
Abstract
Background
Germline variants affecting the proofreading activity of polymerases epsilon and delta cause a hereditary cancer and adenomatous polyposis syndrome characterized by tumors with a high mutational burden and a specific mutational spectrum. In addition to the implementation of multiple pieces of evidence for the classification of gene variants, POLE and POLD1 variant classification is particularly challenging given that non-disruptive variants affecting the proofreading activity of the corresponding polymerase are the ones associated with cancer. In response to an evident need in the field, we have developed gene-specific variant classification recommendations, based on the ACMG/AMP (American College of Medical Genetics and Genomics/Association for Molecular Pathology) criteria, for the assessment of non-disruptive variants located in the sequence coding for the exonuclease domain of the polymerases.
Methods
A training set of 23 variants considered pathogenic or benign was used to define the usability and strength of the ACMG/AMP criteria. Population frequencies, computational predictions, co-segregation data, phenotypic and tumor data, and functional results, among other features, were considered.
Results
Gene-specific variant classification recommendations for non-disruptive variants located in the exonuclease domain of POLE and POLD1 were defined. The resulting recommendations were applied to 128 exonuclease domain variants reported in the literature and/or public databases. A total of 17 variants were classified as pathogenic or likely pathogenic, and 17 as benign or likely benign.
Conclusions
Our recommendations, with room for improvement in the coming years as more information become available on carrier families, tumor molecular characteristics and functional assays, are intended to serve the clinical and scientific communities and help improve diagnostic performance, avoiding variant misclassifications.
Similar content being viewed by others


Background
The major function of polymerases is to replicate the genome, which is performed by polymerases, α, ε and δ in eucaryotes. Unlike α, polymerases ε (Polε) and δ (Polδ) contain an active 3'-5' exonuclease domain (ED) which proofreads newly synthesized DNA for replication errors. Polε and Polδ are comprised of four subunits in humans, the largest of which contains the catalytic polymerase and exonuclease domains and is encoded by the genes POLE and POLD1 respectively [1, 2].
Heterozygous germline pathogenic variants affecting the proofreading activity of Polε and Polδ cause increased risk to develop adenomatous polyposis and colorectal cancer (CRC), as well as endometrial, ovarian, breast, brain and upper gastrointestinal cancers, among other tumors [3,4,5,6]. This autosomal dominant cancer syndrome is called polymerase proofreading-associated polyposis (PPAP; MIM# 615083, 612591). The associated clinical features are usually developed in the adult age, except for rare aggressive cases that present with a constitutional mismatch repair deficiency (CMMRD)-like phenotype in childhood or adolescence [7,8,9]. Somatic POLE ED pathogenic variants occur in 7–15% of endometrial cancers [10,11,12,13], 0.5–8% of colorectal tumors [14,15,16,17], and more rarely in brain tumors (gliomas), extracolonic gastrointestinal cancers, and other tumor types. Somatic POLD1 ED mutations are extremely rare.
As mentioned above, the ED determines the proofreading function of Polε and Polδ, which is essential for replication fidelity. Therefore, Polε and Polδ exonuclease disruption by pathogenic variants, either germline or somatic, leads to the accumulation of thousands of variants in the tumors (> 10 somatic variants per Mb (mut/Mb), and often, > 100) [12, 18,19,20]. Moreover, they present a characteristic variant spectrum, enriched in C > A transversions in the context of TCT, and C > T transitions in the context TCG [15, 21], which corresponds to tumor mutational signatures SBS10a, SBS10b, and SBS28 [22] for Polε proofreading defects, and SBS10d and SBS10c (identified in unaffected tissues) for Polδ proofreading deficiency, of the Catalogue Of Somatic Mutations In Cancer (COSMIC) (https://cancer.sanger.ac.uk/signatures/sbs/Mutational Signatures v3.2) [21, 23]. Occasionally, polymerase proofreading deficiency co-occurs with DNA mismatch repair (MMR) deficiency (dMMR) in the tumors. In that scenario, the tumor mutational signatures present are SBS14 (Polε proofreading deficiency + dMMR), and SBS20 (Polδ proofreading deficiency + dMMR) [15, 21, 24]. Hereditary and sporadic proofreading-deficient tumors, due to the strong immunogenicity elicited by the high mutation rate (strong neoantigen expression), show favorable prognosis and clinical benefit from immune checkpoint blockade [25,26,27,28,29,30].
Constitutional loss-of-function variants and variants located outside the exonuclease domain of POLE and POLD1 do not cause the cancer predisposition syndrome PPAP; however, they may predispose to autosomal recessive or dominant congenital disorders. FILS syndrome (MIM# 615139), a very rare recessive Mendelian disorder characterized by facial dysmorphism, immunodeficiency, livedo, short stature, and variable skin manifestations, is caused by POLE pathogenic variants located outside the exonuclease domain and/or disrupting the encoded protein [31,32,33]. Biallelic POLE pathogenic variants have also been associated with another rare Mendelian syndrome, IMAGE-I (MIM# 618336), characterized by intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, genital anomalies, immunodeficiency, and diffuse large B-cell lymphoma [34, 35]. None of the patients tested with the congenital disorders herein mentioned show complete lack of POLE expression, suggesting that this would be lethal to the embryo. Constitutional heterozygous POLD1 pathogenic variants that impair the polymerase (replicative) activity of Polδ (dominant negative effect), cause an autosomal dominant progeroid syndrome called MDPL (MIM# 615381), characterized by mandibular hypoplasia, deafness, progeroid features, and lipodystrophy [36, 37].
Accurate POLE and POLD1 ED variant classification, which is the focus of this article, is of utmost importance due to the consequences for the correct clinical management of ED variant heterozygotes and their families, impacting clinical surveillance based on specific cancer risks, as well decision making in oncology, based on the predictive value of ED mutations for prognosis and response to immunotherapy.
The American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) developed generic variant classification guidelines that include criteria with varying levels of strength for and against pathogenicity, based on evidence gathered from multiple sources, including population data, computational and predictive data, phenotype/family history information, and functional data [38]. These recommendations allow the classification of variants into five categories: pathogenic (P), likely pathogenic (LP), variant of uncertain significance (VUS), likely benign (LB), and benign (B). Despite their value, these guidelines are generic for any Mendelian disease-causative gene and do not take into consideration gene and/or syndrome-specific particularities. Here we present specific recommendations to apply the ACMG/AMP guidelines for the classification of variants located in the ED of POLE and POLD1, -where the variants associated with cancer predisposition are found-, and the scientific rationale applied for their definition. We also present the curated classification of 128 ED missense variants after applying the recommendations that we propose. These recommendations have been developed to be made available to the scientific and clinical communities until official recommendations from the InSiGHT/ClinGen Hereditary Colorectal Cancer and Polyposis Variant Curation Expert Panel (https://clinicalgenome.org/affiliation/50099/) are published.
Methods
ACMG/AMP variant classification guidelines
Assessment of each ACMG/AMP rule code and evaluation of their utility for the classification of POLE and POLD1 ED variants was performed. Previously published specifications developed by ClinGen Variant Curation Expert Panels (VCEP) were taken into consideration (https://cspec.genome.network/cspec/ui/svi/summary), in particular those defined for cancer predisposition genes [39,40,41,42,43].
ACMG/AMP rules were divided into four types of evidence: (i) population data; (ii) variant nature, variant location and computational predictive data; (iii) segregation and phenotypic data, including tumor mutational data; and (iv) functional data. As per ACMG/AMP guidelines, evidence in each category have varying levels of strength: very strong (PVS), strong (PS), moderate (PM), and supporting (PP) for pathogenic criteria; and stand-alone (BA), strong (BS), and supporting (BP) for benign criteria. All 28 original criteria were evaluated for their application to POLE and POLD1 ED variant classification. Rule codes that were irrelevant to POLE and POLD1 or the syndrome, or for which limited data was available, or that included redundant information with another criterion, or that had been removed by the ClinGen Sequence Variant Interpretation (SVI) working group [44], were excluded. Criteria modifications included gene- or disease-specific modifications, strength-level adjustments, general recommendations, and certain criteria deemed not applicable.
For the final variant classification, recommendations provided in the manuscript have been followed, and the standard ACMG/AMP combination rules to define pathogenic, likely pathogenic, likely benign and benign variants were applied (Additional file 1: Table S1) [38].
PM and LV performed the classification of the 128 variants in parallel, without access to the other researcher’s classification. Complete concordance between the two classifications was reached for all variants.
Variant nomenclature
Variant nomenclature follows HGVS recommendations (v.20.05), with nucleotide 1 corresponding to the A of the ATG translation initiation codon. All variants were annotated according to RefSeq IDs LRG_789; NM_006231.4 (POLE) and LRG_785; NM_001256849.1 (≈NM_002691.4) (POLD1). POLE ED includes amino acids 268–471, and POLD1 ED, amino acids 304–533 (based on NCBI: “region_name DNA_polB_epsilon_exo and DNA_polB_delta_exo").
Population data
The Genome Aggregation Database [45] (gnomAD, non-cancer dataset; https://gnomad.broadinstitute.org/) was used as source of publicly available population control data (as of today, gnomAD non-cancer v.2.1.1, as it is the largest available dataset: 134,187 individuals, 50,913 of whom are non-Finnish Europeans), ignoring the frequencies observed in populations with high potential for founder effects, such as Ashkenazi Jewish or Finnish sub-populations, and the unclear ancestry “Population: other”.
In silico predictions
In silico predictions of pathogenicity were performed with SIFT [46], PolyPhen-2 [47], CADD [48, 49] and the metapredictor REVEL [50], which combines pathogenicity predictions and conservation information obtained from 18 individual scores. Scores were obtained from the Variant Effect Predictor (VEP) web tool [51]. The BLOSUM62 matrix was used to score pairs of aligned residues [52].
3D modeling: DNA binding cleft
3D models based on the crystallographic structure of the homologous yeast proteins Pol2 (PBD ID: 4m8o) and Pol3 (PDB ID: 3iay, chain A), with a single-stranded DNA (ssDNA) from the aligned bacteriophage T4 polymerase complex (PDB ID: 1noy) located in the proper position for exonuclease proofreading, were used to evaluate the location of the affected amino acids in the 3D structure of POLE and POLD1. Structural superpositions, refinement, and manual adjustments to the 3D models of human POLE and POLD1 in complex with ssDNA were performed with COOT [53].
The DNA binding cavity was defined according to CASTp (http://sts.bioe.uic.edu/castp/calculation.html). Interatomic distances were calculated with ContPro (http://procarb.org/contpro/). Direct contact of an amino acid with the ssDNA (positioned for proofreading) was defined when any atom of the amino acid is accessible to the cavity where the DNA binds and at less than 6 Å from the ssDNA. Indirect contact is defined when any atom of the amino acid is accessible to the cavity but at ≥ 6 Å from the ssDNA. No contact was considered when the atoms of an amino acid are, in the 3D models, at > 6 Å from the ssDNA (ContPro) and not accessible to the DNA-binding cavity (CASTp). Additional file 2: Table S2 indicates the predictions for each residue in the ED of POLE and POLD1.
Tumor mutational burden and signatures
Exome or genome sequencing data processing for the calculation of tumor mutational burden and COSMIC mutational signatures was performed as previously specified [5]. Total mutation burden was estimated by considering single nucleotide variants (SNV) from exonic regions and with a variant allele frequency higher than 10%. The number of mutations per megabase (mut/Mb) was calculated as the total mutational burden divided by the genomic exome length (32.95 Mb). The contribution of tumor mutational signatures was calculated with FitMS through the Signal web application (https://signal.mutationalsignatures.com/), not selecting tissue-specific signatures (access date: November 2022). In Signal, COSMIC v.3 signatures were considered when evaluating a POLE variant, since they include, among others, SBS10a, SBS10b, SBS28, SBS14 and SBS20. For POLD1 variants, Cancer Reference Signatures (CRS) were considered, which include, among others, SBS10a, SBS10d, SBS14 and SBS20.
Exome sequencing data (BAM files) or targeted sequencing data (≥ 100 genes analyzed) from tumors harboring the POLE and POLD1 ED variants identified in inherited cases were obtained from TCGA (accessed May 2021) and/or COSMIC v.94 (accessed May 2021).
Analysis of the specificity of mutational signatures associated with proofreading deficiency
Two subgroups of samples, obtained from TCGA, were considered based on the ED mutational status: 68 proofreading deficient TCGA tumor samples, and 70 without mutations in the exonuclease domain of POLE or POLD1, randomly selected (gastric, colorectal, and endometrial cancers). Sequencing data processing was performed as described above.
The clustering of the samples was performed based on the percentages of contribution of polymerase proofreading deficient-associated signatures: SBS10a, SBS10b, SBS10d, SBS28, SBS14 and SBS20 [23, 54]. The distances among samples were computed via R function dist, with Euclidean distance. Subsequently, hclust function was used to generate the clustering based on the distances calculated with the Ward-D2 linkage method. For visualization purposes, data was plotted in a heatmap using the ComplexHeatmap package.
Tumor MMR deficiency
Tumor MMR status was obtained from the data reported in TCGA, whenever available. MMR deficiency (microsatellite instability, MSI) was established in TCGA based on the estimations retrieved from MANTIS [55] (cutoff: 0.4) and MSIsensor [56] (cutoff: 3.5). When the MSI Bethesda panel [57, 58] results were available, this status was prioritized. Only one sample showed discordant results between the MSI panel and the TCGA determinations.
Results and discussion
Training set of pathogenic and benign ED variants to define the usability and strength of the ACMG/AMP criteria
Literature and database searches were performed using PubMed, Mastermind, gnomAD, and ClinVar (accessed February 2021), and variants with strong evidence of pathogenicity or benignity/neutrality were considered to define a training set of variants used to help in the definition of the specifications of the ACMG/AMP guidelines. Variant selection was based on a simplistic model where strong pieces of evidence in favor of pathogenicity or neutrality were considered (Table 1): 17 variants (13 in POLE and 4 in POLD1) were considered pathogenic, and 5 (3 in POLE and 2 in POLD1) benign (Table 2; Additional file 3: Table S3).
POLE and POLD1 ED-specific variant curation criteria
POLE and POLD1 specifications to the ACMG/AMP criteria are shown in Table 3. Of the 28 original criteria, 8 were excluded (PVS1, PM3, PM4, PP2, PP5, BP1, BP3, and BP6). Rules were modified by detailing the content and/or changing the strength level of the original recommendations.
Population data
BA1 and BS1 are criteria against pathogenicity based on the frequency of the variant in general population. To calculate the allele frequency threshold, the prevalence and penetrance of germline pathogenic variants in POLE and POLD1 ED should be considered. Available data indicate that PPAP is a rare syndrome with very low population prevalence: Only one of 17 POLE ED pathogenic variants considered (Table 2) was detected in gnomAD non-cancer individuals (POLD1:c.946G>C; p.Asp316His: 1 in ~ 230,000 alleles). Although accurate unbiased penetrance estimates are still unavailable, available data [5, 6] suggests that the penetrance for POLE and POLD1 ED pathogenic variants might be close to other autosomal dominant cancer syndromes caused by DNA repair defects, such as Lynch syndrome (MLH1, MSH2), with an estimated average CRC risk of ~ 40%—50% by age 70 [60]. By using the Whiffin/Ware calculator [61] (http://cardiodb.org/allelefrequencyapp/), the inferred allele frequency threshold (AFT) (95% CI) obtained for BA1, with allele heterogeneity set at 1, was 0.002%, and for BS1, with allele heterogeneity set at 0.1, 0.0002% (Additional file 1: Supplementary Results). Due to the scarcity of available data, the rough estimation of the syndrome penetrance, and the fact that the number of pathogenic variants is likely underestimated (missense variants are harder to classify than loss-of-function variants), we recommend applying higher AFTs: BA1 to variants with a population allele frequency ≥ 0.02%, and BS1 to variants with a population allele frequency ≥ 0.002%. Data may be obtained from gnomAD (non-cancer), or from any outbred (non-founder) population groups in that repository (non-Finnish European, African/African American, Latino/admixed American, South Asian, or East Asian). The variant must be present in at least 5 alleles.
PM2 uses absence in controls for autosomal dominant diseases. Based on the incomplete penetrance and/or late disease onset, we recommend using PM2 with a supporting level of strength for variants absent, or present in ≤ 1 in 200,000 alleles (≤ 0.0005%) in gnomAD non-cancer dataset (all individuals) (coverage of variant position > 30X) [59]. Supportive of this threshold is the fact that POLE p.Leu424Val, the most recurrent known pathogenic germline variant, is not present in non-cancer gnomAD individuals (~ 200,000 in gnomAD v.2.1.1 and v.3.1.1).
BS2 uses the presence of the variant in healthy adult individuals when full penetrance is expected at an early age. We specified the code to account for the reduced penetrance and later age of PPAP onset. Also, heterozygotes identified among non-cancer individuals could have polyps that have not been detected or reported. Considering the families with the 17 pathogenic variants listed in Table 2, among the 169 carriers reported (POLE n = 128 and POLD1 n = 41), there are 47 cancer-free individuals. Of them, 12 carriers had no polyp information and/or had not undergone colonoscopy screening. Of the cancer-free carriers with polyp information (35/47), 97% (34/35) had polyps (any number). Of these, detailed information on polyp number was specified for 25 individuals: 60% of them (15/25) had been diagnosed with ≥ 10 polyps (median age at diagnosis: 35; age range: 15–53) (Additional file 4: Table S4). Based on the available data, and on the extremely low prevalence of PPAP-associated recurrent pathogenic variants, we recommend using BS2, with a supporting level of strength, for variants that have been identified in ≥ 5 cancer-free individuals aged > 60. If BA1/BS1 is applied and gnomAD data is used for BS2_supp, apply only BA1/BS1. A strong level of strength may be applied if the variant is identified in ≥ 10 cancer-free and adenoma-free individuals aged > 60. To apply this level of strength, the cancer-free individuals must have been subjected to colonoscopy screening (not applicable for gnomAD individuals).
No biallelic germline ED pathogenic variants have been identified in humans. It has been speculated that those could likely be embryonic lethal [3]. Interestingly, depending on the nature of the pathogenic variant, biallelic mutant mice may be viable [62, 63]. While PoleP286R/P286R mice showed embryonic lethality, homozygotes for other ED pathogenic variants survived into adulthood but developed cancer very early in life. In that same line, PoleP286R/+ mice develop more severe phenotypes than heterozygotes for other ED pathogenic variants, which may even be indistinguishable from wildtype animals, suggesting a more severe effect in humans than in mice [62,63,64]. Pold1 homozygous mutant mice die of cancer at extremely early ages [63]. Based on the mice findings, even in the hypothetical case that biallelic ED-mutated humans were identified (viable), we would expect extremely aggressive tumor phenotypes, probably with very early age of onset. Therefore, we propose to apply BS2 to variants identified in homozygous state in one cancer- and adenoma-free adult individual if his/her homozygosity status has been confirmed by genotyping the parents. BS2_supporting may be applied when two homozygous adult cases are identified without available parental confirmation and/or polyp information (e.g. gnomAD non-cancer dataset).
PS4 is based on the statistically significant higher frequency of the variant in patients compared to controls. We recommend applying the case–control criterion, considering PPAP-associated phenotypes (Table 4), when the resulting p-value is ≤ 0.05 and OR ≥ 2 or the lower 95% CI is ≥ 1.5 [43]. Also, we recommend applying PS4, with supporting level of strength, when a CMMRD-like phenotype [65] in absence of germline biallelic MMR (likely) pathogenic variants or VUSs is identified in one proband, and with moderate level of strength, when the CMMRD-like phenotype is identified in ≥ 2 probands. No other PPAP-associated phenotypes are considered due to their non-specificity. See Table 3 for permitted co-usages.
Variant nature and location, and in silico predictions
Evidence suggests that loss-of-function and outside-ED POLE and POLD1 variants are nonpathogenic for PPAP, and only missense and in-frame indel variants within the ED should be considered as potential cause of PPAP and as predictive biomarkers in oncology [3, 5, 66, 67]. Therefore, PVS1, PM4, BP1, and BP3 are not considered due to their irrelevance to the syndrome and its mechanism of pathogenicity.
The PM1 criterion is given to mutational hotspots and/or critical well-established functional domains without benign variation. We recommend applying PM1 (moderate) for: (i) somatic mutational hotspots (observed in ≥ 10 tumors), which currently include: POLE P286R, S297F, V411L, A456P and S459F (somatic hotspot information obtained from TCGA and COSMIC tumors: Additional file 5: Table S5); and (ii) variants affecting the exonuclease catalytic sites POLE D275 and E277, and POLD1 D316 and E318 [4], when the resulting amino acid shows a negative BLOSUM62 when compared to the wildtype residue. Available data indicate that variants affecting the binding of the exonuclease with the DNA, and/or located within the Exo motifs are likely to be pathogenic [68]. In fact, 11 of the 13 non-catalytic pathogenic variants, and none of the benign variants, affect residues of Exo motifs and/or are in contact (distance < 6 Å) with the DNA when the polymerase is in proofreading position (Table 2). We recommend applying PM1 with a supporting level of strength to any variant fulfilling either one of these two conditions, when PM1 (moderate) has not been applied. ED amino acids at < 6 Å from the DNA are listed in Additional file 4: Table S2. See Table 3 for permitted co-usages of PM1 with other criteria.
PP3 and BP4 are related to in silico pathogenicity predictions. Following recent ClinGen indications [69], PP3 should be applied for variants with REVEL scores ≥ 0.644, which occurs for 11 of the 17 pathogenic variants and none of the benign variants, and BP4 for silent (synonymous) variants or intronic variants without predicted splicing effect, and for missense ED variants with a REVEL score ≤ 0.290. Sensitivity/specificity analysis should be performed to set gene-specific cutoff values for POLE and POLD1 ED variants, when enough pathogenic and benign variants are identified to be used for the analysis. Predictions of loss of function or splicing impact (unless it causes an in-frame splicing defect that affects the ED) should not be considered as supporting evidence of pathogenicity or benignity.
BP7 is applied for any synonymous or intronic variant at or beyond +7/-21 for which splicing prediction algorithms predict no impact to the splice consensus sequence nor creation of a new splice site, regardless of nucleotide conservation.
PS1 considers any missense nucleotide change that translates into an amino acid change that has been previously established as (likely) pathogenic with a different nucleotide change (i.e., different nucleotide variant, same amino acid change). Strong level of evidence is recommended for pathogenic variants, and moderate, for likely pathogenic variants. Likewise, PM5 relates to a missense variant at a residue where a different pathogenic missense variant caused a change to a different amino acid. In this case, we recommend using PM5 only when the resulting amino acid shows equal or lower BLOSUM62 score (i.e., equally or more damaging) than the previously classified pathogenic (PM5) or likely pathogenic (PM5_supporting) amino acid [70].
Segregation and phenotypic data
PP1 original criterion uses cosegregation of the variant with the disease in multiple family members affected with the associated phenotype as evidence for pathogenicity. The main PPAP-associated tumor types are colorectal, endometrial, ovarian, breast, brain, and upper gastrointestinal cancers, as well as polyposis (> 10 adenomas), all with prevalence values > 10% among cancer-affected carriers (Table 4; Additional file 4: Table S4). Nevertheless, due to the broad phenotypic spectrum and the relative high population frequency of most PPAP-associated tumor types, which may lead to phenocopies, we recommend considering only the three most prevalent PPAP-associated phenotypes, i.e. adenomatous polyposis (> 10 adenomas), CRC and endometrial cancer, unless tumor mutational data indicate that other tumor types are hyper/ultra-mutated and harbor the gene-specific mutational signature(s).
Based on the gradations considered by ClinGen variant curation expert panels [39, 40, 42, 71], we recommend the system that considers the number of meiosis across one or more families [72]: strong level of evidence when co-segregation is observed in ≥ 7 meiosis in ≥ 2 families; moderate level of evidence when cosegregation is observed in ≥ 5 meioses in ≥ 1 family; and supporting level when cosegregation is observed in 3–4 meioses in ≥ 1 family. The meiosis counting-based system may not be optimal for cosegregation analyses in cancer-related genes [72], particularly when there are variable ages at onset, high probability of phenocopies, and/or incomplete penetrance, as happens for PPAP. When more accurate data on the syndrome are available, this rule code will likely implement a Bayes factor-based approach, which measures the likelihood that cosegregation patterns represent a gene-disease penetrance model [72].
BS4 is used when there is lack of segregation. Due to existence of de novo cases, the wide tumor spectrum observed in PPAP, the expected incomplete penetrance and the -often- late onset of cancer, we recommend considering only non-carrier family members affected with > 10 adenomas, or CRC, or endometrial, or any other hyper- or ultra-mutated tumor (≥ 10 mut/Mb) with the mutational signature(s) associated with the corresponding polymerase proofreading deficiency. BS4 should be applied, with a supporting level of strength, when there is ≥ 1 family with ≥ 2 meiosis with a genotype-negative phenotype-positive situation, in absence of pathogenic or likely pathogenic variants or variants of unknown significance in other known hereditary cancer or polyposis genes that could explain the phenotype. As for PP1, this criterion will likely implement a Bayes factor–based approach [72] in the future.
PS2 and PM6 contemplate the presence of de novo variants. We recommend applying the point-based criteria based on phenotypes indicated in Table 3 to determine the levels of strength. Points are additive per each de novo case.
We recommend applying BP2 when the variant is observed in trans with another (likely) pathogenic ED variant in the same gene in a tumor-free (cancer- and adenoma-free) adult (see comment in “Population data” section; BS2 criterion) or when the variant is identified ≥ 3 times with additional ED (likely) pathogenic variants in the same gene with unknown phase. The other observed ED variant must have been classified as (likely) pathogenic using the herein defined recommendations.
Tumor data: mutational burden and signatures
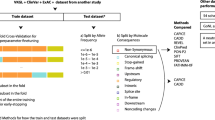
To evaluate the specificity of the proofreading-associated mutational signatures, we analyzed 134 tumor samples (different tumor types) including: i) 50 MMR proficient (pMMR) and 20 dMMR TCGA tumors without ED variants, and ii) 50 pMMR, 12 dMMR tumors and 2 tumors without available MMR status information with somatic pathogenic ED variants (62 tumors with POLE and 2 with POLD1 ED mutations) that represent 9 of the 17 pathogenic variants listed in Table 2 (data source: 59 TCGA tumors and 5 COSMIC tumors with available exome sequencing data). The results are represented as a Heatmap in Fig. 1 (details in Additional file 6: Table S6). SBS10a, SBS10b, SBS28 and SBS14 were highly specific of Polε proofreading deficiency; no trace of those signatures was detected among the tumors without ED variants. SBS14 was mostly, although not exclusively, found among dMMR tumors.

Heatmap showing the clustering of tumors based on the contribution of tumor mutational signatures SBS10a, SBS10b, SBS10d, SBS28, SBS14, SBS20 and “other signatures”. Analysis was performed with 64 tumor samples with somatic pathogenic variants in POLE and POLD1 EDs, 3 tumors belonging to three probands with germline pathogenic variants in POLD1, and 70 TCGA tumor samples without polymerase exonuclease domain variants
Only two POLD1 ED-mutated tumors, both dMMR, could be included in the analysis: one tumor had 10% SBS14 contribution and no trace of Polδ proofreading-deficient signatures (SBS10d or SBS20), and the other had 83% SBS20 contribution. Unlike the other polymerase proofreading-associated signatures, SBS20 was also observed in a subgroup of dMMR tumors (n = 15) without ED variants, at contributions ranging from 18 to 40%. Due to its non-specificity, we recommend not using SBS20 for variant classification. Due to the lack of pMMR, Polδ proofreading-deficient sporadic tumors, we re-analyzed exome/genome sequencing data obtained from three additional proofreading-deficient tumors (two CRCs and one adenoma), developed by heterozygous carriers of germline POLD1 p.Leu474Pro, p.Asp316His, and p.Ser478Asn [54, 73]. All three samples were hypermutated (59, 114 and 36 mut/Mb respectively) and had 34%-68% contribution of SBS10d, highly specific of Polδ proofreading deficiency in tumors (Fig. 1). Moreover, all three tumors had copy-neutral loss of heterozygosity (cnLOH) in the POLD1 region that caused the loss of the wildtype allele [73].
The 50 pMMR Polε proofreading-deficient cancers had an average of 144 mut/Mb (range: 2.6—325), and the 10 dMMR Polε proofreading-deficient cancers, 255 mut/Mb (range: 109 – 531). Only 2 samples, both harboring POLE p.Leu424Val had TMBs < 25 mut/Mb (2.6 and 4.4 mut/Mb). All 62 POLE ED-mutated tumors, regardless of their MMR status, had > 5% contribution of signatures SBS10a and/or 10b (median: 65%; range: 6%– 87%). When considering all Polε proofreading-deficient signatures combined, i.e. SBS10a, SBS10b, SBS28 and SBS14, 100% of samples reached > 20% contribution.
In the generic ACMG/AMP guidelines, PP4 corresponds to highly specific phenotypes or family history of a disease with a single genetic etiology, and BP5, to variants found in cases with an alternate molecular disease basis. We propose to adapt these criteria to the presence or absence of the proofreading deficiency-specific mutational signatures and high TMB. To consider PP4, no other (somatic) ED missense variant classified as (likely) pathogenic or of unknown significance in the same gene (POLE or POLD1) should occur in the tumor, and at least PM2_supporting must be fulfilled. We recommend performing the mutational signature analysis when the tumors are hypermutated (> 10 mut/Mb) or have at least a total of 80 somatic SNVs, to minimize the detection of false (artifact) signatures generated from an extremely small number of variants. Optimally, the use of exome or genome sequencing data is recommended, although the use of sequencing data obtained from panels that include a relevant number of genes may also be used.
We recommend using PP4 with a strong level of strength: For POLE ED variants, when at least two tumors have SBS10a, SBS10b, SBS28, and/or SBS14; and for POLD1 ED variants, when at least two tumors have SBS10d or when one tumor has SBS10d and loss of heterozygosity (LOH) that causes the loss of the wildtype allele. PP4_moderate may be applied for POLE ED variants when one tumor has SBS10a, SBS10b, SBS28, and/or SBS14; and for POLD1 ED variants when there is one tumor with SBS10d (no available 2nd hit information or no LOH). These recommendations are based on the data obtained from fresh/frozen tumor samples. To minimize the potential effect of FFPE sequencing artifacts, a ≥ 5% contribution of the gene-specific signatures will be considered to apply PP4 strong and moderate criteria.
We recommend using BP5 when two or more tumors with the ED variant have ≤ 1 mut/Mb. For POLE variants, BP5 should be used when two or more tumors harboring the variant, with > 1 mut/Mb or at least > 80 total single nucleotide variants, have neither SBS10a, nor SBS10b, nor SBS28, nor SBS14; or when one tumor has ≤ 1 mut/Mb and another one, with > 1 mut/Mb or > 80 single nucleotide variants, has neither SBS10a, nor SBS10b, nor SBS28, nor SBS14. For POLD1 variants, use BP5 when two or more pMMR tumors harboring the variant, with > 1 mut/Mb or at least > 80 total single nucleotide variants, do not have SBS10d; or when one tumor has ≤ 1 mut/Mb and one pMMR tumor, with > 1 mut/Mb or at least > 80 total single nucleotide variants, has no SBS10d. In all instances, at least two tumors are required to minimize the possible analysis of phenocopies and the effect of FFPE-derived sequencing artifacts.
Functional data
Available in vitro assays to test the functionality of POLE and POLD1 ED variants assess the proofreading ability of the polymerases in absence and presence of the variant. The studies reported to date rely mostly on yeast-based assays, although cell-free assays, in vitro human or murine cell line experiments, and in vivo mouse models, have also been used (Additional file 3: Table S3).
PS3 and BS3 rely on well-established in vitro or in vivo functional studies supporting or discarding a damaging effect of the variant. Based on available data and the fact that the performance of the functional studies published so far has not been evaluated, we recommend using PS3_moderate when results from at least 2 independent experiments (at least one in a non-yeast model) that assess, with proper positive and negative controls, the proofreading function of the corresponding polymerase in presence and absence of the variant, show defects and are concordant. If only results from one experiment are available, or the results, even from multiple experiments, are produced exclusively in yeast-based systems [5], we recommend applying a supporting level of strength. We propose to decrease the level of strength for yeast-based evidence because published results show high variability among replicates and experiments (publications in Additional file 3: Table S3), and some concerns have been raised regarding the assessment of variants affecting the DNA binding, which might show an effect in yeast even when the variant is non-pathogenic [5, 10, 68]. We currently recommend using BS3_supporting, when at least two independent experiments (≥ 1 in a non-yeast model) show no proofreading defect. For both PS3 and BS3 criteria, the assayed amino acid change must be the same as the one identified in the patient.
The ClinGen Sequence Variant Interpretation Committee recommends assessing the performance of any functional assay using variants classified as pathogenic or benign according to clinical parameters (cross validation) [74], which has not been done for any of the POLE/POLD1 functional assays reported to date. Calibration according to the cross-validation results is recommended to correctly apply the PS3 and BS3 rules, providing the correct level of strength, or a calibrated quantitative value if Bayesian transformation of the ED-specific ACMG/AMP guidelines is applied.
Classification of reported variants
The defined classification recommendations (Table 3) were applied to 128 variants reported in the literature (reviewed: March 2023) and ClinVar (access date: July 2021), including the 23 variants used for the definition of the guidelines. Of the 128 variants considered, 7 were classified as pathogenic, 10 as likely pathogenic, 7 as benign, and 10 as likely benign. Of the 17 a priori pathogenic variants included in Table 2, all but POLE:c.824A>T; p.(Asp275Val) and POLE:c.830A>G; p.(Glu277Gly), now classified as variants of unknown significance, were classified as P (n = 7) or LP (n = 8). Moreover, two additional variants were classified as likely pathogenic: POLE:c.857C>T; p.(Pro286Leu) and POLE:c.1373A>T; p.(Tyr458Phe). Additional file 3: Table S3 shows the classification of all 128 variants taking into consideration the data available.
Clinical features of reported individuals with constitutional POLE or POLD1 ED pathogenic or likely pathogenic variants
To date, literature reports include 205 individuals heterozygous for the 17 POLE or POLD1 variants classified as pathogenic or likely pathogenic following the defined recommendations. Of the 205 heterozygotes, 149 (73%) were diagnosed with cancer: 120 (58% of the 205 carriers) with CRC (mean age at diagnosis: 41; range: 13–80), 21 (22% of 95 female carriers) with endometrial cancer (age: 50; range: 31–58), 11 (12% of female carriers) with breast cancer (age: 55; range: 38–65); 8 (8% of female carriers) with ovarian cancer (age: 42; range: 33–50), 19 (9%) with extracolonic gastrointestinal cancers (age: 45; range: 35–78), 18 (9%) with brain cancer (age: 28; range: 4–66), and 9 (4%) with other cancer types. The majority of heterozygotes (88%) had reports of cancer, and/or preneoplastic lesions, and/or non-tumoral extracolonic manifestations (e.g. café-au-lait macules). Sixty-four percent of those with polyp information (70/108) were reported to have > 10 gastrointestinal polyps (detailed phenotypes in Additional file 4: Table S4).
While these phenotypes should currently guide clinical surveillance in carriers, future prospective collaborative efforts will provide more accurate (unbiased) estimates of cancer risk and penetrance. Furthermore, oncologic therapeutic decisions in the context of the hereditary cancer syndrome, and for cancers with somatic pathogenic or likely pathogenic POLE or POLD1 exonuclease variants, should consider the good prognosis and response to immune checkpoint inhibitors of polymerase proofreading deficient tumors [75,76,77].
Conclusions
We propose the first recommendations based on the general ACMG/AMP guidelines for the classification of variants in the exonuclease domain of POLE and POLD1, taking into consideration the available evidence (Table 3, Fig. 2). With better phenotypic and molecular characterization of the syndrome and associated tumors, together with access to better and cross validated functional assays, improved recommendations are expected in following years.

Schematic summary of the evidence that supports pathogenicity of ED variants
Availability of data and materials
Data supporting the reported results may be found in the article, supplementary material, public repositories (TCGA, COSMIC, gnomAD, ClinVar) and/or published articles.
References
Shevelev IV, Hübscher U. The 3’ 5’ exonucleases. Nat Rev Mol Cell Biol. 2002;3(5):364–76.
McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18(1):148–61.
Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45(2):136–44.
Rayner E, van Gool IC, Palles C, et al. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer. 2016;16(2):71–81.
Mur P, García-Mulero S, Del Valle J, et al. Role of POLE and POLD1 in familial cancer. Genet Med. 2020;22(12):2089–100.
Palles C, Martin L, Domingo E, et al. The clinical features of polymerase proof-reading associated polyposis (PPAP) and recommendations for patient management. Fam Cancer. 2022;21(2):197–209.
Lindsay H, Scollon S, Reuther J, et al. Germline POLE mutation in a child with hypermutated medulloblastoma and features of constitutional mismatch repair deficiency. Cold Spring Harb Mol Case Stud. 2019;5(5):a004499.
Wimmer K, Beilken A, Nustede R, et al. A novel germline POLE mutation causes an early onset cancer prone syndrome mimicking constitutional mismatch repair deficiency. Fam Cancer. 2017;16(1):67–71.
Sehested A, Meade J, Scheie D, et al. Constitutional POLE variants causing a phenotype reminiscent of constitutional mismatch repair deficiency. Hum Mutat. 2022;43(1):85–96.
Barbari SR, Shcherbakova PV. Replicative DNA polymerase defects in human cancers: Consequences, mechanisms, and implications for therapy. DNA Repair (Amst). 2017;56:16–25.
Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73.
Church DN, Briggs SE, Palles C, et al. DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013;22(14):2820–8.
Church DN, Stelloo E, Nout RA, et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. J Natl Cancer Inst. 2015;107(1):402.
Brannon AR, Vakiani E, Sylvester BE, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014;15(8):454.
Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7.
Seshagiri S, Stawiski EW, Durinck S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488(7413):660–4.
Giannakis M, Mu XJ, Shukla SA, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;17(4):1206.
Campbell BB, Light N, Fabrizio D, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171(5):1042-1056.e1010.
Erson-Omay EZ, Çağlayan AO, Schultz N, et al. Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis. Neuro Oncol. 2015;17(10):1356–64.
Briggs S, Tomlinson I. Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013;230(2):148–53.
Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21.
Cornish AJ, Gruber AJ, Kinnersley B, et al. Whole genome sequencing of 2,023 colorectal cancers reveals mutational landscapes, new driver genes and immune interactions. bioRxiv 2022.11.16.515599.
Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101.
Haradhvala NJ, Kim J, Maruvka YE, et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat Commun. 2018;9(1):1746.
Mehnert JM, Panda A, Zhong H, et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J Clin Invest. 2016;126(6):2334–40.
Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8.
Johanns TM, Miller CA, Dorward IG, et al. Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy. Cancer Discov. 2016;6(11):1230–6.
Wang C, Gong J, Tu TY, Lee PP, Fakih M. Immune profiling of microsatellite instability-high and polymerase ε (POLE)-mutated metastatic colorectal tumors identifies predictors of response to anti-PD-1 therapy. J Gastrointest Oncol. 2018;9(3):404–15.
Picard E, Verschoor CP, Ma GW, Pawelec G. Relationships between immune landscapes, genetic subtypes and responses to immunotherapy in colorectal cancer. Front Immunol. 2020;11:369.
Lau D, Kalaitzaki E, Church DN, et al. Rationale and design of the POLEM trial: avelumab plus fluoropyrimidine-based chemotherapy as adjuvant treatment for stage III mismatch repair deficient or POLE exonuclease domain mutant colon cancer: a phase III randomised study. ESMO Open. 2020;5(1):e000638.
Pachlopnik Schmid J, Lemoine R, Nehme N, et al. Polymerase ε1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (“FILS syndrome”). J Exp Med. 2012;209(13):2323–30.
Thiffault I, Saunders C, Jenkins J, et al. A patient with polymerase E1 deficiency (POLE1): clinical features and overlap with DNA breakage/instability syndromes. BMC Med Genet. 2015;16:31.
Eason C, Aleisa A, Jones JR, Prijoles EJ, Wine LL. Filling in the gaps on FILS syndrome: A case report and literature review. Pediatr Dermatol. 2020;37(5):915–7.
Logan CV, Murray JE, Parry DA, et al. DNA Polymerase Epsilon Deficiency Causes IMAGe Syndrome with Variable Immunodeficiency. Am J Hum Genet. 2018;103(6):1038–44.
Nakano T, Sasahara Y, Kikuchi A, et al. Novel POLE mutations identified in patients with IMAGE-I syndrome cause aberrant subcellular localisation and protein degradation in the nucleus. J Med Genet. 2022;59(11):1116–22.
Shastry S, Simha V, Godbole K, et al. A novel syndrome of mandibular hypoplasia, deafness, and progeroid features associated with lipodystrophy, undescended testes, and male hypogonadism. J Clin Endocrinol Metab. 2010;95(10):E192-197.
Murdocca M, Spitalieri P, D’Apice MR, Novelli G, Sangiuolo F. From cue to meaning: The involvement of POLD1 gene in DNA replication, repair and aging. Mech Ageing Dev. 2023;211:111790.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Mester JL, Ghosh R, Pesaran T, et al. Gene-specific criteria for PTEN variant curation: Recommendations from the ClinGen PTEN Expert Panel. Hum Mutat. 2018;39(11):1581–92.
Lee K, Krempely K, Roberts ME, et al. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum Mutat. 2018;39(11):1553–68.
Luo X, Maciaszek JL, Thompson BA, et al. Optimising clinical care through CDH1-specific germline variant curation: improvement of clinical assertions and updated curation guidelines. J Med Genet. 2023;60(6):568–75.
Luo X, Feurstein S, Mohan S, et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv. 2019;3(20):2962–79.
ClinGen Hereditary Breast, Ovarian and Pancreatic Cancer Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for ATM Version 1.1. URL: https://clinicalgenome.org/docs/clingen-hereditary-breast-ovarian-and-pancreatic-cancer-expert-panel-specifications-to-the-acmg-amp-variant-interpretation-1.1/.
Biesecker LG, Harrison SM, Group CSVIW. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med. 2018;20(12):1687–8.
Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11(1):1–9.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–94.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122.
Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89(22):10915–9.
Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501.
Robinson PS, Coorens THH, Palles C, et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat Genet. 2021;53(10):1434–42.
Kautto EA, Bonneville R, Miya J, et al. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget. 2017;8(5):7452–63.
Niu B, Ye K, Zhang Q, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30(7):1015–6.
Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–57.
Berg KD, Glaser CL, Thompson RE, Hamilton SR, Griffin CA, Eshleman JR. Detection of microsatellite instability by fluorescence multiplex polymerase chain reaction. J Mol Diagn. 2000;2(1):20–8.
Davidson AL, Leonard C, Koufariotis LT, et al. Considerations for using population frequency data in germline variant interpretation: Cancer syndrome genes as a model. Hum Mutat. 2021;42(5):530–6.
Dominguez-Valentin M, Sampson JR, Seppälä TT, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. 2020;22(1):15–25.
Whiffin N, Minikel E, Walsh R, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19(10):1151–8.
Galati MA, Hodel KP, Gams MS, et al. Cancers from Novel Pole-mutant mouse models provide insights into polymerase-mediated hypermutagenesis and immune checkpoint blockade. Cancer Res. 2020;80(24):5606–18.
Albertson TM, Ogawa M, Bugni JM, et al. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A. 2009;106(40):17101–4.
Li HD, Cuevas I, Zhang M, et al. Polymerase-mediated ultramutagenesis in mice produces diverse cancers with high mutational load. J Clin Invest. 2018;128(9):4179–91.
Wimmer K, Kratz CP, Vasen HF, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium “care for CMMRD” (C4CMMRD). J Med Genet. 2014;51(6):355–65.
Mur P, Palles C, Tomlinson I, Valle L. Reply to: “Development of an MSI-positive colon tumor with aberrant DNA methylation in a PPAP patient.” J Hum Gene. 2020;65:513–4 England.
Bonjoch L, Soares de Lima Y, Díaz-Gay M, et al. Unraveling the impact of a germline heterozygous POLD1 frameshift variant in serrated polyposis syndrome. Front Mol Biosci. 2023;10:1119900.
Barbari SR, Kane DP, Moore EA, Shcherbakova PV. Functional Analysis of Cancer-Associated DNA Polymerase ε Variants in Saccharomyces cerevisiae. G3 (Bethesda). 2018;8(3):1019–29.
Pejaver V, Byrne AB, Feng BJ, et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. 2022;109(12):2163–77.
Loong L, Cubuk C, Choi S, et al. Quantifying prediction of pathogenicity for within-codon concordance (PM5) using 7541 functional classifications of BRCA1 and MSH2 missense variants. Genet Med. 2022;24(3):552–63.
Fortuno C, James PA, Spurdle AB. Current review of TP53 pathogenic germline variants in breast cancer patients outside Li-Fraumeni syndrome. Hum Mutat. 2018;39(12):1764–73.
Belman S, Parsons MT, Spurdle AB, Goldgar DE, Feng BJ. Considerations in assessing germline variant pathogenicity using cosegregation analysis. Genet Med. 2020;22(12):2052–9.
Andrianova MA, Seplyarskiy VB, Terradas M, et al. Extended family with an inherited pathogenic variant in polymerase delta provides strong evidence for recessive effect of proofreading deficiency in human cells. bioRxiv 2022.07.20.500591.
Brnich SE, Abou Tayoun AN, Couch FJ, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019;12(1):3.
Das A, Sudhaman S, Morgenstern D, et al. Genomic predictors of response to PD-1 inhibition in children with germline DNA replication repair deficiency. Nat Med. 2022;28(1):125–35.
van Gool IC, Bosse T, Church DN. proofreading mutation, immune response and prognosis in endometrial cancer. Oncoimmunology. 2016;5(3):e1072675.
Strauss JD, Pursell ZF. Replication DNA polymerases, genome instability and cancer therapies. NAR Cancer. 2023;5(3):033.
Acknowledgements
Not applicable.
Funding
This study was funded by the Spanish Ministry of Science and Innovation (Agencia Estatal de Investigación), co-funded by FEDER funds -a way to build Europe- [PID2020-112595RB-I00 (LV), and “Contratos predoctorales para la formación de doctores” (JV-E)]; Instituto de Salud Carlos III [CIBERONC CB16/12/00234; Infraestructura de Medicina de Precisión asociada a la Ciencia y la Tecnología – IMPaCT IMP/00009, co-funded by the European Union]; and Government of Catalonia [AGAUR 2021SGR01112, and CERCA Program for institutional support].
Author information
Authors and Affiliations
Contributions
Conceptualization: PM, LV; Data curation: PM, JV-E, LV; Formal analysis: PM, JV-E, SG-M, LM-P, IGM, TP, LV; Funding acquisition: LV, GC; Investigation: PM, JV-E, SG-M, LM-P, IGM, TP, LV; Methodology: PM, SG-M, TP, MP, LF, LV; Project administration: LV; Resources: LV, GC; Software: JV-E, SG-M, LM-P, IGM, TP; Supervision: LV; Validation: PM, MP, LF, LV; Visualization: PM, JV-E, SG-M, LV; Writing-original draft: PM, LV; Writing-review & editing: LF, MP (main review and editing), and all the other authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study received the approval of the IDIBELL Ethics Committee (Reference number: PR252/21). Consent to participate: not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Results.
Population allele frequency threshold (AFT) calculation. Table S1. Standard ACMG/AMP combination rules to define pathogenic, likely pathogenic, likely benign and benign variants.
Additional file 2: Table S2.
Location of POLE and POLD1 amino acids in the 3D structure and their accessibility to the DNA.
Additional file 3: Table S3.
Characteristics and classification of constitutional (germline) POLE and POLD1 exonuclease domain missense variants.
Additional file 4: Table S4.
Phenotypic features of reported individuals with the POLE and POLD1 ED germline pathogenic variants listed in Table 2 and of heterozygous carriers of two additional variants reclassified as pathogenic or likely pathogenic after the application of the recommendations defined in this article.
Additional file 5: Table S5.
TCGA and COSMIC tumors with the POLE or POLD1 ED variants evaluated in this study.
Additional file 6: Table S6.
Tumors evaluated in this study with available sequencing data for the calculation of tumor mutational burden and mutational signatures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mur, P., Viana-Errasti, J., García-Mulero, S. et al. Recommendations for the classification of germline variants in the exonuclease domain of POLE and POLD1. Genome Med 15, 85 (2023). https://doi.org/10.1186/s13073-023-01234-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-023-01234-y