(Suppl):S27–36.
Goldring MB. Osteoarthritis and cartilage: the role of cytokines. Curr Rheumatol Rep. 2000;2(6):459–65.
Martel-Pelletier J. Pathophysiology of osteoarthritis. Osteoarthritis Cartilage. 2004;12(Suppl A):S31–3.
Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, Cerami A, Bucala R. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377(6544):68–71.
Aeberli D, Leech M, Morand EF. Macrophage migration inhibitory factor and glucocorticoid sensitivity. Rheumatology (Oxford). 2006;45(8):937–43.
Leech M, Metz C, Hall P, Hutchinson P, Gianis K, Smith M, Weedon H, Holdsworth SR, Bucala R, Morand EF. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 1999;42(8):1601–8.
Lang T, Foote A, Lee JP, Morand EF, Harris J. MIF: implications in the Pathoetiology of Systemic Lupus Erythematosus. Front Immunol. 2015;6:577.
Munoz-Valle JF, Oregon-Romero E, Rangel-Villalobos H, Martinez-Bonilla GE, Castaneda-Saucedo E, Salgado-Goytia L, Leyva-Vazquez MA, Illades-Aguiar B, Alarcon-Romero Ldel C, Espinoza-Rojo M, et al. High expression of TNF alpha is associated with -308 and -238 TNF alpha polymorphisms in knee osteoarthritis. Clin Exp Med. 2014;14(1):61–7.
Livshits G, Zhai G, Hart DJ, Kato BS, Wang H, Williams FM, Spector TD. Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: the Chingford Study. Arthritis Rheum. 2009;60(7):2037–45.
Daheshia M, Yao JQ. The interleukin 1beta pathway in the pathogenesis of osteoarthritis. J Rheumatol. 2008;35(12):2306–12.
Ranganathan V, Ciccia F, Zeng F, Sari I, Guggino G, Muralitharan J, Gracey E, Haroon N. Macrophage migration inhibitory factor induces inflammation and predicts spinal progression in ankylosing spondylitis. Arthritis Rheumatol. 2017;69(9):1796–806.
Kontny E, Zielinska A, Ksiezopolska-Orlowska K, Gluszko P. Secretory activity of subcutaneous abdominal adipose tissue in male patients with rheumatoid arthritis and osteoarthritis - association with clinical and laboratory data. Reumatologia. 2016;54(5):227–35.
Rowe MA, Harper LR, McNulty MA, Lau AG, Carlson CS, Leng L, Bucala RJ, Miller RA, Loeser RF. Reduced osteoarthritis severity in aged mice with deletion of macrophage migration inhibitory factor. Arthritis Rheumatol. 2017;69(2):352–61.
Zhang PL, Liu J, Xu L, Sun Y, Sun XC. Synovial fluid macrophage migration inhibitory factor levels correlate with severity of self-reported pain in knee osteoarthritis patients. Med Sci Monit. 2016;22:2182–6.
Liu M, Hu C. Association of MIF in serum and synovial fluid with severity of knee osteoarthritis. Clin Biochem. 2012;45(10–11):737–9.
Kim HR, Kim KW, Jung HG, Yoon KS, Oh HJ, Cho ML, Lee SH. Macrophage migration inhibitory factor enhances osteoclastogenesis through upregulation of RANKL expression from fibroblast-like synoviocytes in patients with rheumatoid arthritis. Arthritis Res Ther. 2011;13(2):R43.
Costello CA, Hu T, Liu M, Zhang W, Furey A, Fan Z, Rahman P, Randell EW, Zhai G. Metabolomics signature for non-responders to total joint replacement surgery in primary osteoarthritis patients: the Newfoundland Osteoarthritis Study. J Orthop Res. 2020;38(4):793–802.
Altman R, Alarcon G, Appelrouth D, Bloch D, Borenstein D, Brandt K, Brown C, Cooke TD, Daniel W, Feldman D, et al. The American College of Rheumatology criteria for the classification and reporting of osteoarthritis of the hip. Arthritis Rheum. 1991;34(5):505–14.
Altman R, Asch E, Bloch D, Bole G, Borenstein D, Brandt K, Christy W, Cooke TD, Greenwald R, Hochberg M, et al. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 1986;29(8):1039–49.
Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569–81.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
Aref-Eshghi E, Liu M, Razavi-Lopez SB, Hirasawa K, Harper PE, Martin G, Furey A, Green R, Sun G, Rahman P, et al. SMAD3 is upregulated in human osteoarthritic cartilage independent of the promoter DNA methylation. J Rheumatol. 2016;43(2):388–94.
Radstake TR, Sweep FC, Welsing P, Franke B, Vermeulen SH, Geurts-Moespot A, Calandra T, Donn R, van Riel PL. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum. 2005;52(10):3020–9.
Chang W. R graphics cookbook. Beijing; Sebastopol, CA: O'Reilly; 2013.
Illescas O, Gomez-Verjan JC, Garcia-Velazquez L, Govezensky T, Rodriguez-Sosa M. Macrophage migration inhibitory factor -173 G/C polymorphism: a global meta-analysis across the disease spectrum. Front Genet. 2018;9:55.
Sreih A, Ezzeddine R, Leng L, LaChance A, Yu G, Mizue Y, Subrahmanyan L, Pons-Estel BA, Abelson AK, Gunnarsson I, et al. Dual effect of the macrophage migration inhibitory factor gene on the development and severity of human systemic lupus erythematosus. Arthritis Rheum. 2011;63(12):3942–51.
Banos-Hernandez CJ, Navarro-Zarza JE, Bucala R, Hernandez-Bello J, Parra-Rojas I, Ramirez-Duenas MG, Garcia-Arellano S, Hernandez-Palma LA, Machado-Sulbaran AC, Munoz-Valle JF. Macrophage migration inhibitory factor polymorphisms are a potential susceptibility marker in systemic sclerosis from southern Mexican population: association with MIF mRNA expression and cytokine profile. Clin Rheumatol. 2019;38(6):1643–54.
De la Cruz-Mosso U, Bucala R, Palafox-Sanchez CA, Parra-Rojas I, Padilla-Gutierrez JR, Pereira-Suarez AL, Rangel-Villalobos H, Vazquez-Villamar M, Angel-Chavez LI, Munoz-Valle JF. Macrophage migration inhibitory factor: association of -794 CATT5-8 and -173 G>C polymorphisms with TNF-alpha in systemic lupus erythematosus. Hum Immunol. 2014;75(5):433–9.
Zhong XB, Leng L, Beitin A, Chen R, McDonald C, Hsiao B, Jenison RD, Kang I, Park SH, Lee A, et al. Simultaneous detection of microsatellite repeats and SNPs in the macrophage migration inhibitory factor (MIF) gene by thin-film biosensor chips and application to rural field studies. Nucleic Acids Res. 2005;33(13):e121.
Zhai G, Zhou J, Woods MO, Green JS, Parfrey P, Rahman P, Green RC. Genetic structure of the Newfoundland and Labrador population: founder effects modulate variability. Eur J Hum Genet. 2016;24(7):1063–70.
Bae SC, Lee YH. Associations between circulating macrophage migration inhibitory factor (MIF) levels and rheumatoid arthritis, and between MIF gene polymorphisms and disease susceptibility: a meta-analysis. Postgrad Med J. 2018;94(1108):109–15.
Onodera S, Tanji H, Suzuki K, Kaneda K, Mizue Y, Sagawa A, Nishihira J. High expression of macrophage migration inhibitory factor in the synovial tissues of rheumatoid joints. Cytokine. 1999;11(2):163–7.
Schindler L, Dickerhof N, Hampton MB, Bernhagen J. Post-translational regulation of macrophage migration inhibitory factor: basis for functional fine-tuning. Redox Biol. 2018;15:135–42.
Weber C, Kraemer S, Drechsler M, Lue H, Koenen RR, Kapurniotu A, Zernecke A, Bernhagen J. Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc Natl Acad Sci U S A. 2008;105(42):16278–83.
Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451(7178):578–82.
Zhang Y, Zhou Y, Chen S, Hu Y, Zhu Z, Wang Y, Du N, Song T, Yang Y, Guo A, et al. Macrophage migration inhibitory factor facilitates prostaglandin E2 production of astrocytes to tune inflammatory milieu following spinal cord injury. J Neuroinflammation. 2019;16(1):85.
Rohner E, Matziolis G, Perka C, Fuchtmeier B, Gaber T, Burmester GR, Buttgereit F, Hoff P. Inflammatory synovial fluid microenvironment drives primary human chondrocytes to actively take part in inflammatory joint diseases. Immunol Res. 2012;52(3):169–75.
Jankauskas SS, Wong DWL, Bucala R, Djudjaj S, Boor P. Evolving complexity of MIF signaling. Cell Signal. 2019;57:76–88.
Leyton-Jaimes MF, Kahn J, Israelson A. Macrophage migration inhibitory factor: A multifaceted cytokine implicated in multiple neurological diseases. Exp Neurol. 2018;301(Pt B):83–91.
Yende S, Angus DC, Kong L, Kellum JA, Weissfeld L, Ferrell R, Finegold D, Carter M, Leng L, Peng ZY, et al. The influence of macrophage migration inhibitory factor gene polymorphisms on outcome from community-acquired pneumonia. FASEB J. 2009;23(8):2403–11.
Heinrichs D, Knauel M, Offermanns C, Berres ML, Nellen A, Leng L, Schmitz P, Bucala R, Trautwein C, Weber C, et al. Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc Natl Acad Sci U S A. 2011;108(42):17444–9.
Fujihara Y, Hikita A, Takato T, Hoshi K. Roles of macrophage migration inhibitory factor in cartilage tissue engineering. J Cell Physiol. 2018;233(2):1490–9.
Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis. 2013;5(2):77–94.
Singh M, Mastana S, Singh S, Juneja PK, Kaur T, Singh P. Promoter polymorphisms in IL-6 gene influence pro-inflammatory cytokines for the risk of osteoarthritis. Cytokine. 2020;127:154985.
Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3(10):791–800.
Acknowledgements
The metabolic profiling was done at The Metabolomics Innovation Centre (TMIC).
Funding
This study was supported by Canadian Institutes of Health Research (grant numbers 132178, 143058, 153298), the Research and Development Corporation of Newfoundland and Labrador (grant number 5404.1423.102), and the Memorial University of Newfoundland Medical Research Fund.
Author information
Authors and Affiliations
Contributions
GZ designed the study. ML, ZX, LC, HZ, and JX performed the experiments. ML and GZ analyzed and interpreted the data. ML, ZX, and GZ drafted the manuscript. GS, DQ, AF, PR, and GL revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Health Research Ethics Authority of Newfoundland and Labrador (HREB # 2011.311), and written consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, M., Xie, Z., Sun, G. et al. Macrophage migration inhibitory factor may play a protective role in osteoarthritis. Arthritis Res Ther 23, 59 (2021). https://doi.org/10.1186/s13075-021-02442-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-021-02442-w