
Abstract
Background
Investigating the pathophysiological mechanisms of early osteoarthritis (OA) is of utmost interest since this stage holds the strongest promise for therapeutic interventions. The aims of this study were to analyze if synovial inflammation is already present in early OA and to characterize the involved cell populations, by investigating synovial fluid (SF) and synovial membrane (SM) of early OA patients for the presence and polarization status of CD4 T cells.
Methods
A quantitative analysis of CD4+ T cell infiltration in SF and SM compared to peripheral blood (PB) was performed in patients with early stages of OA. We further investigated intracellular staining (ICS), surface marker, and chemokine receptor expression profiles of CD4+ T cells in SF, SM, and PB, as well as cytokine expression in native SF and PB. Matched samples of SF, SM, and PB were harvested from 40 patients with early OA at the time of surgery. Early OA was confirmed by independent surgeons intraoperatively. Samples were analyzed by flow cytometry for surface markers and cytokines, which are preferentially expressed by distinct T cell subsets (Th1, Th2, Th17, regulatory T cells). Furthermore, we analyzed native SF and PB supernatants using MACSPlex for multiple cytokine expression profiles.
Results
SF and SM showed a distinct infiltration of CD4+ T lymphocytes, with significantly increased expression of chemokine receptors CXCR3/CCR5, cytokine IFN-γ (preferentially expressed by Th1 cells), and CD161 (preferentially expressed by IL-17 producing Th17 cells) compared to PB. Furthermore, the percentage of CD4+ T cells polarized to Treg was significantly increased in SM compared to SF and PB. No significant differences were observed for CCR3 and CCR4 (preferentially expressed by Th2 cells), although IL-4 values were significantly higher in SM and SF compared to PB. Cytokine analysis showed comparable results between PB and SF, with only IL-6 being significantly increased in SF.
Conclusions
Early OA joints show already significant inflammation through CD4+ T cell infiltration, with predominant Th1 cell polarization. Inflammation seems to be driven by direct proinflammatory cell interaction. Cytokine signaling seems to be negligible at the site of inflammation in early OA, with only IL-6 being significantly increased in SF compared to PB.
Similar content being viewed by others

Background
Treatment of osteoarthritis (OA) continues to be challenging for patients, treatment providers, and health care systems worldwide. With only limited non-surgical options to treat OA symptoms and only surgical options for end-stage OA, it cannot be stated that we have reached a satisfying level. The impact of cellular and molecular pathways contributing to OA pathophysiology is being increasingly recognized to the same amount that the concept of wear and tear is questioned. Synovial inflammation has been identified as an independent factor significantly contributing to OA pathology. Modulation of inflammatory pathways has been successful in delaying OA progression in animal models [1,2,3]. Studies investigating the inflammatory cell infiltrate have identified T cells and macrophages as the main mononuclear cells (MNC) in the synovial fluid (SF) and membrane (SM). Flow cytometry and immunohistological studies revealed that CD4+ T helper cells present an activated phenotype and are polarized distinctly to proinflammatory subsets (Th1), overweighing the immunomodulatory regulatory T cells (Treg) in OA joints. Thus, Th1 infiltration is increasingly recognized as pivotal in OA pathology [4,5,6,7,8]. But these previous studies have mainly focused on joint-derived samples of patients with advanced OA. In contrast, early OA has widely been ignored so far [7,8,9]. Since end-stage OA is the result of chronic progressive disease with outbalanced joint-homeostasis developing over decades, it might be hard to decipher the impact of these inflammatory patterns on OA pathogenesis if we continue to analyze only final disease stages. Thus, it is pivotal to investigate the cellular and molecular inflammatory processes in early OA joints. Knowledge of the initial inflammatory processes in early OA might prove essential for establishing a new treatment window specifically in view of the limited regenerative capability of hyaline cartilage and the irreversible chronic changes that occur in the whole joint during OA progression. Assessment of early OA is indispensable in the search for biomarkers as a diagnostic tool. OA pathology has a temporal pattern, and cartilage, bone, and synovial matrix biomarkers show a positive association with the progression of knee OA [10]. Changes in cartilage and synovial fluid metabolism occur before any quantifiable structural damage can be recognized [11]. Although several publications suggest markers of early OA, an established diagnostic tool is still missing [12,13,14]. This is to our best knowledge the first study to perform flow cytometry analysis of tissue samples from early OA patients that represent a clearly defined group as categorized by independent surgeons intraoperatively. Furthermore, analysis of matched SF, SM, and peripheral blood (PB) samples from early OA patients is still missing.
Methods
Study population
A total of 40 patients with arthroscopic or MRI findings of early osteoarthritis of the knee (29 women, 11 men) were enrolled in this study. All patients included had a Kellgren-Lawrence grade 0–I, as confirmed by two-plane radiographs, and were scheduled for arthroscopic surgery at our hospital. No patient included underwent surgery directly after trauma. The minimum time after trauma/onset of symptoms was 6 weeks, and the median was 8 months (Table 1). International Cartilage Repair Society (ICRS) grade was assessed by independent surgeons intraoperatively. All patients included had ICRS grades I–IV in at least two compartments (5%) or grades II–IV in one compartment (95%) and met the criteria of early OA as previously suggested by Luyten et al. [15, 16]. None of the patients had a history of underlying inflammatory disease, intake of DMARD, intra-articular injection of corticosteroids and hyaluronic acid, or regular intake of NSAID. Systemic inflammatory parameters (CRP, WBC) were within the physiological range at the time of surgery, except for one patient (CRP 13.4 mg/dl). The mean age of the study population was 41.7 ± 14.3 years. Underlying pathologies for arthroscopic knee surgery were meniscal tear (40%), traumatic anterior cruciate ligament (ACL) tear (5%), and localized cartilage damage (55%) (Table 1). Subgroups were comparable concerning clinical data. Due to the methodological setup, e.g., limitations concerning cell number, not all patients could be included in every subgroup analysis. The local ethics committee approved this study (S-333/2007). Written informed consents from all patients were obtained prior to study enrolment.
Sample collection
The synovial fluid (SF), synovial membrane (SM), and peripheral blood (PB) were harvested at the time of surgery. SF was aspirated prior to the establishment of the arthroscopic portals into sterile tubes and further processed as described below. SM biopsies were performed intra-operatively from the suprapatellar pouch as previously described [7]. Heparinized PB samples were taken concurrently at the time of surgery.
Sample preparation
Two hundred microliters of native SF prior to further processing and 200 μl of PB supernatants after density gradient centrifugation (see below) were harvested for MACSPlex analysis (Miltenyi Biotec, Germany) and deep-frozen at − 80 °C with a maximum time between sample collection and cryopreservation of 30 min. The remaining volume of SF samples was treated with bovine testicular hyaluronidase (10 mg/ml, Sigma-Aldrich, USA) for 30 min at 37 °C and washed twice with PBS. SM samples were rinsed twice with phosphate-buffered saline (PBS), minced finely with sterilized scissors, and digested with collagenase B (1 mg/ml; Roche, USA) and bovine testicular hyaluronidase IV (2 mg/ml; Sigma-Aldrich, USA) at 37 °C for 2 h in RPMI 1640 culture medium (Invitrogen, USA) supplemented with 10 μg/ml penicillin-streptomycin (Invitrogen, USA) and 5% FCS (Biochrom AG, Germany). The cell suspension was filtered through a 100-μm (BD Biosciences, USA) and a 40-μm pore-size cell strainer (EMD Millipore, USA) to remove any undigested tissue. The filtered cell suspension was washed twice with PBS. Mononuclear cells were isolated from heparin anti-coagulated whole blood, SF, and SM cell suspensions using Ficoll-PaqueTM PLUS (GE Healthcare, USA) density gradient centrifugation. T cells were isolated from PB, SF, and SM mononuclear cells by CD3 MACS bead separation (Miltenyi Biotec, Germany).
Flow cytometry analysis of cell surface markers and intracellular staining
Multicolor flow cytometry was used to identify CD3+CD4+ T cell subsets by their preferential expression of surface and intracellular markers. In brief, for the staining of regulatory T cell (Treg) and T helper (Th1, Th2, Th17) cell surface markers, CD3+ MACS-isolated T cells from PB, SF, and SM were washed twice in staining buffer, blocked with FcR blocking reagent (Miltenyi Biotec, Germany), and then stained for 30 min at 4 °C with the following monoclonal antibodies (mAb) in two setups: VioBlue-labeled mAb against CD4 (clone VIT4, Miltenyi Biotec, Germany), Alexa Fluor 488-labeled mAb against CXCR3 (clone 1C6), PE-labeled mAb against CCR4 (clone 1G1), PE-Cy7-labeled mAB against CD25 (clone 2A3), Alexa Fluor 647-labeled mAb against CCR3 (clone 2A3), and APC-Cy7 labeled mAb against CCR5 (clone 2D7) in one setup; E-Fluor 450-labeled mAb against CD127 (clone eBioRDR5, E-Bioscience, USA), FITC-labeled mAb against CD161 (clone DX12), PE-Cy7-labeled mAb against CCR6 (clone 11A9), APC-labeled mAb against CD25 (clone 2A3), PE-labeled mAb against CCR4 (clone 1G1), and APC-Cy7-labeled mAb against CD4 (clone RPA-T4) in another setup. Before flow cytometric detection, 0.5 μg/ml 7-AAD (eBioscience; USA) was added to the cell suspensions, to exclude cell debris and dead cells.
For the staining of intracellular markers, MACS CD3-isolated T cells from PB, SF, and SM were taken into culture at a final density of 106 T cells/ml and incubated for 10 h at 37 °C/5% CO2. Cell cultures were stimulated with phorbol-myristate-acetate (PMA) (50 ng/ml) and ionomycin (1 μg/ml). After 4 h of activation, brefeldin A (5 μg/ml, Sigma-Aldrich, Germany) was added for another 6 h. After a total of 10 h of activation, T cells were collected, washed twice in FACS buffer, and blocked with FCS blocking reagent. For surface marker expression, PE-Cy7-conjugated mAb against CD3 (clone SK7), APC-Cy7-conjugated mAb against CD4 (clone RPA-T4), and VioBlue-labeled mAb against CD8 (clone BW135/80, Miltenyi Biotec, Germany) were used, and staining was performed at 4 °C for 30 min. Following surface staining, T cells were fixed and permeabilized using cytofix/cytoperm reagent and then stained with APC labeled anti-IFN-γ (clone B27), FITC-labeled anti-IL-4 (clone MP4-25D2), and PE-labeled anti-IL-17A (clone N49-653). Cytokine values for CD3+CD4+CD8− T cells were measured by flow cytometry.
Flow analysis was performed using a MACSQuant Analyzer (Miltenyi Biotec, Germany), which is a 7-channel flow cytometer. The total number of surface markers analyzed exceeded the channel number. Therefore, two antibody panels were needed. Data analysis was performed using FlowJo 9.6 (Treestar, USA). The cutoff for all cell surface markers was defined based on fluorescence minus one (FMO) controls. Antibodies and cell preparation solutions were purchased from BD Biosciences, USA, if not stated otherwise.
Cytokine analysis
Native SF and PB sera were analyzed by the MACSPlex 12 Kit (Miltenyi Biotec, Germany). All working steps were carried out according to the manufacturer’s instructions, and washing procedures were performed with a centrifuge. Flow analysis was performed using MACSQuant Analyzer (Miltenyi Biotec, Germany). Data analysis was performed using Flow Logic™ (V7.2.1) and BeadLogic™ (V7) (Miltenyi Biotec, Germany).
Statistical analysis
Data are presented as arithmetic median (IQR), if not stated otherwise. Gaussian distribution was assessed by the Kolmogorov-Smirnov test. To compare concurrent SF, SM, and PB samples, we used the Friedman test for non-parametric data with Wilcoxon signed-rank post hoc test and Bonferroni correction. Wilcoxon signed-rank test with Bonferroni correction was used for MACSPlex analysis. p values < 0.05 were considered significant. Statistical analysis was performed using SPSS 22.0 (IBM SPSS Inc., USA).
Results
Presence of T cells in OA joints and peripheral blood
Frequencies of MACS-isolated T lymphocytes of joint-derived samples (SF, SM) were analyzed by flow cytometry and compared to concurrent PB samples. PB revealed higher cell numbers with a mean of 39.9 × 105 ± 6 × 105 T cells compared to 0.96 × 105 ± 0.3 × 105 (SF) and 0.08 × 105 ± 0.02 × 105 (SM), as expected (Table 2). To further estimate the T cell percentage in SF and SM samples, the relation between T cell numbers and acquired sample volume or sample weight was calculated. Although still lower than PB (558.1 ± 88.5 cells/μl), SF and SM showed a distinct infiltration with a mean of 51.3 ± 21.3 T lymphocytes per microliter (SF) and 42.5 ± 12.7 T lymphocytes per milligram (SM), respectively. SF and SM contained comparable proportions of T lymphocytes that stained positive for CD4 (38.8% in SF and 44.3% in SM), albeit significantly lower than PB (71.2%).
Comparison of T cell surface marker and chemokine receptor status in OA joints and peripheral blood
To evaluate T cell surface marker expression quantitatively, we isolated CD4+ T cells from PB, SF, and SM and used flow cytometry for detection of CXCR3, CCR5 (preferentially expressed by Th1 cells), CCR3, CCR4 (preferentially expressed by Th2 cells), CCR6, and CD161 (preferentially expressed by Th17 cells) as well as Treg-specific CD25 and CD127. All analyzed surface markers (chemokine and cytokine receptors) could be detected on synovial T cell surfaces. Table 3 presents the median (IQR) for the surface markers investigated in this study.
Synovial T cells in SF and SM showed a predominant and in comparison with PB significantly increased expression of CXCR3 (p < 0.01), CCR5 (p < 0.01), and CD161 (p < 0.01 [SM] and p < 0.05 [SF]). No significant differences were found between SF and SM for these markers. CCR4 showed significantly higher expression rates on T cells isolated from SM, compared to SF (p = 0.021). No significant differences were shown for chemokine receptors CCR3 and CCR6 between the groups. CD4+CD25+/highCD127low/− regulatory T cells were significantly enriched in SM compared to PB (p < 0.01).
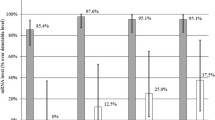
Furthermore, analysis of CD4+ T cells co-expressing CXCR3 and CCR5 showed that the majority of CD4+CXCR3+ T cells in SF and SM stained also positive for CCR5 and vice versa (55.4% [SF] and 67.1% [SM]), whereas only 2.6 out of 10.0% of CD4+CXCR3+ and CD4+CCR5+ T cells showed that expression pattern in PB. In contrast to PB, our data revealed highly significant polarization towards CD4+CXCR3+CCR5+ T cells with 55.4% (SF) and 67.2% (SM), compared to 2.6% in PB (p < 0.01). Co-expression of CCR3 and CCR4 was significantly higher in SM with 48.4% compared to SF (12.0%; p = 0.038). No significant differences were found, when compared to PB (14.9%; p = 0.051). Also, analyzing the co-expression of CD161 and CCR6, SM (16.8%) and SF (12.2%) had significantly higher values than PB (3.2%) (Fig. 1, Table 4).

Flow cytometry analysis of CD4+ T cell surface marker expression. Representative flow cytometry histograms of CD4+ T cell surface marker expression are shown. In short, mononuclear cells were isolated from PB, SF, and SM by density gradient centrifugation. CD3+ MACS-isolated T cells from PB, SF, and SM were stained with monoclonal antibodies (mAb) for the following surface markers: CD4, CD25, CD127, CXCR3, CCR5, CCR3, CCR4, CD161, and CCR6. Before flow cytometric detection, 7-AAD was added to the cell suspensions to exclude cell debris and dead cells. The cutoff for all cell surface markers was defined based on fluorescence minus one (FMO) controls (shown as gray overlay population). mAb, monoclonal antibody; MACS, magnetic-activated cell sorting
To further evaluate the expression pattern of the mentioned chemokine receptors and surface markers on CD4+ T cells, we analyzed how many double-positive CD4+CXCR3+CCR5+-stained T cells did not express CCR3/CCR4 and vice versa. Interestingly, in SM and SF as well as in PB, a big proportion of CD4+CXCR3+CCR5+ T cells stained also positive for CCR3 and CCR4. Whereas in PB 0.6% out of 2.6% (23.1%) of CD4+CXCR3+CCR5+ T cells stained negative for CCR3/CCR4, SF showed a higher percentage with 31.5% out of 55.4% (56.7%) and SM showed a lower fraction of 5.8% out of 67.2% (8.6%). Similar results were obtained for CD4+CD161+CCR6+ T cells with 1.2% out of 3.2% (37.5%) staining negative for CCR4 in PB, whereas 5.1% out of 12.2% (41.8%) in SF and 2.2 out of 16.8% (13.1%) in SM showed no co-expression of CCR4 (Table 4, (Fig. 2).

Co-expression of CXCR3/CCR5, CCR3/CCR4, and CD161/CCR6 on CD4+ T lymphocytes. Percentage rates of CXCR3/CCR5, CCR3/CCR4, and CD161/CCR6 co-expression on CD4+ T lymphocytes in the peripheral blood (PB), synovial fluid (SF), and synovial membrane (SM) of patients with early knee OA are shown. Surface marker expression was analyzed by flow cytometry, and the results are presented as a percentage of total CD4+ T cells. Data is shown as mean (SEM) (n = 12 paired samples). Significant differences between SF/PB and SM/PB are marked with asterisks (*); significant differences between SF/SM are marked with a hash (#): *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.05
CD4+CCR3+CCR4+ and CD4+CD161+CCR6+ T cells presented a clear difference between peripheral and synovial T cells regarding the exclusive expression of chemokine receptors. While 11.2 out of 14.9% of CD4+CCR3+CCR4+ T cells in PB did not co-express CXCR3 and CCR5, very few T cells showed this exclusive expression pattern in SM and SF.
Cytokine secretion of activated CD3+CD4+CD8− T cells in OA joints and peripheral blood
We further examined the polarization of the T cell population by utilizing flow detection of Th cell-specific intracellular markers to define the following T helper cell subsets: Th1 (IFN-γ), Th2 (IL-4), and Th17 (IL-17A), and compared synovial samples to concurrent samples of PB (Table 5). Whereas in PB only a small proportion of T cells stained positive for T helper cell subsets, synovial T cells isolated from SF and SM showed a significantly increased polarization towards Th1 cells with 55.8% in SF and 42.8% in SM compared to 14.2% in PB and Th2 cells with 2.9% in SF and 2.0% in SM compared to 0.6% in PB. No significant differences were detected for Th17. Interestingly, polarization towards Th2 and Th17 was much lower, than expected from single surface marker analysis.
MACSPlex analysis of cytokine expression pattern of native peripheral blood and synovial fluid supernatants
Matched samples of native SF and PB supernatants were analyzed for cytokine expression, using MACSPlex (Miltenyi, Germany).
Overall, the analyzed set of cytokines showed comparable results between PB and SF (Table 6). Significantly higher values were only shown for IL-6 in SF (19.45 ng/ml) compared to PB (1.18 ng/ml).
Discussion
OA is a multifactorial disease affecting the whole joint. Reflecting recent literature, the model of sole mechanical “wear and tear” causing cartilage degradation and thus OA progression is obsolete. Overwhelming evidence ascribes inflammatory processes a pivotal role in the generation of OA symptoms as well as OA onset and progression. Nevertheless, expertise about T cell polarization and molecular mechanisms during the onset of the disease is limited. To our best knowledge, there is no study utilizing flow cytometry to analyze T cell subtypes in SF, SM, and PB of a clearly defined early OA group.
This study analyzed the expression of a broad variety of cytokines, chemokines, and other surface markers on CD4+ T cells in paired samples of SF, SM, and PB of patients with clearly defined early OA as proposed by Luyten et al. [15, 16], as well as cytokine expression in native SF compared to PB. Although staining of CD4 showed significantly lower percentages in SM and SF compared to PB, our results reveal a significantly increased expression of CCR5, CXCR3, and CD161 among joint-derived CD4+ T cells (SM and SF) compared to PB in early OA. The expression of CCR5 and CXCR3 were previously identified as biomarkers correlating with OA-related knee pain and OA severity in a group of end-stage OA patients, respectively [17].
Korchi et al. found signs of synovial inflammation and hypervascularization already 3 months after OA onset in an animal model of early OA by analyzing radiological and histological changes [18]. Interestingly, the amount of CD4+ T cells expressing CCR5 in SM and SF of our early OA patients is comparably high as previously described in SF of patients with different forms of rheumatic joint diseases such as RA, psoriatic arthritis, and ankylosing spondylitis [19, 20]. Nevertheless, one limitation of our study is that CD4 staining of MACS-isolated CD3+ T lymphocytes might be compounded by surface marker cleavage due to the performed enzymatic digestion, as previously described [21]. Other groups showed lower expression for CCR4 and CCR5 in end-stage OA compared to RA. CCR5 expression was also remarkably lower in these end-stage OA data than in our early OA group [22, 23]. Moreover, studies comparing the synovial membrane from early and late OA patients by histology and immunohistochemistry found a higher inflammatory potential in early OA compared to late OA [6, 24]. These findings indicate that inflammatory disease activity in OA joints is highest during the onset of the disease, possibly promoting cartilage degradation [25, 26]. Of course, the underlying pathology itself (ACL tear, meniscus injury, etc.) might induce inflammatory pathways to a certain extent, leading to a selection bias in our study. Several groups investigated inflammation in meniscal injury or ACL tear cohorts that might probably overlap with our study group. Synovial inflammation was found to be present in 43% of patients with meniscal injuries [27]. Interestingly, the report of Clair et al. also showed significantly increased synovial concentration of IL-6, but also MCP-1, MIP-1β, and MMP-3, which were not examined in our study [28]. Another group investigating the synovial inflammatory biomarkers 2 years after ACL tear showed no correlation to patient-reported outcomes after 5 years, concluding chronic inflammation is not mainly responsible for the clinical results [29]. However, obtaining tissue samples from early OA patients without a certain pathology is justifiably not possible due to ethical considerations.
Furthermore, our data presents an increased percentage of CD4+CD25+/highCD127low/− Treg in SM compared to SF and PB, which is even higher than we previously published for end-stage OA [30]. This is highly interesting, and we hypothesize that these elevated Treg numbers in early OA are an attempt of the immune system to contain the ongoing immune response and redress balance between pro- and anti-inflammatory immune cells.
In general, conflicting results have been reported regarding cellular inflammation in end-stage OA. It seems that a number of different cells contribute to inflammatory processes. Some groups consider cells of the innate immune system to be of utmost importance in OA inflammation. Monocytes and macrophages were ascribed to have the highest impact on inflammation in end-stage OA [31]. Neutrophil granulocytes were shown to be the major source of cartilage-degrading enzymes in a murine model of early OA [32]. Other groups declared cells of the adaptive immune system, especially T cells to be in an activated status and highly involved in generating a proinflammatory milieu in OA [30, 33]. Possible reasons for these diverging results might be that specimens of end-stage OA patients are heterogenous regarding inflammatory activity and that experimental setups differ. In our study, we focused on T cells and T cell-specific surface markers as well as analyzing a broad spectrum of cytokines. It cannot be ruled out that cells of innate immunity interact with T cells and thereby influence synovial inflammation in early OA.
Besides, differing results regarding inflammation in early OA could be caused not only by a different experimental setup, but also by an accentuated bias. One limitation of our study is that due to ethical concerns and experimental setup, not all performed experiments were applicable to the whole study population. Above all, limited cell numbers isolated from SM samples were a bottleneck for some subanalyses.
Further, until today, the term “early OA” itself is not clearly defined and used for various and heterogenous joint pathologies. Patients with isolated cartilage lesions but otherwise healthy joints, as well as symptomatic patients with multiple joint surface defects who are at higher risk to develop manifest OA, were previously lumped together in several studies. The overall problem is that OA criteria, based on radiographic changes and clinical findings, have not been changed since 1986 [34], although it is commonly accepted that early OA (Kellgren and Lawrence 0–I) can rarely be diagnosed on plain radiographs. That is why we used anamnestic information, as well as arthroscopic assessment of knee joints by independent surgeons to ensure that the criteria of early OA, as proposed by recent publications, were fulfilled [15, 16].
Due to the vast plasticity of T cell surface marker expression, it is hard to define a distinct T cell-specific pattern, especially given that surface marker expression varies during migration and activation processes [35]. Nevertheless, in vitro data indicating a preferential expression pattern associated with T cell differentiation do exist: virtually, all IL-17-producing CD4+ T cells (Th17) were found to express CCR6 [36]. CXCR3 and CCR5 are preferentially expressed on activated Th1 cells [37, 38] and Th17 cells [39], but CXCR3+ and CCR5+ T cells were shown to produce the Th1-specific cytokine IFN-γ upon stimulation ex vivo [40, 41]. CCR4+ and CCR3+ T cells were shown to secrete Th2-specific IL-4 [40,41,42]. How stable these surface marker expression patterns actually remain in vivo is not known. To further validate our surface marker results, we performed ICS staining. This data confirmed significantly increased polarization to Th1 in SM and SF compared to PB, but also significantly increased detection of Th2-specific cytokine IL-4, albeit with a much lower percentage rate than expected (Table 5).
Chemokine receptors have a crucial role in leukocyte migration to sites of inflammation and thus regulation of immune responses. CCR5 is an essential chemokine mediating T cell recruitment to inflammatory sites, but also seems to play a role in the resolution of inflammation [43]. Diapedeses of T cells into inflamed SM or SF is thought to happen by selective migration via endothelial P- and E-selectin and CXCR3/CCR5-activating chemokines [20, 44, 45]. Besides these adhesion molecules, chemotactic cytokines CCL (CC chemokine ligand) 2, CCL3, and RANTES (regulated and normal T cell expressed and secreted) are thought to have the greatest impact on T cell infiltration into sites of inflammation. All of these have been detected in OA joints [23]. Furthermore, chemokine receptor expression is not a unique feature of T cells. Chemokines are abundantly present at inflammatory sites, also in chondrocytes and fibroblasts in the sublining synovial membrane and seem to perpetuate MMP release in the process of cartilage degradation [46]. In the study presented, we focused solely on chemokine expression of CD4+ T cells. Our data reveals a significant increase in the co-expression of CXCR3/CCR5 (CD4+CXCR3+CCR5+ T cell) in SM and SF compared to PB, which are preferentially expressed by (activated) Th1 cells. Hence, we hypothesize that this proinflammatory T cell subset plays the main role in early OA inflammation.
The MACSPlex results presented in our study showed a significantly elevated level only for IL-6 in SF compared to PB. No significant differences were found for the other tested cytokines. The heightened IL-6 level could be due to increased secretion of synovial fibroblasts as an effect of RANTES-induced stimulation [47]. Elevated serum levels of IL-6 were associated with knee cartilage loss in humans [48], and IL-6 levels were shown to be significantly increased in SF of end-stage OA compared to controls [49, 50]. Since anti-inflammatory drugs blocking the IL-6-receptor have been utilized in the second-line treatment of rheumatoid arthritis (RA) for more than a decade [51], it appears peculiar that research focusing on manipulation of IL-6 cascade in OA is scarce. One group demonstrated that IL-6 acts catabolic by inducing cartilage-degrading enzymes mainly via Stat3 (signal transducer and activator of transcription 3) signaling in a murine model of OA, and the administration of mAb against IL-6R was shown to alleviate osteoarthritic cartilage lesions [52]. Also, the application of tocilizumab (IL-6R blocker) intrathecally was shown to attenuate pain symptoms in a murine OA model, although the study setup is not transferable to humankind [53]. Further, several phytotherapeutics were shown to decrease IL-6 chondrocyte-secretion and thereby MMP13 expression [54, 55], postulating that this pathway is possibly a lever that could be used to develop new therapies, although with moderate validity.
Taken together, our data shows that cellular and humoral inflammation is present already in early OA. Assuming that these inflammatory patterns contribute to progressive joint damage by downstream activation of catabolic enzymes, as previously shown for end-stage OA [26], appears conclusive. However, to date, there is no therapeutic strategy aiming at inhibition of synovitis by modulating the molecular characteristics of proinflammatory Th1 cells.
Conclusion
To our best knowledge, this is the first flow cytometry study implementing a complete set of SF, SM, and PB specimens of a clearly defined early OA group. It has to be stated that the literature focusing on patients with early OA is scarce, but even scarcer are studies comparing different stages of OA. Further studies comparing inflammatory parameters in early and late OA are warranted for a better understanding of the changes in inflammatory activity during disease progression. Only then can we achieve the possibility to discover new treatment strategies aimed at halting disease progression. Our data demonstrates that infiltration of inflammatory Th1 cells occurs already in early OA stages and in combination with the solitary increase of IL-6 ascribes direct cellular interaction in combination with humoral immunity and involvement in early OA pathogenesis. Therapeutic studies should focus on blocking IL-6 secretion and function, as well as modulation of Th1 cells to facilitate not only symptomatic, but also causal therapies for OA patients.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACL:
-
Anterior cruciate ligament
- BMI:
-
Body mass index
- CCL:
-
CC chemokine ligand
- CCR:
-
CC chemokine receptor
- CXCR:
-
CXC chemokine receptor
- CD:
-
Cluster of differentiation
- CRP:
-
C-reactive protein
- DMARD:
-
Disease-modifying anti-rheumatic drug
- ICRS:
-
International Cartilage Repair Society grade
- IFN-γ:
-
Interferon gamma
- IL:
-
Interleukin
- IP-10:
-
Interferon γ-induced protein 10 (CXCL10)
- IQR:
-
Interquartile range
- K & L score:
-
Kellgren and Lawrence score
- mAb:
-
Monoclonal antibody
- MDC:
-
Macrophage-derived chemokine (CCL22)
- n.a.:
-
Not applicable
- NSAID:
-
Non-steroidal anti-inflammatory drugs
- OA:
-
Osteoarthritis
- PB:
-
Peripheral blood
- PDGF:
-
Platelet-derived growth factor
- RA:
-
Rheumatoid arthritis
- RANTES:
-
Regulated on activation, normal T cell expressed and secreted (CCL5)
- SF:
-
Synovial fluid
- SM:
-
Synovial membrane
- STAT3:
-
Signal transducer and activator of transcription 3
- Th:
-
T helper cell
- TNF-α:
-
Tumor necrosis factor alpha
- Treg:
-
Regulatory T cells
References
Wang Q, Rozelle AL, Lepus CM, Scanzello CR, Song JJ, Larsen DM, et al. Identification of a central role for complement in osteoarthritis. Nat Med. 2011;17(12):1674–9.
Little CB, Barai A, Burkhardt D, Smith SM, Fosang AJ, Werb Z, et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60(12):3723–33.
Little CB, Meeker CT, Golub SB, Lawlor KE, Farmer PJ, Smith SM, et al. Blocking aggrecanase cleavage in the aggrecan interglobular domain abrogates cartilage erosion and promotes cartilage repair. J Clin Invest. 2007;117(6):1627–36.
Lindblad S, Hedfors E. Arthroscopic and immunohistologic characterization of knee joint synovitis in osteoarthritis. Arthritis Rheum. 1987;30(10):1081–8.
Revell PA, Mayston V, Lalor P, Mapp P. The synovial membrane in osteoarthritis: a histological study including the characterisation of the cellular infiltrate present in inflammatory osteoarthritis using monoclonal antibodies. Ann Rheum Dis. 1988;47(4):300–7.
Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64(9):1263–7.
Rosshirt N, Hagmann S, Tripel E, Gotterbarm T, Kirsch J, Zeifang F, et al. A predominant Th1 polarization is present in synovial fluid of end-stage osteoarthritic knee joints: analysis of peripheral blood, synovial fluid and synovial membrane. Clin Exp Immunol. 2019;195(3):395–406.
Moradi B, Rosshirt N, Tripel E, Kirsch J, Barie A, Zeifang F, et al. Unicompartmental and bicompartmental knee osteoarthritis show different patterns of mononuclear cell infiltration and cytokine release in the affected joints. Clin Exp Immunol. 2015;180(1):143–54.
Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi J, Oyamada A, et al. Preferential accumulation of activated Th1 cells not only in rheumatoid arthritis but also in osteoarthritis joints. J Rheumatol. 2011;38(8):1569–75.
Van Spil WE, Welsing PM, Bierma-Zeinstra SM, Bijlsma JW, Roorda LD, Cats HA, et al. The ability of systemic biochemical markers to reflect presence, incidence, and progression of early-stage radiographic knee and hip osteoarthritis: data from CHECK. Osteoarthr Cartil. 2015;23(8):1388–97.
Makela JT, Han SK, Herzog W, Korhonen RK. Very early osteoarthritis changes sensitively fluid flow properties of articular cartilage. J Biomech. 2015;48(12):3369–76.
Li W, Du C, Wang H, Zhang C. Increased serum ADAMTS-4 in knee osteoarthritis: a potential indicator for the diagnosis of osteoarthritis in early stages. Genet Mol Res. 2014;13(4):9642–9.
Bernard NJ. Osteoarthritis: circulating miRNAs-early osteoarthritis biomarkers? Nat Rev Rheumatol. 2014;10(4):197.
Scanzello CR, Umoh E, Pessler F, Diaz-Torne C, Miles T, Dicarlo E, et al. Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthr Cartil. 2009;17(8):1040–8.
Luyten FP, Denti M, Filardo G, Kon E, Engebretsen L. Definition and classification of early osteoarthritis of the knee. Knee Surg Sports Traumatol Arthrosc. 2012;20(3):401–6.
Luyten FP, Bierma-Zeinstra S, Dell’Accio F, Kraus VB, Nakata K, Sekiya I, et al. Toward classification criteria for early osteoarthritis of the knee. Semin Arthritis Rheum. 2018;47(4):457–63.
Nees TA, Rosshirt N, Zhang JA, Platzer H, Sorbi R, Tripel E, et al. T helper cell infiltration in osteoarthritis-related knee pain and disability. J Clin Med. 2020;9(8).
Korchi AM, Cengarle-Samak A, Okuno Y, Martel-Pelletier J, Pelletier JP, Boesen M, et al. Inflammation and hypervascularization in a large animal model of knee osteoarthritis: imaging with pathohistologic correlation. J Vasc Intervent Radiol. 2019;30(7):1116–27.
Mack M, Bruhl H, Gruber R, Jaeger C, Cihak J, Eiter V, et al. Predominance of mononuclear cells expressing the chemokine receptor CCR5 in synovial effusions of patients with different forms of arthritis. Arthritis Rheum. 1999;42(5):981–8.
Issekutz AC, Quinn PJ, Lang B, Ramsey S, Huber AM, Rowter D, et al. Coexpression of chemokine receptors CCR5, CXCR3, and CCR4 and ligands for P- and E-selectin on T lymphocytes of patients with juvenile idiopathic arthritis. Arthritis Rheum. 2011;63(11):3467–76.
Labinsky H, Panipinto PM, Ly KA, Khuat DK, Madarampalli B, Mahajan V, et al. Multiparameter analysis identifies heterogeneity in knee osteoarthritis synovial responses. Arthritis Rheumatol (Hoboken, NJ). 2020;72(4):598–608.
Flytlie HA, Hvid M, Lindgreen E, Kofod-Olsen E, Petersen EL, Jorgensen A, et al. Expression of MDC/CCL22 and its receptor CCR4 in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Cytokine. 2010;49(1):24–9.
Haringman JJ, Smeets TJ, Reinders-Blankert P, Tak PP. Chemokine and chemokine receptor expression in paired peripheral blood mononuclear cells and synovial tissue of patients with rheumatoid arthritis, osteoarthritis, and reactive arthritis. Ann Rheum Dis. 2006;65(3):294–300.
Ene R, Sinescu RD, Ene P, Cirstoiu MM, Cirstoiu FC, et al. Rom J Morphol Embryol. 2015;56(1):169–73.
Goldring MB, Otero M. Inflammation in osteoarthritis. Curr Opin Rheumatol. 2011;23(5):471–8.
Platzer H, Nees TA, Reiner T, Tripel E, Gantz S, Hagmann S, et al. Impact of mononuclear cell infiltration on chondrodestructive MMP/ADAMTS production in osteoarthritic knee joints-an ex vivo study. J Clin Med. 2020;9(5).
Scanzello CR, McKeon B, Swaim BH, DiCarlo E, Asomugha EU, Kanda V, et al. Synovial inflammation in patients undergoing arthroscopic meniscectomy: molecular characterization and relationship to symptoms. Arthritis Rheum. 2011;63(2):391–400.
Clair AJ, Kingery MT, Anil U, Kenny L, Kirsch T, Strauss EJ. Alterations in synovial fluid biomarker levels in knees with meniscal injury as compared with asymptomatic contralateral knees. Am J Sports Med. 2019;47(4):847–56.
Struglics A, Turkiewicz A, Larsson S, Lohmander LS, Roemer FW, Frobell R, et al. Molecular and imaging biomarkers of local inflammation at 2 years after anterior cruciate ligament injury do not associate with patient reported outcomes at 5 years. Osteoarthr Cartil. 2020;28(3):356–62.
Moradi B, Schnatzer P, Hagmann S, Rosshirt N, Gotterbarm T, Kretzer JP, et al. CD4+CD25+/highCD127low/- regulatory T cells are enriched in rheumatoid arthritis and osteoarthritis joints - analysis of frequency and phenotype in synovial membrane, synovial fluid and peripheral blood. Arthritis Res Ther. 2014;16(2):R97.
Bondeson J, Wainwright SD, Lauder S, Amos N, Hughes CE. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis Res Ther. 2006;8(6):R187.
Wang EC, Newton Z, Hayward OA, Clark SR, Collins F, Perks WV, et al. Regulation of early cartilage destruction in inflammatory arthritis by death receptor 3. Arthritis Rheumatol (Hoboken, NJ). 2014;66(10):2762–72.
Klein-Wieringa IR, de Lange-Brokaar BJ, Yusuf E, Andersen SN, Kwekkeboom JC, Kroon HM, et al. Inflammatory cells in patients with endstage knee osteoarthritis: a comparison between the synovium and the infrapatellar fat pad. J Rheumatol. 2016;43(4):771–8.
Altman R, Asch E, Bloch D, Bole G, Borenstein D, Brandt K, et al. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 1986;29(8):1039–49.
Chen J, Vistica BP, Takase H, Ham DI, Fariss RN, Wawrousek EF, et al. A unique pattern of up- and down-regulation of chemokine receptor CXCR3 on inflammation-inducing Th1 cells. Eur J Immunol. 2004;34(10):2885–94.
Singh SP, Zhang HH, Foley JF, Hedrick MN, Farber JM. Human T cells that are able to produce IL-17 express the chemokine receptor CCR6. J Immunol. 2008;180(1):214–21.
Liu L, Callahan MK, Huang D, Ransohoff RM. Chemokine receptor CXCR3: an unexpected enigma. Curr Top Dev Biol. 2005;68:149–81.
Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187(1):129–34.
Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81(5):1258–68.
Rivino L, Messi M, Jarrossay D, Lanzavecchia A, Sallusto F, Geginat J. Chemokine receptor expression identifies pre-T helper (Th)1, pre-Th2, and nonpolarized cells among human CD4+ central memory T cells. J Exp Med. 2004;200(6):725–35.
Yamamoto J, Adachi Y, Onoue Y, Adachi YS, Okabe Y, Itazawa T, et al. Differential expression of the chemokine receptors by the Th1- and Th2-type effector populations within circulating CD4+ T cells. J Leukoc Biol. 2000;68(4):568–74.
Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277(5334):2005–7.
Doodes PD, Cao Y, Hamel KM, Wang Y, Rodeghero RL, Kobezda T, et al. CCR5 is involved in resolution of inflammation in proteoglycan-induced arthritis. Arthritis Rheum. 2009;60(10):2945–53.
Xie JH, Nomura N, Lu M, Chen SL, Koch GE, Weng Y, et al. Antibody-mediated blockade of the CXCR3 chemokine receptor results in diminished recruitment of T helper 1 cells into sites of inflammation. J Leukoc Biol. 2003;73(6):771–80.
Wedderburn LR, Robinson N, Patel A, Varsani H, Woo P. Selective recruitment of polarized T cells expressing CCR5 and CXCR3 to the inflamed joints of children with juvenile idiopathic arthritis. Arthritis Rheum. 2000;43(4):765–74.
Mazzetti I, Magagnoli G, Paoletti S, Uguccioni M, Olivotto E, Vitellozzi R, et al. A role for chemokines in the induction of chondrocyte phenotype modulation. Arthritis Rheum. 2004;50(1):112–22.
Tang CH, Hsu CJ, Fong YC. The CCL5/CCR5 axis promotes interleukin-6 production in human synovial fibroblasts. Arthritis Rheum. 2010;62(12):3615–24.
Stannus O, Jones G, Cicuttini F, Parameswaran V, Quinn S, Burgess J, et al. Circulating levels of IL-6 and TNF-alpha are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthr Cartil. 2010;18(11):1441–7.
Beekhuizen M, Gierman LM, van Spil WE, Van Osch GJ, Huizinga TW, Saris DB, et al. An explorative study comparing levels of soluble mediators in control and osteoarthritic synovial fluid. Osteoarthr Cartil. 2013;21(7):918–22.
Siqueira MB, Frangiamore S, Klika AK, Gajewski N, Barsoum WK, Higuera CA. Comparison of synovial fluid cytokine levels between traumatic knee injury and end-stage osteoarthritis. J Knee Surg. 2017;30(2):128–33.
Biggioggero M, Crotti C, Becciolini A, Favalli EG. Tocilizumab in the treatment of rheumatoid arthritis: an evidence-based review and patient selection. Drug Design Dev Ther. 2019;13:57–70.
Latourte A, Cherifi C, Maillet J, Ea HK, Bouaziz W, Funck-Brentano T, et al. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann Rheum Dis. 2017;76(4):748–55.
Lin Y, Liu L, Jiang H, Zhou J, Tang Y. Inhibition of interleukin-6 function attenuates the central sensitization and pain behavior induced by osteoarthritis. Eur J Pharmacol. 2017;811:260–7.
Haseeb A, Ansari MY, Haqqi TM. Harpagoside suppresses IL-6 expression in primary human osteoarthritis chondrocytes. J Orthop Res. 2017;35(2):311–20.
Laavola M, Leppanen T, Hamalainen M, Vuolteenaho K, Moilanen T, Nieminen R, et al. IL-6 in osteoarthritis: effects of pine stilbenoids. Molecules (Basel, Switzerland). 2018;24(1).
Acknowledgements
Not applicable
Funding
The University of Heidelberg funded this study. The funding source did not have any involvement in the study design, data collection, analysis and interpretation of the data, writing of the report, and the decision to submit the paper for publication. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
B.M., T.T., H.M.L., and N.R. made substantial contributions to the study conception and design. N.R. and T.R. carried out the flow cytometry and multiplex experiments. N.R., B.M., and E.T. made substantial contributions to the data acquisition. N.R., B.M., H.P., and S.H. made substantial contributions to the data analysis and interpretation. N.R. and B.M. drafted the manuscript, and E.T., S.H., T.A.N., H.M.L, T.T., and T.A.N. revised it critically. N.R. and B.M. performed the statistical analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Local Ethics Committee at Heidelberg University (S-333/2007).
Consent for publication
Not applicable
Competing interests
No benefits in any form have been or will be received from a commercial party related directly or indirectly to the subject of this manuscript. The authors have no potential or apparent conflicts of interest with regard to this work.
The manuscript has not been submitted or is not simultaneously being submitted elsewhere, and no portion of the data has been or will be published in proceedings or transactions of meetings or symposium volumes.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Antibody panel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rosshirt, N., Trauth, R., Platzer, H. et al. Proinflammatory T cell polarization is already present in patients with early knee osteoarthritis. Arthritis Res Ther 23, 37 (2021). https://doi.org/10.1186/s13075-020-02410-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-020-02410-w