
Abstract
Animal models of osteoarthritis are extensively used for investigating disease pathways and for preclinical testing of novel therapies. Their predictive utility, however, has often been questioned, mainly because preclinical efficacy of novel therapeutics is poorly translated in clinical trials. In the current narrative review, we consider the preclinical models that were used to support undertaking clinical trials for disease-modifying osteoarthritis drugs, and compare outcomes between clinical and preclinical studies. We discuss this in light of the 1999 Food and Drug Administration draft guidelines for industry for use in the development of drugs, devices, and biological products intended for the treatment of osteoarthritis, which raised five considerations on the usefulness of osteoarthritis models. We systematically discuss what has been learnt regarding these five points since 1999, with emphasis on replicating distinct risk factors and subtypes of human osteoarthritis, and on comprehensive evaluation of the disease in animals, including pathology of all joint tissues, biomarker analysis, and assessment of pain and joint function. Finally, we discuss lessons learnt and propose some recommendations for how the evidence from preclinical research might be strengthened with a view to improving success in clinical translation.
Similar content being viewed by others
Introduction
The current practice of translational biomedical research is failing its end-users, with as much as 90 % of ‘highly promising basic science discoveries failing to enter routine clinical use within 20 years’ [1]. As a result, it has been estimated that 85 % of research resources are ‘wasted’ [2]. That the majority of basic and preclinical medical research does not move to clinical trials, let alone lead to substantive improvements in patient health, challenges the relevance of the present translational discovery and development model [3, 4]. In an attempt to rationalize selection of promising therapeutic targets, metrics to evaluate ‘translatability’ have been developed [5, 6] and retrospectively validated [7]. Interestingly, in this proposed scoring system, ‘starting evidence’, which includes data from in vitro research, genetic modification in mice, and preclinical animal models of disease, only contributes a maximum of up to 11 % of the total translatability score. Despite this, a detailed analysis by one large pharmaceutical company of failures in their drug development pipeline showed that the most common reason (40 % versus 29 % for next most common) was inadequate linkage of the molecular/cellular target with the disease and no validated models [8]. Thus, while other metrics such as human genetic data, availability of biomarkers, and early phase clinical trial outcomes are clearly critical for successful translation, drug development programs will fail if their biological basis (that is, ‘the starting evidence’) is not sound. A cornerstone of strong scientific foundation for therapeutic development in all areas of medicine is the use of valid preclinical animal models of human disease, with predictive utility for research into disease pathways as well as for drug testing.
In 1999, the US Food and Drug Administration (FDA) provided draft guidelines for industry for use in the development of drugs, devices, and biological products intended for the treatment of osteoarthritis (OA) [9]. This nine-page document contains a paragraph on the use of preclinical models in OA, which surprisingly is lacking in the equivalent European guidelines [10]. The brief section (excerpt in Box 1) in the FDA document raises a number of specific issues that speak largely to how well any proposed animal model of OA mimics the human disease, and will therefore be predictive of therapeutic outcome in clinical trials and ultimately medical practice. Since the publication of the FDA document, 15 years ago with no further updates, OA research has witnessed many advances, both basic and clinical. For one, the concept of OA has broadened from a cartilage-driven disease to ‘a whole joint disease’ (Fig. 1). Magnetic resonance imaging (MRI) of painful OA knees clearly shows involvement of all joint tissues [11] and detailed molecular studies reveal that different joint tissues may all contribute to pathogenesis [12]. Consequently, many new targets that act on distinct aspects of OA pathology have been identified, leading to several clinical trials of putative disease-modifying OA drugs (DMOADs) that target various pathways in different joint tissues (Table 1). In general, outcomes of these trials have been disappointing, and there are still no FDA-approved DMOADs on the market. One question that arises is whether we could have predicted trial outcomes better using more appropriate preclinical models? Therefore, in the current narrative review, we systematically discuss the five considerations raised by the FDA regarding preclinical models of OA (Box 1), and review what, if anything, has been learnt since 1999. We consider the preclinical models that were used to support undertaking clinical trials for DMOADs, and compare outcomes between clinical and preclinical studies. Finally, we discuss lessons learnt and propose some recommendations for how the evidence from preclinical OA research might be strengthened with a view to improving success in this area of translational medicine.

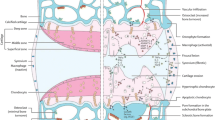
Osteoarthritis (OA) is a disease of the whole joint with pathology in all articular tissues and associated skeletal muscle. Pain in the OA joint can only arise directly from innervated tissues, but sensory neurons may be activated by factors released from aneural joint tissues
How accurately does the model replicate human osteoarthritis?
OA is no longer considered a single disorder; rather, it is a collection of different disease phenotypes that share certain clinical and pathological features [13]. Consequently the responses to treatment, and indeed the appropriate therapeutic targets in these different OA phenotypes, may be quite distinct [14, 15]. Suggested clinically relevant stratification approaches into OA phenotypes that may warrant distinct therapeutic strategies include: (i) cause or mechanism of onset (post-traumatic, age-associated, metabolic, or genetic); (ii) tissues affected (cartilage erosion, bone erosion or formation, synovitis/inflammation, muscle atrophy); (iii) progression (stage and rate); and (iv) symptoms (pain, disability) [16–20]. In light of these recent refinements in OA stratification, the choice of an ‘appropriate’ animal model will strongly depend on which type of human OA one wishes to replicate. Indeed, studies in genetically modified mice have revealed that in approximately one-third of cases the therapeutic outcome of an identical ‘intervention’ was dissimilar in experimental models mimicking different OA subtypes [21, 22]. This difference in outcome varied from an intervention being effective in one subtype but not another (for example, ablation of S100A8/9 inhibits cartilage erosion and osteophyte development only in experimental OA with a significant inflammatory component [23, 24]) to actually having opposite effects (for example, IL-6 ablation is protective in post-traumatic OA in young mice [25] but worsens age-associated OA [26]).
Age is one of the strongest risk factors for OA. Ageing causes changes in chondrocytes and articular cartilage, as well as in other joint tissues (meniscus, ligaments, bone, and synovium) and these age-related changes contribute to OA pathogenesis (reviewed in [27]). Radiographic changes in the joint become more common with age and OA often becomes symptomatic [27]. In contrast, most preclinical studies are conducted in young animals. For example, in the most widely used murine model of OA, the destabilization of the medial meniscus (DMM) model [28], the surgery is usually performed on 10- to 12-week-old mice. However, comparison of 12-week-old and 12-month-old mice revealed that age affects the basal pattern of gene expression in joint tissues [29], and when DMM is performed on 12-month-old animals, the ensuing OA is more severe than in young mice [29]. Rodents also develop spontaneous OA with age [30]. Thus, when modeling molecular mechanisms to define therapeutic targets in mice and rats, animal age should be a critical consideration. Response to specific compounds, such as putative DMOADs or novel OA analgesics, has not yet been directly compared in young versus old animals using the same OA model. There has been some comparison of the effect of a given genetic modification on induced post-traumatic versus spontaneous age-associated OA (reviewed in [21]) but there have been no studies that directly compare the effect of a specific gene on post-traumatic OA (induced by DMM, for example) in young versus old mice. Such studies would likely enhance our appreciation of the predictive potential of specific models with respect to different OA phenotypes.
While age remains perhaps the strongest predictor of OA development, overweight and obesity not only significantly increase the risk of incident hip and knee OA, particularly in women, but also its radiographic progression (reviewed in [31]). Studies in experimental animals have similarly shown increased OA incidence and severity with obesity, and it is apparent that this is not simply driven by increased mechanical loading of joints [32]. High-fat diets in the absence of obesity, and elevation of specific lipid components increase OA to a similar extent in laboratory animals, highlighting the complexity of the metabolic syndrome and the role of local and systemic inflammation and specific cytokines, chemokines and adipokines [33–36]. In addition to being a primary risk factor, obesity/metabolic syndrome also exacerbates post-traumatic OA in mice [37, 38], and the OA that occurs spontaneously in Trpv4 −/− mice [39]. To our knowledge, no studies have evaluated whether high-fat diet/obesity offsets the protective effect of other genetic modifications in mice such as ablation of Adamts5 or Mmp13. As suggested above with ageing, such studies would be of value in improving knowledge of the interaction of clinically important OA risk factors and improve the translational utility of data from preclinical models.
Not only does diet-induced obesity increase OA joint pathology in a mouse model, but it also induces anxiety and hyperalgesia, and decreases muscle function and locomotor activity [40]. These global effects are typical of the metabolic OA syndrome in patients, supporting the relevance of this preclinical model to the human condition [41]. In the few reported pharmacological interventions in high-fat diet-induced OA, a statin and a peroxisome proliferator-activated receptor γ agonist reduced pathology in C-reactive protein-transgenic mice [34] and a statin but not another cholesterol lowering agent reduced OA in the APOE*3 L.CETP transgenic hyperlipidemia mouse model [36]. The effect of statins on spontaneous OA in the STR/Ort mouse, which also displays obesity, have been inconsistent [42, 43]. While these divergent effects suggest caution in extrapolation of outcomes to humans, they actually reflect the variability seen in patients. Thus, a population-based study suggested there may be a disease-modifying effect of statins in knee but not hip OA [44]. However, a recent report [45] could not demonstrate association between nodal OA, hip OA or knee OA and use of statins after adjusting for confounders, although use of statins was associated with a lower prevalence of the generalized OA phenotype. In addition to effects of pharmacological agents, moderate exercise reduced OA severity in obese mice, and interestingly this was not associated with altered weight or body fat [46]. These results confirm that increased joint loading is not the primary driver of obesity-associated OA, consistent with outcomes of weight-loss and exercise programs in patients [47]. Weight loss relieves pain in obese OA patients, with a weight loss of at least 10 % providing significant pain reduction [48]. A recent study in individuals with symptomatic knee OA suggested a dose–response relationship between changes in body weight and corresponding changes in pain and physical function [49]. To date, no small animal models of obesity-induced OA have evaluated the effect of specific interventions on pain, but trials in overweight dogs confirm that exercise and weight loss may have a positive effect on pain and gait [50, 51].
Despite sex (in association with age and obesity) being a significant confounding factor for OA risk (reviewed in [52]), this has not been systematically studied and compared in preclinical research, either for pathophysiological or drug studies. Female sex, particularly post-menopausally, is a risk factor for prevalence and severity of OA [53]. As in humans, OA in baboons is more common in males in the younger population, but disease progresses more quickly following menopause so it is more prevalent and severe with age in females [54]. In contrast, in mice [55] and guinea pigs [56], spontaneous/age-associated OA is more common and severe in males than females. This may reflect greater weight gain in older males in these species, and that in animals other than primates, natural menopause and associated changes in bone turnover do not occur, necessitating ovariectomy to mimic the increased OA risk in older female patients (reviewed in [57]). Induced OA in animals may also be more progressive in males such as following DMM in mice [58], and as a result the majority of murine DMM studies are done in males with direct comparison of OA outcome measures or interventions (including genetic modification) in females being uncommon (for an example, see [59]). Increasingly, researchers are testing therapeutic intervention in ovariectomized rodents [60–64]. Pain researchers have long recognized that females are at greater risk for chronic pain, and the International Association for the Study of Pain has recommended the use of female experimental animals [65]. A recent meta-analysis showed that female sex is one of the main risk factors associated with onset of knee pain [66]. Despite the clear importance of sex in OA risk, progression and symptoms in patients, we are not aware of any preclinical studies directly comparing outcomes of therapeutic trials in males versus female animals.
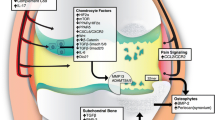
OA can be stratified according to affected tissues (Fig. 1). For instance, a subset of OA patients has high bone turnover and/or low subchondral bone (SCB) density, and as such may be responsive to bone-specific therapies [67]. Variable bone remodeling phenotypes and response to anti-resorptive therapy are also evident in preclinical OA models, depending on: (i) induction method (for example, intra-articular mono-iodoacetate (MIA) induces profound vascular invasion and SCB loss [68, 69]); (ii) stage of disease (early SCB loss followed by formation in a surgical rat model [70]); and (iii) species (for example, OA induced by meniscal injury in mice [61, 71] has a very limited bone resorption phase compared with rats [70, 72]). Like OA patients [17], different preclinical models may show different degrees of inflammation, and this can determine how an experimental treatment affects joint pathology or pain. For example, intra-articularly deposited adipose stem cells protect against cartilage damage and osteophyte size in inflammatory-driven collagenase-induced arthritis but not in the DMM model where synovitis is less pronounced [73]. In a side-by-side comparison in rats [74], meniscectomy resulted in more inflammation than MIA, and this was reflected by a greater analgesic effect of intra-articular triamcinolone, consistent with a greater contribution of synovitis to pain in the surgical model. This sort of side-by-side preclinical study highlights the necessity for selecting the animal model based on the pathological feature of OA one wishes to interrogate.
What are the structural determinants of pain and loss of function?
In knee OA, population studies support a substantial discordance between radiographic changes and knee pain [75]. Many knee MRI studies, both cross-sectional and longitudinal, have suggested associations of specific structural changes with OA pain, the strongest for synovitis and SCB (Fig. 1) - in particular, MRI-detected bone marrow lesions (reviewed in [76]). Recent clinical trial data suggest that targeting SCB in OA may indeed have an effect on pain (Table 1). A randomized trial with intravenous zoledronic acid, a bisphosphonate used for the treatment of osteoporosis, demonstrated a reduction in the volume of bone marrow lesions, and this was associated with reduced pain in subjects with knee OA [77]. Another osteoporosis drug, strontium ranelate, recently showed disease modification in a large 3-year placebo-controlled trial, and this was associated with a beneficial effect on pain, further supporting the role of bone in OA pathogenesis and symptoms [78, 79]. It has been reported that worsening of MRI-detected synovitis is associated with increased risk of frequent knee pain, but improvement of synovitis is not associated with decreased risk of pain or pain severity [80]. Therapeutic studies have demonstrated significant association between reduced synovitis on MRI and reduced pain in patients with rheumatoid arthritis [81] but not OA [82], suggesting there may be a poorer structure-function relationship in the latter.
To date, the relationship between pain/disability and specific joint structural changes have not been extensively explored across different OA models and species. Yet, targeted pharmacological modulation of specific facets of OA pathology in experimental animals permits dissecting the contribution of different aspects of joint disease to pain. Such studies require measuring pain on the one hand, which can be approached using different assays, including evoked pain responses and spontaneous pain behaviors (reviewed in [83]), and joint pathology on the other hand. In order to determine specific structural correlates to pain, pathology has to be carefully evaluated in all joint tissues, as opposed to a limited evaluation of the cartilage. While cartilage damage remains the primary outcome in most OA models, there has been a clear shift in recent years to include evaluation of other pathological changes in the joint such as osteophytes, SCB remodeling and synovitis [22]. Such comprehensive histopathology can reveal differential effects of a specific gene on joint tissues; for instance, ablation of Adamts5 or Mmp13 protects against cartilage damage and SCB sclerosis but not osteophyte formation after DMM [84, 85], while Ccr5 null mice develop less cartilage degeneration but show no differences in bone or synovial response to surgery [86]. This comprehensive approach to joint pathology is, however, far from the norm across different species and models, and methods for quantifying histopathology in joint tissues other than cartilage have not been standardized [87]. In addition, while muscle weakness and/or wasting is typical in OA patients, potentially playing a role in disease onset and progression and being a target for therapeutic intervention [88, 89], changes in skeletal muscle either as a consequence or cause of OA and associated pain in preclinical models remain largely understudied [90].
The contribution of SCB to OA pain has been examined in some detail in preclinical models of OA treated with different bone-remodeling agents. In rat MIA, pre-emptive alendronate treatment preserved SCB trabecular microarchitecture, decreased bone turnover and had moderate effects on cartilage degradation. These structural effects were accompanied by a positive effect on weight-bearing asymmetry, an indicator of pain [69]. In a canine study where OA was induced by transection of the anterior cruciate ligament, tiludronate treatment from time of surgery for 8 weeks had no effect on the severity of cartilage lesions or osteophytes, but treated dogs had a greater SCB surface and less synovitis than the vehicle-control group. These effects on bone and synovium were accompanied by improved pain behaviors (video-captured) and less gait disability [91]. These studies suggest that SCB bone may be a source of OA pain and targeting the bone may represent a strategy for OA analgesia. Several studies have compared therapeutic efficacy of targeting SCB in early versus later stages of disease, and found that the beneficial effect is greatest in early stages of disease when remodeling is most active. The effects of zoledronate in attenuating bone and cartilage loss and the accompanying weight-bearing asymmetry were more pronounced in prophylactic and early treatment protocols than in a delayed therapeutic setting [92]. Zoledronate had similar effects in the rat medial meniscal tear model [70], but only if administered early in the disease. Interestingly, an independent study recently confirmed the effect of prophylactic treatment with zoledronate in the rat MIA model on joint protection and concomitant partial reversal of weight-bearing deficits, and found this was associated with fewer channels crossing the osteochondral junction [93]. These osteochondral channels, containing blood vessels and small nociceptive neurons, have also been demonstrated in human OA, and may contribute to pain [94]. While pre-emptive administration of the osteoclastogenesis inhibitor osteoprotegerin in rat MIA reduced the number of subchondral osteoclasts, synovitis, and asymmetric weight-bearing, therapeutic treatment prevented further changes in weight-bearing and attenuated osteoclast numbers but not other structural changes in the joint [93].
Do structural changes correlate with clinical or biochemical markers?
When characterizing a preclinical model, assessing the effect of a particular gene, or evaluating therapeutic efficacy, emphasis has traditionally been on structural assessment of the joint, mostly through macroscopic assessment and histological scoring. The Osteoarthritis Research Society International (OARSI) has published guidelines for histological assessment of OA joints in different species in an attempt to standardize the assessment and reporting of animal studies (see collected papers in Osteoarthritis and Cartilage 2010, Supplement 3). Histology, however, does not allow for longitudinal assessment of the joint, and recent years have witnessed tremendous efforts to improve joint imaging in animals. In rodents, microCT, high-field MRI, and an array of contrast, luminescent and fluorescent bio-imaging methods have been developed (for reviews, see [95–97]). Discussion of these methods is beyond the scope of the current review, but preclinical models may play a critical role in identifying imaging markers that are predictive for onset and progression of OA.
One of the great challenges of clinical trials in OA has been the slow progression of disease and a lack of biomarkers that are predictive of OA structural disease onset and progression, and associated pain and disability. Since the writing of the FDA guidelines, emphasis has shifted to diagnosis and treatment of patients with early (preclinical) OA prior to significant/irreversible structural change and/or chronic pain where central sensitization may confound symptom modification [98]. With recent advances in imaging modalities, it has also become apparent that patients may have early osteochondral damage and meniscal tears that are asymptomatic but represent significant risk factors for progressive OA. Thus, the emphasis may shift even further to identifying, and perhaps treating, those that are at higher risk of developing OA. The search for predictive imaging or biological/biochemical biomarkers for structural and symptomatic OA risk, onset, progression and response to therapy remains paramount [99], and more slowly progressive preclinical models could play a critical role in this. Longitudinal assessment of biomarkers in serum, urine, or synovial fluid can further contribute to unraveling correlations between changes in imaging and biochemical biomarkers with disease stage (time after onset) and structural pathology/histopathology. Much of the biomarker work focuses on specific matrix degradation products that are also commonly used in clinical trials [100]. Recently some interesting studies have attempted to cast a broader net for predictive markers, for instance through white blood cell microarrays in a horse model [101] and metabolic profiling of serum [102] or synovial fluid in sheep [103].
For patients and clinicians, clearly the outcomes that matter are pain relief and preservation of joint function. In large animal models, for instance in dogs and horses, it has been a longstanding practice to assess joint function, mainly through evaluating gait. Increasingly in small animal models, methods to assess disease sequelae of OA, including pain, sensitization, locomotion, gait, and even anxiety or depression, are incorporated as outcomes [104]. For instance, in a cruciate ligament transection model in mice, significant correlation was found between cartilage structural damage and motor function, gait, and thermal hyperalgesia [105]. These behavioral outcomes have yet to be standardized in the context of OA but it can be anticipated that comprehensive longitudinal assessment of structural disease, imaging and/or biochemical markers and pain, sensitization, and/or functional outcomes - as exemplified in recent clinical [106] and preclinical [107] studies - will increase our appreciation of the correlation between specific aspects of OA (and this may be different for different models mimicking distinct OA phenotypes).
Is the model useful for studying prophylactic strategies or for studying structural arrest or reversal?
Many induced models of OA, particularly in rodents, are relatively rapidly progressive; thus, there may be a limited window for differentiating prophylactic from therapeutic effects. Progression of OA is generally slower in large animals, providing scope for evaluating timing of intervention, but they require more drug and are more expensive. The same OA induction method may have a very different time course and severity of disease in different species - for example, anterior cruciate ligament transection in sheep (mild, slow progression), goats (moderate, slow progression), dogs (moderate to severe, slow progression with phases of stability), and mice (severe, very rapid progression) (reviewed in [108]). Within a given species the severity and progression of OA will vary with the OA model, as demonstrated with different surgical induction methods in the mouse knee [109]. Interpretation of the prophylactic or therapeutic efficacy of an intervention, and how the findings translate to humans, requires in-depth understanding of the preclinical model used.
Spontaneous OA develops not only with advanced ageing (for instance, in male C57BL/6 mice) but also precociously (that is, during maturation rather than ageing) in a number of animals (for example, male Hartley guinea pigs and male STR/Ort mice). These spontaneous models are slowly progressive and amenable to evaluation of therapies for both prevention and structural arrest (for examples in guinea pig, see [110–112], and in STR/Ort mice, see [43, 113]). However, the variable onset and progression compared with induced models of OA may necessitate greater animal numbers to sufficiently power studies to detect therapeutic effect, and longer treatment times for prophylactic studies and thus more test compound and cost. It is also important to recognize that the underlying mechanisms that drive the onset and progression of spontaneous OA in these animals are not well defined, and may reflect specific subtypes of idiopathic human OA - for example, early SCB remodeling, meniscal ossification and ligament changes in Hartley guinea pigs [114–117] and systemic inflammatory/immune response in STR/Ort mice [118, 119].
Most preclinical OA studies test the efficacy of a given intervention on structural protection and usually in ‘prophylactic protocols’ - that is, treatment starts at the time or immediately after disease is induced. The direct translational relevance of this approach into the clinical situation is questionable, since even ‘early OA’ has structural changes in the joint at the time of diagnosis [98]. A prophylactic approach may be valid as an initial preclinical therapeutic trial given the current interest in specifically targeting post-traumatic OA [22], and it may provide the best opportunity to detect a therapeutic effect of the test compound or class of compounds - that is, proof of principle. However, when considering a broader patient population including those with established OA, testing interventions at different stages of disease in preclinical models is critical, as exemplified by the bisphosphonate [69, 70, 92] and osteoprotegerin [93] studies in rat OA models, where efficacy was quite different depending on when therapy was initiated. Intra-articular mesenchymal stem cells were found to be effective only when administered early in the mouse collagenase-induced arthritis model [73, 120]. Pharmacological augmentation of Runx1 on the other hand was effective in both prophylactic and therapeutic protocols in a surgically induced OA model in mice [121]. Clearly, to appreciate the therapeutic potential of any new agent, it will be necessary to test both prophylactic and therapeutic protocols across different models.
Can the model be used to assess long-term toxicity?
Since the chronic course of OA will require prolonged therapy aimed at improving quality of life, drug safety is a particularly important issue [99]. The single most common reason for closure of drug development programs is safety, usually during discovery phase mandatory toxicity testing [8] (Fig. 2). Toxicology studies are usually performed in healthy animals rather than in concert with the preclinical disease model, and may not involve use of animals with typical risks and co-morbidities associated with OA (such as age or obesity). It could be proposed that for a chronic pathology like OA, it may be advisable to test long-term blockade of a specific target in a disease-specific chronic model. With rapidly progressive disease models, such as rat MIA, often only evaluated for 3 to 4 weeks, long-term side effects or toxicity of therapy are unlikely to become apparent. The experience with unexpected musculoskeletal side effects in patients on prolonged broad-spectrum matrix metalloproteinase inhibitors, however, suggests that even routine use of long-term preclinical OA models may not have been predictive. To induce the syndrome in rats required high doses administered by osmotic pumps and careful histopathological screening or specific functional testing [122, 123]. More recently, the occurrence of rapidly progressive OA in non-target joints in patients enrolled in a clinical trial of anti-nerve growth factor [124] highlights the need for disease- and drug-specific toxicity models. In this case concomitant administration of nonsteroidal anti-inflammatory drugs (NSAIDs) has been implicated in the risk for side effects. It seems very unlikely that a preclinical model would routinely involve multi-drug therapy and model target and non-target joint disease.

Drug development pipeline showing the translational phases (T1 to T4) from basic science discovery through to measurement of impact on population health. Failure of drug development programs can occur at all stages, but the major reasons differ with translational phase. **Efficacy failure is primarily due to poor biological rationale for the clinical trial: target linkage to disease not established or no validated models available (40 %) and indication selected does not fit the strongest preclinical evidence (20 %) [8]. 1[8], 2[142], 3[143], 4[144]
Predicting efficacy in preclinical models: how well have we done and what can we improve?
Placebo-controlled DMOAD trials for knee OA published in peer-reviewed journals since 1999 are listed in Table 1, juxtaposing clinical trial outcomes and findings in preclinical studies. A cursory evaluation of this table leads to the simple conclusion that preclinical models were poor predictors of clinical trial outcomes. However, when interpreted in light of the above discussion, it emerges that animal study design and use has been largely suboptimal. Preclinical testing was typically performed by treating prophylactically or early in induced models (mostly post-traumatic OA) in young and normal-weight animals, whereas clinical trials mostly focus on age/obesity-associated, established/late-stage (Kellgren-Lawrence grade 2 to 3) OA. Thus, the OA target population and preclinical phenotype are mismatched. Further, most preclinical studies reported are restricted to limited time points in one study in one animal model, in one species, and in one laboratory - that is, there is no testing for reproducibility. The animal studies usually evaluate a limited set of outcome parameters, and these parameters typically interrogate the mode of action of the drug more than assessing the overall joint health and animal well-being (that is, ‘exploratory’ rather than ‘confirmatory’ studies [125]). This limits translational value to the clinical DMOAD trial, where the primary outcome is joint space narrowing and there is usually a secondary symptomatic outcome.
Evidence for the predictive validity of OA animal models for testing efficacy of novel analgesics under development for OA pain is extremely limited. Recent clinical trials with antibodies against nerve growth factor showed clinical benefit, with reduction in joint pain and improved function [124], but results of preclinical testing of anti-nerve growth factor in OA models are not yet available in the public domain. Duloxetine, recently approved for the treatment of chronic musculoskeletal pain, was efficacious in a 13-week, randomized, double-blind, placebo-controlled trial in patients with symptomatic knee OA [126]. It has been tested in rat MIA, where it had a moderate effect on hind limb grip force deficit [127], and corrected weight-bearing deficit but not gait imbalance [128]. In the same model, local administration of a fatty acid amide hydrolase-1 inhibitor, which modulates endocannabinoids, significantly reduced hindlimb incapacitance [129], but a randomized, placebo-controlled clinical trial with an irreversible fatty acid amide hydrolase-1 inhibitor failed to induce effective analgesia in patients with painful knee OA [130]. The potential reasons for the different predictive utility of the model for patient outcomes are manifold. A recent, first of its kind, meta-analysis of preclinical studies of the effects of existing analgesic drugs (NSAIDs and opioids) in models of OA pain provides an analysis of factors that may impact the magnitude of the analgesic treatment effect in animal models of OA pain [131]. Improved understanding of how the analgesic effects of existing and novel compounds observed in various OA animal models relate to pain and disability in patients will be paramount to advance translation of symptom-modifying therapies.
Conclusion
Design and use of animal models of OA can be deemed suboptimal, mainly with respect to OA phenotype, risk factors, and endpoints under evaluation. Suggestions for improvements that will enable translation in OA research are similar in other diseases [132] and include:
-
1.
Better aligning of preclinical models and the clinical trial population - including OA disease phenotype, stage, age, sex and confounding co-morbidities.
-
2.
Increasing the ‘translatability score’ of tested therapies [5, 6] by including evaluation in multiple OA models and species, and improving reproducibility (for example, replicating data in different laboratories or using ‘multi-institutional synchronous co-clinical trials in mice’ [132, 133]).
-
3.
Standardizing preclinical outcome measures for both OA pathology and pain (for example, see collected papers in Osteoarthritis and Cartilage 2010, Supplement 3; and see [134]). This will not only enable comparisons between studies and research groups, but also permit evaluation of the relative efficacy of different interventions/treatments.
-
4.
Making measured outcomes more clinically relevant by evaluating whole joint pathology (not just cartilage) and including measures of pain/disability in addition to structural pathology. Understanding how the outcomes measured in the animal model relate to the human disease may be improved through using clinically relevant imaging modalities in preclinical studies [135]. Changing emphasis from ‘statistically significant’ to ‘clinically significant’ effects is an issue pertinent to both preclinical and clinical research, requiring the establishment of ‘minimally clinically important differences’ (MCIDs) [136, 137]. The MCIDs for different outcomes in patients need to be translated to an equivalent ‘effect size’ in animal models, and then more universally applicable and standardized measures of efficacy in preclinical studies can be used, such as the ‘percentage responders’ (that is, that reach the MCID/effect size) and ‘number needed to treat’ [138].
-
5.
Increasing the rigor of preclinical science and its reporting, including use of ARRIVE or other guidelines [139] enabling better use of meta-analysis of preclinical OA studies of structure and pain therapeutic modification [131, 140].
-
6.
Embracing and making use of the One Health Initiative [141]. The concept behind this worldwide strategy is that interdisciplinary collaboration and communication between medical and veterinary health care and health research will provide multi-directional flow of knowledge and synergistic gains for both disciplines. Thus, beyond the use of induced OA models in laboratory animals to investigate pathophysiology and treatment, studying OA onset, progression and management in clinical veterinary practice provides a ‘real-world laboratory’ of naturally occurring disease cohorts with similar co-morbidities to humans (for example, obesity and ageing in dogs or exercise-induced trauma in athletic horses) for robust trials of newly developed therapies. This offers the potential to both benefit animals with OA and to inform human clinical trials and patient care.
Rather than concluding that preclinical models are not useful in translational medical research [4], this review has highlighted a number of issues that could be addressed to improve the predictive utility of OA animal models. Increasingly, researchers are incorporating these considerations into the design and reporting of studies in OA models. As both clinical and preclinical researchers improve comprehensive longitudinal assessment of structural disease, imaging and/or biochemical markers and pain, sensitization, and/or functional outcomes, our appreciation of OA phenotypes and their appropriate modeling will increase. It can be expected that this will result in better predictivity of preclinical findings for human translation and reduce failures.
Abbreviations
- DMM:
-
Destabilization of the medial meniscus
- DMOAD:
-
Disease-modifying osteoarthritis drug
- FDA:
-
Food and drug administration
- MCID:
-
Minimally clinically important difference
- MIA:
-
Mono-iodoacetate
- MRI:
-
Magnetic resonance imaging
- NSAID:
-
Nonsteroidal anti-inflammatory drug
- OA:
-
Osteoarthritis
- OARSI:
-
Osteoarthritis research society international
- SCB:
-
Subchondral bone
References
Contopoulos-Ioannidis DG, Ntzani E, Ioannidis JP. Translation of highly promising basic science research into clinical applications. Am J Med. 2003;114:477–84.
Macleod MR, Michie S, Roberts I, Dirnagl U, Chalmers I, Ioannidis JP, et al. Biomedical research: increasing value, reducing waste. Lancet. 2014;383:101–4.
Wehling M. Drug development in the light of translational science: shine or shade? Drug Discov Today. 2011;16:1076–83.
Pound P, Bracken MB. Is animal research sufficiently evidence based to be a cornerstone of biomedical research? BMJ. 2014;348:g3387.
Wehling M. Assessing the translatability of drug projects: what needs to be scored to predict success? Nat Rev Drug Discov. 2009;8:541–6.
Wendler A, Wehling M. The translatability of animal models for clinical development: biomarkers and disease models. Curr Opin Pharmacol. 2010;10:601–6.
Wendler A, Wehling M. Translatability scoring in drug development: eight case studies. J Transl Med. 2012;10:39.
Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, et al. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13:419–31.
FDA. Clinical development programs for drugs, devices, and biological products intended for the treatment of osteoarthritis (OA). 1999. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071577.pdf.
EMEA: Guideline On Clinical Investigation Of Medicinal Products Used In The Treatment Of Osteoarthritis. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003443.pdf.
Wenham CY, Conaghan PG. Imaging the painful osteoarthritic knee joint: what have we learned? Nat Clin Pract Rheumatol. 2009;5:149–58.
Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–707.
Lane NE, Brandt K, Hawker G, Peeva E, Schreyer E, Tsuji W, et al. OARSI-FDA initiative: defining the disease state of osteoarthritis. Osteoarthritis Cartilage. 2011;19:478–82.
Bierma-Zeinstra SM, Verhagen AP. Osteoarthritis subpopulations and implications for clinical trial design. Arthritis Res Ther. 2011;13:213.
Conaghan PG. Osteoarthritis in 2012: Parallel evolution of OA phenotypes and therapies. Nat Rev Rheumatol. 2013;9:68–70.
Herrero-Beaumont G, Roman-Blas JA, Castaneda S, Jimenez SA. Primary osteoarthritis no longer primary: three subsets with distinct etiological, clinical, and therapeutic characteristics. Semin Arthritis Rheum. 2009;39:71–80.
Driban JB, Sitler MR, Barbe MF, Balasubramanian E. Is osteoarthritis a heterogeneous disease that can be stratified into subsets? Clin Rheumatol. 2010;29:123–31.
McGonagle D, Tan AL, Carey J, Benjamin M. The anatomical basis for a novel classification of osteoarthritis and allied disorders. J Anat. 2010;216:279–91.
Bijlsma JW, Berenbaum F, Lafeber FP. Osteoarthritis: an update with relevance for clinical practice. Lancet. 2011;377:2115–26.
Knoop J, van der Leeden M, Thorstensson CA, Roorda LD, Lems WF, Knol DL, et al. Identification of phenotypes with different clinical outcomes in knee osteoarthritis: data from the Osteoarthritis Initiative. Arthritis Care Res (Hoboken). 2011;63:1535–42.
Little CB, Zaki S. What constitutes an ‘animal model of osteoarthritis’ - the need for consensus? Osteoarthritis Cartilage. 2012;20:261–7.
Little CB, Hunter DJ. Post-traumatic osteoarthritis: from mouse models to clinical trials. Nat Rev Rheumatol. 2013;9:485–97.
van Lent PL, Blom AB, Schelbergen RF, Sloetjes A, Lafeber FP, Lems WF, et al. Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum. 2012;64:1466–76.
Schelbergen RF, de Munter W, van den Bosch MH, Lafeber FP, Sloetjes A, Vogl T, et al. Alarmins S100A8/S100A9 aggravate osteophyte formation in experimental osteoarthritis and predict osteophyte progression in early human symptomatic osteoarthritis. Ann Rheum Dis. 2014; doi:10.1136/annrheumdis-2014-205480.
Ryu JH, Yang S, Shin Y, Rhee J, Chun CH, Chun JS. Interleukin-6 plays an essential role in hypoxia-inducible factor 2alpha-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2011;63:2732–43.
de Hooge AS, van de Loo FA, Bennink MB, Arntz OJ, de Hooge P, van den Berg WB. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthritis Cartilage. 2005;13:66–73.
Loeser RF. Aging processes and the development of osteoarthritis. Curr Opin Rheumatol. 2013;25:108–13.
Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15:1061–9.
Loeser RF, Olex AL, McNulty MA, Carlson CS, Callahan MF, Ferguson CM, et al. Microarray analysis reveals age-related differences in gene expression during the development of osteoarthritis in mice. Arthritis Rheum. 2012;64:705–17.
Bendele AM. Animal models of osteoarthritis. J Musculoskelet Neuronal Interact. 2001;1:363–76.
Johnson VL, Hunter DJ. The epidemiology of osteoarthritis. Best Pract Res Clin Rheumatol. 2014;28:5–15.
Issa RI, Griffin TM. Pathobiology of obesity and osteoarthritis: integrating biomechanics and inflammation. Pathobiol Aging Age Relat Dis. 2012;2:17470.
Brunner AM, Henn CM, Drewniak EI, Lesieur-Brooks A, Machan J, Crisco JJ, et al. High dietary fat and the development of osteoarthritis in a rabbit model. Osteoarthritis Cartilage. 2012;20:584–92.
Gierman LM, van der Ham F, Koudijs A, Wielinga PY, Kleemann R, Kooistra T, et al. Metabolic stress-induced inflammation plays a major role in the development of osteoarthritis in mice. Arthritis Rheum. 2012;64:1172–81.
Iwata M, Ochi H, Hara Y, Tagawa M, Koga D, Okawa A, et al. Initial responses of articular tissues in a murine high-fat diet-induced osteoarthritis model: pivotal role of the IPFP as a cytokine fountain. PLoS One. 2013;8, e60706.
Gierman LM, Kuhnast S, Koudijs A, Pieterman EJ, Kloppenburg M, van Osch GJ, et al. Osteoarthritis development is induced by increased dietary cholesterol and can be inhibited by atorvastatin in APOE*3Leiden.CETP mice - a translational model for atherosclerosis. Ann Rheum Dis. 2014;73:921–7.
Mooney RA, Sampson ER, Lerea J, Rosier RN, Zuscik MJ. High-fat diet accelerates progression of osteoarthritis after meniscal/ligamentous injury. Arthritis Res Ther. 2011;13:R198.
Louer CR, Furman BD, Huebner JL, Kraus VB, Olson SA, Guilak F. Diet-induced obesity significantly increases the severity of posttraumatic arthritis in mice. Arthritis Rheum. 2012;64:3220–30.
O’Conor CJ, Griffin TM, Liedtke W, Guilak F. Increased susceptibility of Trpv4-deficient mice to obesity and obesity-induced osteoarthritis with very high-fat diet. Ann Rheum Dis. 2013;72:300–4.
Griffin TM, Fermor B, Huebner JL, Kraus VB, Rodriguiz RM, Wetsel WC, et al. Diet-induced obesity differentially regulates behavioral, biomechanical, and molecular risk factors for osteoarthritis in mice. Arthritis Res Ther. 2010;12:R130.
van der Kraan PM. Osteoarthritis and a high-fat diet: the full ‘OA syndrome’ in a small animal model. Arthritis Res Ther. 2010;12:130.
Yudoh K, Karasawa R. Statin prevents chondrocyte aging and degeneration of articular cartilage in osteoarthritis (OA). Aging (Albany NY). 2010;2:990–8.
Wei W, Clockaerts S, Bastiaansen-Jenniskens YM, Gierman LM, Botter SM, Bierma-Zeinstra SM, et al. Statins and fibrates do not affect development of spontaneous cartilage damage in STR/Ort mice. Osteoarthritis Cartilage. 2014;22:293–301.
Clockaerts S, Van Osch GJ, Bastiaansen-Jenniskens YM, Verhaar JA, Van Glabbeek F, Van Meurs JB, et al. Statin use is associated with reduced incidence and progression of knee osteoarthritis in the Rotterdam study. Ann Rheum Dis. 2012;71:642–7.
Valdes AM, Zhang W, Muir K, Maciewicz RA, Doherty S, Doherty M. Use of statins is associated with a lower prevalence of generalised osteoarthritis. Ann Rheum Dis. 2014;73:943–5.
Griffin TM, Huebner JL, Kraus VB, Yan Z, Guilak F. Induction of osteoarthritis and metabolic inflammation by a very high-fat diet in mice: effects of short-term exercise. Arthritis Rheum. 2012;64:443–53.
Messier SP, Mihalko SL, Legault C, Miller GD, Nicklas BJ, DeVita P, et al. Effects of intensive diet and exercise on knee joint loads, inflammation, and clinical outcomes among overweight and obese adults with knee osteoarthritis: the IDEA randomized clinical trial. JAMA. 2013;310:1263–73.
Christensen R, Astrup A, Bliddal H. Weight loss: the treatment of choice for knee osteoarthritis? A randomized trial. Osteoarthritis Cartilage. 2005;13:20–7.
Riddle DL, Stratford PW. Body weight changes and corresponding changes in pain and function in persons with symptomatic knee osteoarthritis: a cohort study. Arthritis Care Res (Hoboken). 2013;65:15–22.
Marshall W, Bockstahler B, Hulse D, Carmichael S. A review of osteoarthritis and obesity: current understanding of the relationship and benefit of obesity treatment and prevention in the dog. Vet Comp Orthop Traumatol. 2009;22:339–45.
Marshall WG, Hazewinkel HA, Mullen D, De Meyer G, Baert K, Carmichael S. The effect of weight loss on lameness in obese dogs with osteoarthritis. Vet Res Commun. 2010;34:241–53.
Neogi T, Zhang Y. Epidemiology of osteoarthritis. Rheum Dis Clin North Am. 2013;39:1–19.
Srikanth VK, Fryer JL, Zhai G, Winzenberg TM, Hosmer D, Jones G. A meta-analysis of sex differences prevalence, incidence and severity of osteoarthritis. Osteoarthritis Cartilage. 2005;13:769–81.
Macrini TE, Coan HB, Levine SM, Lerma T, Saks CD, Araujo DJ, et al. Reproductive status and sex show strong effects on knee OA in a baboon model. Osteoarthritis Cartilage. 2013;21:839–48.
Uchida K, Urabe K, Naruse K, Kozai Y, Onuma K, Mikuni-Takagaki Y, et al. Differential age-related bone architecture changes between female and male STR/Ort mice. Exp Anim. 2012;61:59–66.
Kraus VB, Huebner JL, DeGroot J, Bendele A. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the guinea pig. Osteoarthritis Cartilage. 2010;18:S35–52.
Sniekers YH, Weinans H, Bierma-Zeinstra SM, van Leeuwen JP, van Osch GJ. Animal models for osteoarthritis: the effect of ovariectomy and estrogen treatment - a systematic approach. Osteoarthritis Cartilage. 2008;16:533–41.
Ma HL, Blanchet TJ, Peluso D, Hopkins B, Morris EA, Glasson SS. Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage. 2007;15:695–700.
Malfait AM, Ritchie J, Gil AS, Austin JS, Hartke J, Qin W, et al. ADAMTS-5 deficient mice do not develop mechanical allodynia associated with osteoarthritis following medial meniscal destabilization. Osteoarthritis Cartilage. 2010;18:572–80.
Chang TK, Huang CH, Chen HC, Cheng CK. The influence of long-term treadmill exercise on bone mass and articular cartilage in ovariectomized rats. BMC Musculoskelet Disord. 2010;11:185.
Funck-Brentano T, Lin H, Hay E, Ah Kioon MD, Schiltz C, Hannouche D, et al. Targeting bone alleviates osteoarthritis in osteopenic mice and modulates cartilage catabolism. PLoS One. 2012;7, e33543.
Zhu S, Chen K, Lan Y, Zhang N, Jiang R, Hu J. Alendronate protects against articular cartilage erosion by inhibiting subchondral bone loss in ovariectomized rats. Bone. 2013;53:340–9.
Yang PY, Tang CC, Chang YC, Huang SY, Hsieh SP, Fan SS, et al. Effects of tibolone on osteoarthritis in ovariectomized rats: association with nociceptive pain behaviour. Eur J Pain. 2014;18:680–90.
Cui Z, Xu C, Li X, Song J, Yu B. Treatment with recombinant lubricin attenuates osteoarthritis by positive feedback loop between articular cartilage and subchondral bone in ovariectomized rats. Bone. 2015;74C:37–47.
Greenspan JD, Craft RM, LeResche L, Arendt-Nielsen L, Berkley KJ, Fillingim RB, et al. Studying sex and gender differences in pain and analgesia: a consensus report. Pain. 2007;132:S26–45.
Silverwood V, Blagojevic-Bucknall M, Jinks C, Jordan JL, Protheroe J, Jordan KP. Current evidence on risk factors for knee osteoarthritis in older adults: a systematic review and meta-analysis. Osteoarthritis Cartilage. 2015;23:507–15.
Roman-Blas JA, Castaneda S, Largo R, Lems WF, Herrero-Beaumont G. An OA phenotype may obtain major benefit from bone-acting agents. Semin Arthritis Rheum. 2014;43:421–8.
Morenko BJ, Bove SE, Chen L, Guzman RE, Juneau P, Bocan TM, et al. In vivo micro computed tomography of subchondral bone in the rat after intra-articular administration of monosodium iodoacetate. Contemp Top Lab Anim Sci. 2004;43:39–43.
Mohan G, Perilli E, Parkinson IH, Humphries JM, Fazzalari NL, Kuliwaba JS. Pre-emptive, early, and delayed alendronate treatment in a rat model of knee osteoarthritis: effect on subchondral trabecular bone microarchitecture and cartilage degradation of the tibia, bone/cartilage turnover, and joint discomfort. Osteoarthritis Cartilage. 2013;21:1595–604.
Yu DG, Yu B, Mao YQ, Zhao X, Wang XQ, Ding HF, et al. Efficacy of zoledronic acid in treatment of teoarthritis is dependent on the disease progression stage in rat medial meniscal tear model. Acta Pharmacol Sin. 2012;33:924–34.
Kadri A, Funck-Brentano T, Lin H, Ea HK, Hannouche D, Marty C, et al. Inhibition of bone resorption blunts osteoarthritis in mice with high bone remodelling. Ann Rheum Dis. 2010;69:1533–8.
Iijima H, Aoyama T, Ito A, Tajino J, Nagai M, Zhang X, et al. Destabilization of the medial meniscus leads to subchondral bone defects and site-specific cartilage degeneration in an experimental rat model. Osteoarthritis Cartilage. 2014;22:1036–43.
Schelbergen RF, van Dalen S, ter Huurne M, Roth J, Vogl T, Noel D, et al. Treatment efficacy of adipose-derived stem cells in experimental osteoarthritis is driven by high synovial activation and reflected by S100A8/A9 serum levels. Osteoarthritis Cartilage. 2014;22:1158–66.
Mapp PI, Sagar DR, Ashraf S, Burston JJ, Suri S, Chapman V, et al. Differences in structural and pain phenotypes in the sodium monoiodoacetate and meniscal transection models of osteoarthritis. Osteoarthritis Cartilage. 2013;21:1336–45.
Hannan MT, Felson DT, Pincus T. Analysis of the discordance between radiographic changes and knee pain in osteoarthritis of the knee. J Rheumatol. 2000;27:1513–7.
Hunter DJ, Guermazi A, Roemer F, Neogi T. Structural correlates of pain in joints with osteoarthritis. Osteoarthritis Cartilage. 2013;21:1170–8.
Laslett LL, Dore DA, Quinn SJ, Boon P, Ryan E, Winzenberg TM, et al. Zoledronic acid reduces knee pain and bone marrow lesions over 1 year: a randomised controlled trial. Ann Rheum Dis. 2012;71:1322–8.
Reginster JY. Efficacy and safety of strontium ranelate in the treatment of knee osteoarthritis: results of a double-blind randomised, placebo-controlled trial. Ann Rheum Dis. 2014;73, e8.
Bruyere O, Reginster JY, Bellamy N, Chapurlat R, Richette P, Cooper C. Clinically meaningful effect of strontium ranelate on symptoms in knee osteoarthritis: a responder analysis. Rheumatology (Oxford). 2014;53:1457–64.
Zhang Y, Nevitt M, Niu J, Lewis C, Torner J, Guermazi A, et al. Fluctuation of knee pain and changes in bone marrow lesions, effusions, and synovitis on magnetic resonance imaging. Arthritis Rheum. 2011;63:691–9.
Ostergaard M, Stoltenberg M, Gideon P, Sorensen K, Henriksen O, Lorenzen I. Changes in synovial membrane and joint effusion volumes after intraarticular methylprednisolone. Quantitative assessment of inflammatory and destructive changes in arthritis by MRI. J Rheumatol. 1996;23:1151–61.
Wenham CY, Balamoody S, Grainger AJ, Hensor EM, Draycott S, Hodgson R, et al. The responsiveness of novel, dynamic, contrast-enhanced magnetic resonance measures of total knee synovitis after intra-articular corticosteroid for painful osteoarthritis. Osteoarthritis Cartilage. 2014;22:1614–8.
Malfait AM, Little CB, McDougall JJ. A commentary on modeling osteoarthritis pain in small animals. Osteoarthritis Cartilage. 2013;21:1316–26.
Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434:644–8.
Little CB, Barai A, Burkhardt D, Smith SM, Fosang AJ, Werb Z, et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60:3723–33.
Takebe K, Farooq Rai M, Schmidt E, Sandell L. The chemokine receptor CCR5 plays a role in post-traumatic cartilage loss in mice, but does not affect synovium and bone. Osteoarthritis Cartilage. 2015;23:454–61.
Aigner T, Cook JL, Gerwin N, Glasson SS, Laverty S, Little CB, et al. Histopathology atlas of animal model systems - overview of guiding principles. Osteoarthritis Cartilage. 2010;18:S2–6.
Bennell K, Hinman RS, Wrigley TV, Creaby MW, Hodges P. Exercise and osteoarthritis: cause and effects. Compr Physiol. 2011;1:1943–2008.
Roos EM, Herzog W, Block JA, Bennell KL. Muscle weakness, afferent sensory dysfunction and exercise in knee osteoarthritis. Nat Rev Rheumatol. 2011;7:57–63.
Egloff C, Sawatsky A, Leonard T, Hart DA, Valderrabano V, Herzog W. Effect of muscle weakness and joint inflammation on the onset and progression of osteoarthritis in the rabbit knee. Osteoarthritis Cartilage. 2014;22:1886–93.
Moreau M, Rialland P, Pelletier JP, Martel-Pelletier J, Lajeunesse D, Boileau C, et al. Tiludronate treatment improves structural changes and symptoms of osteoarthritis in the canine anterior cruciate ligament model. Arthritis Res Ther. 2011;13:R98.
Strassle BW, Mark L, Leventhal L, Piesla MJ, Jian Li X, Kennedy JD, et al. Inhibition of osteoclasts prevents cartilage loss and pain in a rat model of degenerative joint disease. Osteoarthritis Cartilage. 2010;18:1319–28.
Sagar DR, Ashraf S, Xu L, Burston JJ, Menhinick MR, Poulter CL, et al. Osteoprotegerin reduces the development of pain behaviour and joint pathology in a model of osteoarthritis. Ann Rheum Dis. 2014;73:1558–65.
Mapp PI, Walsh DA. Mechanisms and targets of angiogenesis and nerve growth in osteoarthritis. Nat Rev Rheumatol. 2012;8:390–8.
Piscaer TM, van Osch GJ, Verhaar JA, Weinans H. Imaging of experimental osteoarthritis in small animal models. Biorheology. 2008;45:355–64.
Goebel JC, Pinzano A, Grenier D, Perrier AL, Henrionnet C, Galois L, et al. New trends in MRI of cartilage: advances and limitations in small animal studies. Biomed Mater Eng. 2010;20:189–94.
Tremoleda JL, Khalil M, Gompels LL, Wylezinska-Arridge M, Vincent T, Gsell W. Imaging technologies for preclinical models of bone and joint disorders. EJNMMI Res. 2011;1:11.
Felson DT, Hodgson R. Identifying and treating preclinical and early osteoarthritis. Rheum Dis Clin North Am. 2014;40:699–710.
Hunter DJ, Nevitt M, Losina E, Kraus V. Biomarkers for osteoarthritis: current position and steps towards further validation. Best Pract Res Clin Rheumatol. 2014;28:61–71.
Kraus VB, Burnett B, Coindreau J, Cottrell S, Eyre D, Gendreau M, et al. Application of biomarkers in the development of drugs intended for the treatment of osteoarthritis. Osteoarthritis Cartilage. 2011;19:515–42.
Kamm JL, Frisbie DD, McIlwraith CW, Orr KE. Gene biomarkers in peripheral white blood cells of horses with experimentally induced osteoarthritis. Am J Vet Res. 2013;74:115–21.
Maher AD, Coles C, White J, Bateman JF, Fuller ES, Burkhardt D, et al. 1H NMR spectroscopy of serum reveals unique metabolic fingerprints associated with subtypes of surgically induced osteoarthritis in sheep. J Proteome Res. 2012;11:4261–8.
Mickiewicz B, Heard BJ, Chau JK, Chung M, Hart DA, Shrive NG, et al. Metabolic profiling of synovial fluid in a unilateral ovine model of anterior cruciate ligament reconstruction of the knee suggests biomarkers for early osteoarthritis. J Orthop Res. 2015;33:71–7.
Allen KD, Adams SB, Setton LA. Evaluating intra-articular drug delivery for the treatment of osteoarthritis in a rat model. Tissue Eng Part B Rev. 2010;16:81–92.
Ruan MZ, Patel RM, Dawson BC, Jiang MM, Lee BH. Pain, motor and gait assessment of murine osteoarthritis in a cruciate ligament transection model. Osteoarthritis Cartilage. 2013;21:1355–64.
Arendt-Nielsen L, Eskehave TN, Egsgaard LL, Petersen KK, Graven-Nielsen T, Hoeck HC, et al. Association between experimental pain biomarkers and serologic markers in patients with different degrees of painful knee osteoarthritis. Arthritis Rheumatol. 2014;66:3317–26.
Bowles RD, Mata BA, Bell RD, Mwangi TK, Huebner JL, Kraus VB, et al. In vivo luminescence imaging of NF-kappaB activity and serum cytokine levels predict pain sensitivities in a rodent model of osteoarthritis. Arthritis Rheumatol. 2014;66:637–46.
Little CB, Smith MM. Animal models of osteoarthritis. Curr Rheumatol Rep. 2008;4:175–82.
Kamekura S, Hoshi K, Shimoaka T, Chung U, Chikuda H, Yamada T, et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthritis Cartilage. 2005;13:632–41.
McDougall JJ, Schuelert N, Bowyer J. Cathepsin K inhibition reduces CTXII levels and joint pain in the guinea pig model of spontaneous osteoarthritis. Osteoarthritis Cartilage. 2010;18:1355–7.
Taniguchi S, Ryu J, Seki M, Sumino T, Tokuhashi Y, Esumi M. Long-term oral administration of glucosamine or chondroitin sulfate reduces destruction of cartilage and up-regulation of MMP-3 mRNA in a model of spontaneous osteoarthritis in Hartley guinea pigs. J Orthop Res. 2012;30:673–8.
Horcajada MN, Sanchez C, Membrez Scalfo F, Drion P, Comblain F, Taralla S, et al. Oleuropein or rutin consumption decreases the spontaneous development of osteoarthritis in the Hartley guinea pig. Osteoarthritis Cartilage. 2015;23:94–102.
Chiusaroli R, Visentini M, Galimberti C, Casseler C, Mennuni L, Covaceuszach S, et al. Targeting of ADAMTS5’s ancillary domain with the recombinant mAb CRB0017 ameliorates disease progression in a spontaneous murine model of osteoarthritis. Osteoarthritis Cartilage. 2013;21:1807–10.
Quasnichka HL, Anderson-MacKenzie JM, Bailey AJ. Subchondral bone and ligament changes precede cartilage degradation in guinea pig osteoarthritis. Biorheology. 2006;43:389–97.
Muraoka T, Hagino H, Okano T, Enokida M, Teshima R. Role of subchondral bone in osteoarthritis development: a comparative study of two strains of guinea pigs with and without spontaneously occurring osteoarthritis. Arthritis Rheum. 2007;56:3366–74.
Thomsen JS, Straarup TS, Danielsen CC, Oxlund H, Bruel A. Relationship between articular cartilage damage and subchondral bone properties and meniscal ossification in the Dunkin Hartley guinea pig model of osteoarthritis. Scand J Rheumatol. 2011;40:391–9.
Wang T, Wen CY, Yan CH, Lu WW, Chiu KY. Spatial and temporal changes of subchondral bone proceed to microscopic articular cartilage degeneration in guinea pigs with spontaneous osteoarthritis. Osteoarthritis Cartilage. 2013;21:574–81.
Poulet B, Ulici V, Stone TC, Pead M, Gburcik V, Constantinou E, et al. Time-series transcriptional profiling yields new perspectives on susceptibility to murine osteoarthritis. Arthritis Rheum. 2012;64:3256–66.
Uchida K, Naruse K, Satoh M, Onuma K, Ueno M, Takano S, et al. Increase of circulating CD11b(+)Gr1(+) cells and recruitment into the synovium in osteoarthritic mice with hyperlipidemia. Exp Anim. 2013;62:255–65.
ter Huurne M, Schelbergen R, Blattes R, Blom A, de Munter W, Grevers LC, et al. Antiinflammatory and chondroprotective effects of intraarticular injection of adipose-derived stem cells in experimental osteoarthritis. Arthritis Rheum. 2012;64:3604–13.
Yano F, Hojo H, Ohba S, Fukai A, Hosaka Y, Ikeda T, et al. A novel disease-modifying osteoarthritis drug candidate targeting Runx1. Ann Rheum Dis. 2013;72:748–53.
Renkiewicz R, Qiu L, Lesch C, Sun X, Devalaraja R, Cody T, et al. Broad-spectrum matrix metalloproteinase inhibitor marimastat-induced musculoskeletal side effects in rats. Arthritis Rheum. 2003;48:1742–9.
Mazurek SG, Li J, Nabozny GH, Reinhart GA, Muthukumarana AC, Harrison PC, et al. Functional biomarkers of musculoskeletal syndrome (MSS) for early in vivo screening of selective MMP-13 inhibitors. J Pharmacol Toxicol Methods. 2011;64:89–96.
Seidel MF, Lane NE. Control of arthritis pain with anti-nerve-growth factor: risk and benefit. Curr Rheumatol Rep. 2012;14:583–8.
Kimmelman J, Mogil JS, Dirnagl U. Distinguishing between exploratory and confirmatory preclinical research will improve translation. PLoS Biol. 2014;12, e1001863.
Chappell AS, Desaiah D, Liu-Seifert H, Zhang S, Skljarevski V, Belenkov Y, et al. A double-blind, randomized, placebo-controlled study of the efficacy and safety of duloxetine for the treatment of chronic pain due to osteoarthritis of the knee. Pain Pract. 2011;11:33–41.
Chandran P, Pai M, Blomme EA, Hsieh GC, Decker MW, Honore P. Pharmacological modulation of movement-evoked pain in a rat model of osteoarthritis. Eur J Pharmacol. 2009;613:39–45.
Ishikawa G, Nagakura Y, Takeshita N, Shimizu Y. Efficacy of drugs with different mechanisms of action in relieving spontaneous pain at rest and during movement in a rat model of osteoarthritis. Eur J Pharmacol. 2014;738:111–7.
Schuelert N, Johnson MP, Oskins JL, Jassal K, Chambers MG, McDougall JJ. Local application of the endocannabinoid hydrolysis inhibitor URB597 reduces nociception in spontaneous and chemically induced models of osteoarthritis. Pain. 2011;152:975–81.
Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837–46.
Suokas AK, Sagar DR, Mapp PI, Chapman V, Walsh DA. Design, study quality and evidence of analgesic efficacy in studies of drugs in models of OA pain: a systematic review and a meta-analysis. Osteoarthritis Cartilage. 2014;22:1207–23.
McGonigle P, Ruggeri B. Animal models of human disease: challenges in enabling translation. Biochem Pharmacol. 2014;87:162–71.
Howells DW, Sena ES, Macleod MR. Bringing rigour to translational medicine. Nat Rev Neurol. 2014;10:37–43.
Rice AS, Cimino-Brown D, Eisenach JC, Kontinen VK, Lacroix-Fralish ML, Machin I, et al. Animal models and the prediction of efficacy in clinical trials of analgesic drugs: a critical appraisal and call for uniform reporting standards. Pain. 2008;139:243–7.
Panahifar A, Jaremko JL, Tessier AG, Lambert RG, Maksymowych WP, Fallone BG, et al. Development and reliability of a multi-modality scoring system for evaluation of disease progression in pre-clinical models of osteoarthritis: celecoxib may possess disease-modifying properties. Osteoarthritis Cartilage. 2014;22:1639–50.
McGlothlin AE, Lewis RJ. Minimal clinically important difference: defining what really matters to patients. JAMA. 2014;312:1342–3.
Naci H, Ioannidis JP. How good is ‘evidence’ from clinical studies of drug effects and why might such evidence fail in the prediction of the clinical utility of drugs? Annu Rev Pharmacol Toxicol. 2015;55:169–89.
Whiteside GT, Pomonis JD, Kennedy JD. An industry perspective on the role and utility of animal models of pain in drug discovery. Neurosci Lett. 2013;557:65–72.
Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. Osteoarthritis Cartilage. 2012;20:256–60.
Sena ES, Currie GL, McCann SK, Macleod MR, Howells DW. Systematic reviews and meta-analysis of preclinical studies: why perform them and how to appraise them critically. J Cereb Blood Flow Metab. 2014;34:737–42.
One Health Initiative. http://www.onehealthinitiative.com/index.php.
Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40–51.
Lexchin J. Post-market safety warnings for drugs approved in Canada under the Notice of Compliance with conditions policy. Br J Clin Pharmacol. 2015;79:847–59.
Lexchin J. How safe are new drugs? Market withdrawal of drugs approved in Canada between 1990 and 2009. Open Med. 2014;28:e14–9.
Karsdal MA, Byrjalsen I, Alexandersen P, Bihlet A, Andersen JR, Riis BJ, et al. Treatment of symptomatic knee osteoarthritis with oral salmon calcitonin: results from two phase 3 trials. Osteoarthritis Cartilage. 2015;23:532–43.
Nielsen RH, Bay-Jensen AC, Byrjalsen I, Karsdal MA. Oral salmon calcitonin reduces cartilage and bone pathology in an osteoarthritis rat model with increased subchondral bone turnover. Osteoarthritis Cartilage. 2011;19:466–73.
El Hajjaji H, Williams JM, Devogelaer JP, Lenz ME, Thonar EJ, Manicourt DH. Treatment with calcitonin prevents the net loss of collagen, hyaluronan and proteoglycan aggregates from cartilage in the early stages of canine experimental osteoarthritis. Osteoarthritis Cartilage. 2004;12:904–11.
Sondergaard BC, Catala-Lehnen P, Huebner AK, Bay-Jensen AC, Schinke T, Henriksen K, et al. Mice over-expressing salmon calcitonin have strongly attenuated osteoarthritic histopathological changes after destabilization of the medial meniscus. Osteoarthritis Cartilage. 2012;20:136–43.
Lohmander LS, Hellot S, Dreher D, Krantz EF, Kruger DS, Guermazi A, et al. Intraarticular sprifermin (recombinant human fibroblast growth factor 18) in knee osteoarthritis: a randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2014;66:1820–31.
Moore EE, Bendele AM, Thompson DL, Littau A, Waggie KS, Reardon B, et al. Fibroblast growth factor-18 stimulates chondrogenesis and cartilage repair in a rat model of injury-induced osteoarthritis. Osteoarthritis Cartilage. 2005;13:623–31.
Reginster JY, Badurski J, Bellamy N, Bensen W, Chapurlat R, Chevalier X, et al. Efficacy and safety of strontium ranelate in the treatment of knee osteoarthritis: results of a double-blind, randomised placebo-controlled trial. Ann Rheum Dis. 2013;72:179–86.
Pelletier JP, Kapoor M, Fahmi H, Lajeunesse D, Blesius A, Maillet J, et al. Strontium ranelate reduces the progression of experimental dog osteoarthritis by inhibiting the expression of key proteases in cartilage and of IL-1β in the synovium. Ann Rheum Dis. 2013;72:250–7.
Yu DG, Ding HF, Mao YQ, Liu M, Yu B, Zhao X, et al. Strontium ranelate reduces cartilage degeneration and subchondral bone remodeling in rat osteoarthritis model. Acta Pharmacol Sin. 2013;34:393–402.
le Graverand MP H, Clemmer RS, Redifer P, Brunell RM, Hayes CW, Brandt KD, et al. A 2-year randomised, double-blind, placebo-controlled, multicentre study of oral selective iNOS inhibitor, cindunistat (SD-6010), in patients with symptomatic osteoarthritis of the knee. Ann Rheum Dis. 2013;72:187–95.
Pelletier JP, Jovanovic D, Fernandes JC, Manning P, Connor JR, Currie MG, et al. Reduced progression of experimental osteoarthritis in vivo by selective inhibition of inducible nitric oxide synthase. Arthritis Rheum. 1998;41:1275–86.
Pelletier JP, Jovanovic D, Fernandes JC, Manning P, Connor JR, Currie MG, et al. Reduction in the structural changes of experimental osteoarthritis by a nitric oxide inhibitor. Osteoarthritis Cartilage. 1999;7:416–8.
van den Berg WB, van de Loo F, Joosten LA, Arntz OJ. Animal models of arthritis in NOS2-deficient mice. Osteoarthritis Cartilage. 1999;7:413–5.
Connor J, Rogers K, Sunyer T, Hellio Le Graverand-Gastineau MP, Manning PT. Efficacy of the selective inducible nitric oxide synthase inhibitor sd-6010 in nonclinical inflammatory, neuropathic, and osteoarthritis pain. Osteoarthritis Cartilage. 2012;20:S64–5.
McAlindon T, LaValley M, Schneider E, Nuite M, Lee JY, Price LL, et al. Effect of vitamin D supplementation on progression of knee pain and cartilage volume loss in patients with symptomatic osteoarthritis: a randomized controlled trial. JAMA. 2013;309:155–62.
Castillo EC, Hernandez-Cueto MA, Vega-Lopez MA, Lavalle C, Kouri JB, Ortiz-Navarrete V. Effects of vitamin D supplementation during the induction and progression of osteoarthritis in a rat model. Evid Based Complement Alternat Med. 2012;2012:156563.
Jefferies D, Farquharson C, Thomson J, Smith W, Seawright E, McCormack H, et al. Differences in metabolic parameters and gene expression related to osteochondrosis/osteoarthrosis in pigs fed 25-hydroxyvitamin D3. Vet Res. 2002;33:383–96.
Raynauld JP, Martel-Pelletier J, Bias P, Laufer S, Haraoui B, Choquette D, et al. Protective effects of licofelone, a 5-lipoxygenase and cyclo-oxygenase inhibitor, versus naproxen on cartilage loss in knee osteoarthritis: a first multicentre clinical trial using quantitative MRI. Ann Rheum Dis. 2009;68:938–47.
Moreau M, Boileau C, Martel-Pelletier J, Brunet J, Laufer S, Pelletier JP. Licofelone reduces progression of structural changes in a canine model of osteoarthritis under curative conditions: effect on protease expression and activity. J Rheumatol. 2006;33:1176–83.
Thomsen JS, Straarup TS, Danielsen CC, Oxlund H, Brüel A. No effect of risedronate on articular cartilage damage in the Dunkin Hartley guinea pig model of osteoarthritis. Scand J Rheumatol. 2013;42:408–16.
Permuy M, Guede D, López-Peña M, Muñoz F, González-Cantalapiedra A, Caeiro JR. Effects of glucosamine and risedronate alone or in combination in an experimental rabbit model of osteoarthritis. BMC Vet Res. 2014;10:97.
Spector TD, Conaghan PG, Buckland-Wright JC, Garnero P, Cline GA, Beary JF, et al. Effect of risedronate on joint structure and symptoms of knee osteoarthritis: results of the BRISK randomized, controlled trial [ISRCTN01928173]. Arthritis Res Ther. 2005;7:R625–33.
Bingham 3rd CO, Buckland-Wright JC, Garnero P, Cohen SB, Dougados M, Adami S, et al. Risedronate decreases biochemical markers of cartilage degradation but does not decrease symptoms or slow radiographic progression in patients with medial compartment osteoarthritis of the knee: results of the two-year multinational knee osteoarthritis structural arthritis study. Arthritis Rheum. 2006;54:3494–507.
Buckland-Wright JC, Messent EA, Bingham 3rd CO, Ward RJ, Tonkin C. A 2 yr longitudinal radiographic study examining the effect of a bisphosphonate (risedronate) upon subchondral bone loss in osteoarthritic knee patients. Rheumatology (Oxford). 2007;46:257–64.
Krzeski P, Buckland-Wright C, Bálint G, Cline GA, Stoner K, Lyon R, et al. Development of musculoskeletal toxicity without clear benefit after administration of PG-116800, a matrix metalloproteinase inhibitor, to patients with knee osteoarthritis: a randomized, 12-month, double-blind, placebo-controlled study. Arthritis Res Ther. 2007;9:R109.
Janusz MJ, Hookfin EB, Heitmeyer SA, Woessner JF, Freemont AJ, Hoyland JA, et al. Moderation of iodoacetate-induced experimental osteoarthritis in rats by matrix metalloproteinase inhibitors. Osteoarthritis Cartilage. 2001;9:751–60.
Brewster M, Lewis EJ, Wilson KL, Greenham AK, Bottomley KM. Ro 32–3555, an orally active collagenase selective inhibitor, prevents structural damage in the STR/ORT mouse model of osteoarthritis. Arthritis Rheum. 1998;41:1639–44.
Brandt KD, Mazzuca SA, Katz BP, Lane KA, Buckwalter KA, Yocum DE, et al. Effects of doxycycline on progression of osteoarthritis: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2005;52:2015–25.
Yu LP, Smith Jr GN, Brandt KD, Myers SL, O’Connor BL, Brandt DA. Reduction of the severity of canine osteoarthritis by prophylactic treatment with oral doxycycline. Arthritis Rheum. 1992;35:1150–9.
Bowyer J, Heapy CG, Flannelly JK, Waterton JC, Maciewicz RA. Evaluation of a magnetic resonance biomarker of osteoarthritis disease progression: doxycycline slows tibial cartilage loss in the Dunkin Hartley guinea pig. Int J Exp Pathol. 2009;90:174–81.
Silva JA, Fanning PJ, O’Connell SL, Mason-Savas A, Walcott M, Hays P, et al. Effects of disease-modifying osteoarthritis drugs in an in-vivo animal model [abstract]. Las Vegas, NV: Orthopedic Research Society Annual Meeting; 2009. Poster Presentation.
Acknowledgements
AMM acknowledges funding by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR064251 and R01AR060364). CBL acknowledges funding from the National Health and Medical Research Council of Australia (APP384414, APP1045890, and APP1063133), Arthritis Australia and The Hillcrest Foundation through Perpetual Philanthropies. The funding sources had no role in this publication.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
CBL participated in the development of the OARSI recommendations for histological evaluation of preclinical models of OA, as a member of the mouse committee and chair of the sheep and goat committee; he is an author on the publications of recommendations arising from these committees. AMM had no role in OARSI recommendations. The authors have no other competing interests to declare.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Malfait, AM., Little, C.B. On the predictive utility of animal models of osteoarthritis. Arthritis Res Ther 17, 225 (2015). https://doi.org/10.1186/s13075-015-0747-6
Published:
DOI: https://doi.org/10.1186/s13075-015-0747-6