
Abstract
Background
The multiple de novo copy number variant (MdnCNV) phenotype is described by having four or more constitutional de novo CNVs (dnCNVs) arising independently throughout the human genome within one generation. It is a rare peri-zygotic mutational event, previously reported to be seen once in every 12,000 individuals referred for genome-wide chromosomal microarray analysis due to congenital abnormalities. These rare families provide a unique opportunity to understand the genetic factors of peri-zygotic genome instability and the impact of dnCNV on human diseases.
Methods
Chromosomal microarray analysis (CMA), array-based comparative genomic hybridization, short- and long-read genome sequencing (GS) were performed on the newly identified MdnCNV family to identify de novo mutations including dnCNVs, de novo single-nucleotide variants (dnSNVs), and indels. Short-read GS was performed on four previously published MdnCNV families for dnSNV analysis. Trio-based rare variant analysis was performed on the newly identified individual and four previously published MdnCNV families to identify potential genetic etiologies contributing to the peri-zygotic genomic instability. Lin semantic similarity scores informed quantitative human phenotype ontology analysis on three MdnCNV families to identify gene(s) driving or contributing to the clinical phenotype.
Results
In the newly identified MdnCNV case, we revealed eight de novo tandem duplications, each ~ 1 Mb, with microhomology at 6/8 breakpoint junctions. Enrichment of de novo single-nucleotide variants (SNV; 6/79) and de novo indels (1/12) was found within 4 Mb of the dnCNV genomic regions. An elevated post-zygotic SNV mutation rate was observed in MdnCNV families. Maternal rare variant analyses identified three genes in distinct families that may contribute to the MdnCNV phenomenon. Phenotype analysis suggests that gene(s) within dnCNV regions contribute to the observed proband phenotype in 3/3 cases. CNVs in two cases, a contiguous gene duplication encompassing PMP22 and RAI1 and another duplication affecting NSD1 and SMARCC2, contribute to the clinically observed phenotypic manifestations.
Conclusions
Characteristic features of dnCNVs reported here are consistent with a microhomology-mediated break-induced replication (MMBIR)-driven mechanism during the peri-zygotic period. Maternal genetic variants in DNA repair genes potentially contribute to peri-zygotic genomic instability. Variable phenotypic features were observed across a cohort of three MdnCNV probands, and computational quantitative phenotyping revealed that two out of three had evidence for the contribution of more than one genetic locus to the proband’s phenotype supporting the hypothesis of de novo multilocus pathogenic variation (MPV) in those families.
Similar content being viewed by others

Background
De novo copy number variants (dnCNVs) that occur during gametogenesis or early post-zygotic development are present in all or most cells of a multicellular organism. Genome-wide surveys of large populations estimate the de novo mutation rate for structural variants to be 0.16–0.29 events per generation in humans [1, 2]. The rate for dnCNVs with a length over 100 kb is lower, around 0.012 per haploid genome [3]. A unique mutational phenomenon described by Liu et al. highlighted individuals with variable congenital abnormalities and multiple (n ≥ 4) dnCNVs (MdnCNV) throughout their genome [4]. The MdnCNV event, or phenomenon, can encompass several genes at each CNV locus [4].
MdnCNV is a rare mutational phenomenon, identified in only 5/60,000 individuals referred for genome-wide chromosomal microarray analysis [4]. MdnCNV has likely been under-appreciated due to the limited genomic resolution in clinical testing. The prominent features of dnCNVs shared between MdnCNV cases include (1) a predominance of copy number gains across multiple chromosomes, (2) tandem duplications forming the majority of copy number gains, (3) the presence of sequence microhomology or microhomeology at breakpoint junctions, and (4) other mutational signatures of SV mutagenesis such as the DUP-TRP-DUP pattern of Complex Genomic Rearrangement (CGR) [4].
The variable congenital abnormalities observed in individuals exhibiting the MdnCNV phenomenon are thought to be caused by the copy number change of different critical driver gene(s) in each proband. The Human Phenotype Ontology (HPO) represents a structured language database of human phenotype terms that allows for numerical coding of clinical phenotypes as HPO terms; such phenotypes may be observed in a proband or described in association with a gene or rare disease trait clinical synopsis in OMIM [5]. This numerical coding enables quantitative, computational analyses of a patient’s phenotypic features and comparison with phenotype associations within the literature to inform genomic variant prioritization.
Here we report a new family with multiple dnCNVs and leverage multiple genomic and phenotypic methodologies combined with visualization tools to extend our understanding of the MdnCNV mutational phenomenon. Rare variant and mutational signature analyses on the newly described and four previously characterized MdnCNV families suggest a maternal genetic variant contributing to peri-zygotic genome instability. Gene content of the affected genomic regions was analyzed using HPO to identify potential driver gene(s) and explore the hypothesis that the observed trait manifestation may be driven by multilocus pathogenic variation (MPV).
Methods
Subjects
The newly identified MdnCNV family (HOU3579) was initially ascertained through clinical chromosomal microarray analysis (CMA) performed at Baylor Genetics. Written consents were obtained for the proband (BAB9637), unaffected siblings (BAB9640, BAB9641, and BAB9642), and parents (BAB9638 and BAB9639) to perform further genomic studies. Oligo array-based comparative genomic hybridization (aCGH) was performed on all family members (“Methods” section). Illumina short-read (SR) whole-genome sequencing (WGS) and long-read (LR) WGS with Oxford Nanopore Technologies (ONT) were performed on the proband and parents’ blood leukocyte-derived DNA (Additional file 1: Supplementary methods). PacBio LR genomic sequencing was performed on the proband alone (“Methods”). Illumina short-read WGS was performed on nine anonymized families under a separate IRB protocol with a waiver of consent, including four previously reported MdnCNV families (BAB3097, BAB3596, mCNV3/BAB9484, and mCNV7) [4] and five additional families without a MdnCNV phenotype as controls (Additional file 1: Supplementary methods).
Array comparative genomic hybridization
The family’s DNA samples were initially analyzed by a clinical chromosomal microarray analysis (CMA) designed and performed by Baylor Genetics (BG) [6, 7]. Subsequently, high-resolution aCGH, using a 1 million probe whole-genome oligonucleotide microarray (Agilent microarray design ID:085903), was performed on all family members. All array-based experiments were implemented according to the Agilent aCGH protocol for probe labeling and hybridization with minor modifications [8].
Illumina SR sequencing
Genome sequencing was performed on proband and parents with an average read depth of 35 × at the Baylor College of Medicine Human Genome Sequencing Center. Please refer to supplementary material for the details.
Pacific Biosciences (PacBio) LR sequencing data
PacBio LR genomic sequencing was performed on the proband alone. The sequencing library was constructed with 5 µg genomic DNA using the SMRTbell Express Template Preparation Kit with an average insert size of 7.5 kb. The library was sequenced with five SMRTcells using the PacBio Sequel I instrument, which yielded 42 Gb of data.
Nanopore trio LR sequencing and mapping
LR sequencing libraries were generated according to standard Oxford Nanopore Technologies (ONT) protocols. Detailed sequencing process and metrics are provided (Additional file 1: Supplementary methods and Table S1). Average coverage of 25 × was achieved for the trio.
De novo single-nucleotide variant (SNV) and indel variant calling
Individual germline SNVs and indels were called using GATK (v.4.1.3) haplotypecaller and the HGSC xAtlas variant calling pipelines [9]. The “-GVCF” option was used for the GATK haplotypecaller, which outputs a gVCF file that includes reference or variant sequence information for all nucleotide positions. Using recalibrated posterior genotype probabilities to allow rigorous calling, de novo variants (GQ ≥ 20 for all trio members) were annotated. All possible de novo variants were further annotated using DNM (de novo mutation)-Finder (https://github.com/BCM-Lupskilab/DNM-Finder) [10], and manual inspection with Integrative Genomics Viewer (IGV) software was applied to confirm or reject the variant. It was performed with the following criteria per variant: (1) supported by at least 5 uniquely mapped reads; (2) supported by both forward and reverse strand reads; (3) variant did not derive from misalignment at indel variants; (4) not located at highly repetitive regions masked by RepeatMasker file for reference genome GRCh38 extracted from UCSC browser. In addition, potential de novo SNVs mapping within 20 bp of each other (clustered SNVs) was error-prone and thus removed from this study. De novo substitutions with variant read to total read ratio less than 0.35 or more than 0.65 (VR/TR < 0.35 or VR/TR > 0.65 and no read detected in any parental samples to exclude potential mosaicism in the parental genome) were classified as possible post-zygotic variant allele events.
Mutational pattern analysis
The R/Bioconductor package MutationalPatterns [11] was used for mutational signatures analysis on dnSNVs. The “cos_sim_matrix” function within the MutationPatterns package was used to calculate the cosine similarity between known COSMIC (v3.2) signatures [12] with the base substitution profile of MdnCNV families (Additional file 2: Table S2). To avoid overfitting, an unbiased refitting procedure was used to select the optimal combination of signatures using “fit_to_signatures_strict” function with “best_subset” method option. In short, the refitting process starts with a subset of signatures and then removes the signature that has the least contribution. The removal happens iteratively until it gets to the optimal subset.
Phasing SNVs and CNVs with combined read-based and pedigree-based method
The read-based phasing was performed using the publicly available WhatsHap tool [13]. In-house developed R scripts (https://github.com/BCM-Lupskilab/PhaseDenovo) were used to perform pedigree-based genetic phasing of physical haplotype blocks. The script assigns parental origin to the physical haplotype block if there are ≥ 20 informative SNPs present, and ≥ 90% of them are consistent with a single parental origin. The dnSNVs were assigned to the parental chromosome based on segregated haplotype blocks. The dnCNVs were phased using the method as previously described [14].
Structural variant (SV) calling and analyses
Binary sequence alignment (BAM) files from SR and LR sequencing were used for SV calling. For Illumina SR, SVs were called and genotyped using Parliament2 [15]. Sniffles [16] was used for LR SV calling on the proband using the following parameters to maximize sensitivity: “ -s 8”. The SV calls from the proband were genotyped based on parental BAM files using Sniffles with the option “–Ivcf”. Subsequently, SVs called exclusively in the proband were filtered with SURVIVOR allowing a maximum distance of 1000 bp between pairwise breakpoint junction calls from each algorithm. For dnCNVs, the log2 ratio of read depth was visualized and manually examined (See “Visualization of genomics data for dnCNVs”) to minimize false-positive calls.
Breakpoint junction amplification and sequencing analysis
Soft-clipped reads overlapping breakpoint junctions were extracted from LR sequence alignment files and remapped to the human genome (GRCh38) with the UCSC BLAT tool to single base-pair resolution. The amplified breakpoint junctions were confirmed by Sanger dideoxynucleotide sequencing. The presence of non-B DNA-forming sequence motifs, including Z-DNA, G-quadruplex, A-phased repeats, inverted repeats, mirror repeats, and direct repeats, were screened for within 50 bp to either side of the breakpoint junctions using the reference genome and the nBMST tool [17].
De novo mutation rate estimation and statistical analysis for SNV clustering
The unphased genome-wide mutation rate per base-pair was estimated by taking the total number of DNMs divided by the size of the mappable diploid human reference genome (GRCh38). The phased genome-wide mutation rate was estimated by taking the total number of phased DNMs divided by the size of the mappable haploid human reference genome. The mappable human genome size (2.91 Tb/haploid genome) was computed using faCount from Kent’s tool (http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/). The number of phased DNMs to each haploid type was extrapolated based on the ratio of successfully phased DNMs. The interval defined was centered on the ~ 1 Mb dnCNV with an additional 4 Mb window size flanking either side, representing ~ 9 Mb intervals in total for an individual dnCNV. The dynamic window size ranged from 1 to 10 Mb. The observed count of DNMs in eight dnCNV regional intervals was calculated in reference to the size of the window range used. The corresponding Poisson probability was calculated by multiplying the genome-wide DNM density by the corresponding dnCNV interval length. The average count of DNMs at the same genomic intervals from 2976 genome sequenced trios [18] was calculated as the regional density control. The Poisson probability of observing the same or more DNMs was calculated using the ppois function from the R base package to determine the p-value.
Visualization of genomics data for dnCNVs
The average read depth from SR sequencing was calculated using mosdepth v0.2.3 with the “– by 1000” option. We used the median read depth of each chromosome to normalize the read depth of that chromosome and calculated the log2 ratio for each 1000-bp window. The log2 ratio profiles were segmented using the Circular Binary Segmentation (CBS) algorithm [19] implemented in the DNAcopy Bioconductor package. The individual and segmented ratios were visualized together across genomic coordinates using KaryoploteR (v.1.16.0) [20] with log2 ratios of 0 representing normal copy number state, > 0.58 representing copy number gains, and < − 1 representing copy number loss.
Quantitative phenotyping analyses
To perform quantitative phenotype analysis, we used a similar method to that previously published [21,22,23] with modification; a detailed description follows. The patient’s clinical description was translated to HPO terms using Doc2Hpo [24] and manually verified. HPO encoded phenotypes are available for known disease genes through Online Mendelian Inheritance in Man (OMIM.org) [25]. HPO encoded phenotypes for known diseases were extracted from OMIM and Orphanet. Using the ontologyX suite of R packages [26], a pairwise Lin semantic similarity score [27] was calculated between the patient’s HPO term set and the HPO term sets of all known genes encompassed by proband dnCNVs. To assess for multilocus pathogenic variation, the phenotypic similarity score of the proband was compared to the combined phenotype associated with sets of known disease-associated genes encompassed by dnCNVs. Due to the limited sample size, a cutoff of 5% was arbitrarily selected to aid in the determination of multilocus pathogenic variation in conjunction with phenotypic overlap assessed by grid comparison of proband and disease gene-associated phenotypes.
Results
Ascertainment and identification of a new individual with genomic MdnCNV phenotype
Since the identification of the five MdnCNV families at Baylor Genetics (BG) [4], another individual (BAB9637) with potential MdnCNV was identified at BG. Clinical CMA performed on proband DNA revealed seven large, ~ 1 Mb, rare variant copy number gains mapping to seven different chromosomes. In addition, an apparent 7 Mb absence of heterozygosity (AOH) genomic region was observed in exome sequencing data mapping to chromosome 15q14q21.1. To capture the full spectrum of dnCNVs, the subject, siblings, and parents’ DNA was analyzed further in the research setting using a high-resolution aCGH. Short- and long-read genome sequencing was performed on proband and parental DNA with an average depth of coverage of 35 × and 25 × , respectively. This multimodal genomic analysis approach (Fig. 1a, b, Additional file 1: Table S3) demonstrated eight tandem duplications, confirming the seven duplications previously identified on clinical CMA and revealing an eighth duplication not previously detected. The eight duplications mapped to different chromosomes and showed their sizes ranged from 899.1 to 1041.6 kb, i.e., ~ 1 Mb.

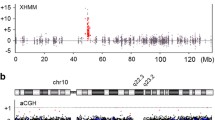
dnCNV and dnSNV identified with multiple genomic approaches. a Pedigree (left) of the MdnCNV family HOU3579. In the middle, the sequencing platform and variant calling pipeline are illustrated. Shown on the right, from top to bottom, is the visualization of an example of dnCNV in CMA, 1 M aCGH, short-read genome sequencing read depth, short-read genome sequencing B-allele frequency, and IGV view a high-quality dnSNV call. b Log2 ratio of phased dnCNV in genome-wide view with chromosomes along the x-axis. Gains present on chromosomes 4, 6, 12, and 14 are each indicated with a green dot representing duplication on the paternally inherited chromosome. Gains present on chromosomes 5, 10, 13, and 21 are each indicated with a pink dot representing duplication on the maternally inherited chromosome. The text adjacent to each dot denotes the size (in Mb) of each dnCNV. c Pedigree of MdnCNV family (top) with aCGH result for each dnCNV region. Parental origin of each chromosome harboring a dnCNV in the proband is indicated by a “P” (paternal) or “M” (maternal) on each array
The high-resolution aCGH and breakpoint PCR confirmed that these eight duplications were only present in the proband (Fig. 1c and Additional file 1: Figures S1, S2). However, the apparent 7 Mb AOH region observed in chr15q14q21.1 by clinical CMA was not supported by the B-allele frequency calculated from genome sequencing data. This is likely due to genome sequencing interrogating a higher density of SNP sites (n = 9080) compared to clinical CMA (n = 50) (Additional file 1: Figure S3). An apparently homozygous ~ 60.2 kb deletion was identified within a rare de novo duplication on chromosome 14. Both parents are heterozygous for the deletion. The allele frequency of the deletion in the control population is 0.139 based on an allele count of 2990/21518 alleles in the gnomAD SV v2.1 database. The deletion allele frequency ranges from 0.043 in the African population to 0.339 in the Latino population (Additional file 1: Figure S4). We explored the potential association between both replication time and CNV regions by overlapping the duplication region to the replication time map of four embryonic stem cell lines [28]. Our analysis did not suggest a preference of dnCNV occurring regarding replication timing, i.e., dnCNVs were mapped to either late or early replication regions. Only microhomologies were found at the breakpoint junction for five dnCNVs (Additional file 1: Figure S1) which suggests a replicative instead of homologous recombination-based mechanism for the dnCNV formation in this case.
With SR sequencing data, we identified 91 DNMs including 79 SNVs (transition to transversion ratio [Ti: Tv] = 2.0) and 12 indels (Fig. 2 and Additional file 3: Table S4). By combining short- and long-read sequencing data, we were able to phase 50 dnSNVs, of which 80% were of paternal origin (Additional file 3: Table S4). The predominance of paternal inheritance of dnSNVs was anticipated due to the accumulation of variants in aging male gametes [29, 30]. The genomic mapping of dnSNVs on the haploid human genome reference did not show any obvious clustering of variants. We calculated the distance of DNM to the nearest dnCNV breakpoint and found 7 (8.2%) DNMs within 4 Mb of the nearest breakpoint (Additional file 3: Table S4 and Fig. 2). The density of DNMs within 4 Mb of the breakpoint junctions was significantly higher than expected based on the number of DNMs and size of the genome (Additional file 1: Table S5, Fig. 2d) and was also significantly higher than the density of DNMs at the same genomic location in approximately 3000 control genomes [18]. We considered only the phased DNMs in cis with dnCNVs, which revealed that the observed density of DNMs was significantly higher than expected for either the maternal or paternal inherited haploid genome consistent with the hypermutation hypothesis [14, 31] of an underlying error-prone repair mechanism, microhomology-mediated break-induced replication (MMBIR) producing both CNVs and local SNVs (Additional file 1: Table S5).

De novo variants detected in BAB9637. a Ratio of transition to transversion is shown at the top of the bar graph. The bar graph represents the relative contribution of types of SNV. b Horizontal red bars represent each dnCNV that is associated with DNMs in proximity to the breakpoint. All seven DNMs found within 4 Mb of the breakpoints are highlighted with a star at the relative location. Maternal and paternal DNMs are highlighted in pink and green, respectively. The DNM at chr4, chr6, chr12, chr10, and chr14 are in cis with dnCNVs. c Sanger traces are visualized (proband, mother, and father) for DNMs. * The variant at the breakpoint junction. d Density of unphased DNM corresponding to the window size highlighting continuous drop-off of observed DNM after 4 Mb
Elevated post-zygotic de novo substitution rate in MdnCNV families
In addition to the new MdnCNV family (BAB9637), four previously reported MdnCNV families (BAB3097, BAB3596, mCNV3/BAB9484, and mCNV7, Table 1) were genome sequenced with an average depth of coverage of 40 × . Through clinical array analysis, five anonymized families ascertained without MdnCNV were included as controls. We identified 470 (transition to transversion ratio [Ti:Tv] = 2.2, 95% confidence interval (CI) = 1.62–2.30) and 361 ([Ti:Tv] = 2.2, CI = 1.29–3.1) high-confidence de novo substitutions in MdnCNV and control families, respectively. SNV substitution mutations that occur during early development can lead to mosaicism in peripheral blood leukocyte-derived DNA, which will change the expectation of variant allele fractions (VAFs), e.g., somatic mutation arising during first cell division will have expected VAFs of 25%. The de novo substitutions were classified into potential germline mutation, or post-zygotic mutation based on the variant read to total read ratio (Fig. 3a, b, and “Methods”). The number of potential germline substitutions in MdnCNV families appears to be comparable (p = 0.1, F-test) to control families with an average paternal age effect of 1.9 (95% confidence interval 1.32–2.54) (Fig. 3c, d). However, the proportion of potential post-zygotic substitutions appears to be higher (z-test, p = 0.004) in MdnCNV (8.0%, n = 36) versus control families (3.2%, n = 11). The paternal age effect on the number of potential post-zygotic variants was not significant for either group.

The number and mutational pattern of pre-zygotic and post-zygotic de novo mutations in MdnCNV families (blue) versus controls (orange). a The VAF distribution of de novo substitutions in MdnCNV (blue) and control (orange) families. b The proportion of DNMs that are predicted to be post-zygotic mutations (dark orange/blue). c The number of pre-zygotic (germline) mutations is positively correlated with paternal age. The gray area denotes the region covered by the 95% confidence interval of the slope and intercept of the linear regression lines. d The number of post-zygotic mutations shows no correlation with age
Mutation pattern analysis for DNMs
We cataloged 466 de novo substitutions into seven different mutation types, including the six possible single base-pair substitutions and one category representing CpG deamination. The mutation pattern of germline substitutions in MdnCNV families does not appear to be different from controls, with ~ 40% of the germline mutations being C > T transition variants (Additional file 1: Figure S5). This is consistent with the elevated rate of spontaneous deamination of 5-methyl-cytosine, which occurs at CpG dinucleotides [32].
A higher-resolution mutation substitution pattern analysis was performed on genome-wide dnSNVs to investigate the contribution of validated mutational signatures extracted from somatic mutation analysis of the cancer genome [12]. We found that SBS5 and SBS1 explain the majority of genome-wide dnSNVs in all the control and three of MdnCNV probands (BAB9484, BAB9637, and mCNV7, Fig. 4d, Additional file 2: Table S2). In contrast, the other mutational signatures (SBS10b, SBS26, and SBS39) were observed in two MdnCNV cases (BAB3097 and BAB3596) (Fig. 4d). To exclude potential context bias around the dnCNV region, e.g., C > T at CpG for TCG context, mutation signatures were reassessed on dnSNV not present within dnCNV and 1 Mb distance flanking the breakpoints. After reassessment, SBS1, SBS26, and SBS39 are still associated with BAB3596. SBS39, SBS10b, and SBS37 instead of SBS39, SBS10b, and SBSB26 are associated with BAB3097 (Fig. 4d and Additional file 1: Figure S6), which suggests a nonspecific association of SBS26 with BAB3097.

Maternal variants potentially contributing to genome instability. a–c MdnCNV pedigrees with identified rare VUS maternal variants affecting genes involved in DNA repair or replicaion. d Bar plot shows the contribution of SBS signatures refitted by on genome-wide dnSNV. Predicted protein structure plots show the amino acid change in proximity to previously reported variants in protein ERCC4 (e) and MSH3 (f). Molecular modeling images were acquired from Varsite [33], with pathogenetic variants from ClinVar mapped. The amino acid residues in red reveal the change caused by variants reported here and the ones in purple or gray reveal the reported pathogenetic variants from ClinVar
Rare variant analyses in the maternal genome identified deleterious variants in three genes involving DNA repair pathways from three MdnCNV families (HOU1209, HOU3425, and HOU3579, Fig. 4a, b and c). The POLE variant (NM_006231:exon39:c.5347G > A:p.D1783N) identified in HOU1209 family (Fig. 4a) is ultrarare in a population database (rs149893630, gnomAD 0.001%). In silico analysis supports that this missense variant has a deleterious effect on protein structure/function (Polyphen2: Probably Damaging; SIFT: Deleterious; CADD v1.6 = 29.4). We also identified variants in ERCC4 and MSH3 from two other MdnCNV families (HOU3425 and HOU3579, Fig. 4b,c). Both variants (ERCC4:NM_005236:exon3:c.476G > A:p.G159D and MSH3:NM_002439:exon6:c.938C > A:p.A313E) are absent from the gnomAD database and are predicted to be pathogenic (Polyphen2: Damaging; Damaging; SIFT: Deleterious, Deleterious; CADD v1.6 = 29.5 and 33.0, respectively). The amino acid residue at both sites are highly conserved from the 161-aligned and 144-aligned protein sequence based on predictions from the tool Varsite [33]. The mutated residues are in proximity to the reported sequences with predicted functional consequences based on 3D protein models (Fig. 4e,f) from Varsite [33] and MichelaNglo [34].
Phenotypic variability derived from variable regions affected by dnCNV in the MdnCNV cohort
While probands described with the MdnCNV phenomenon have a similar molecular finding on analysis for CNV in their genomes, their phenotypic features observed at clinical evaluation are variable. Within the cohort of five individuals with MdnCNV phenotype, three patients have detailed clinical information available (Supplementary text) for genotype and phenotype analysis. To discern whether phenotypic variability results from the variable regions of the genome affected by the MdnCNV phenomenon, HPO-based evaluation was also performed on the three MdnCNV probands (BAB9637, mCNV3/BAB9484, and BAB3097).
Concerning BAB9637, we classified the de novo duplication encompassing NSD1 associated with Sotos syndrome (MIM#117550) as a pathogenic variant, based on the most recent consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) (Table 2) [35]. The remaining dnCNVs (7/8) were classified as variants of unknown significance (VUS). To investigate the contribution of the other dnCNV encompassing genes to patient phenotype, we performed Lin semantic similarity analysis of BAB9637’s phenotype to OMIM/Orphanet HPO annotated gene and disease-associated phenotypes. The Lin semantic similarity scores [27] suggest that, of the genes encompassed by dnCNV regions, NSD1 and SMARCC2 were the two with the highest phenotypic similarity score (0.60 and 0.59, respectively) to the proband’s phenotype (Additional file 1: Table S6). We compared the phenotype similarity of BAB9637 with previously reported probands with only NSD1 variants (30 probands with NSD1 duplication, 30 probands with NSD1 deletion/LoF variant) or only SMARCC2 variants. Three clusters were observed, one consisting of all 30 included NSD1 deletion/LoF probands, the second of 25/30 (81%) of reported NSD1 duplication probands, and the third containing 16/16 SMARCC2 LoF probands, 5/30 of NSD1 duplication probands, and BAB9637 (Fig. 5a).

Phenotype similarity score analysis for disease-associated genes and potential gene combinations for the multiple pathogenic variant case (BAB9637). a Heatmap representing color-coded Lin semantic similarity scores of BAB9637 and database annotated phenotypes included. Both rows and columns are clustered using pairwise similarity scores and the Ward's method. The dendrogram is present at the top and to the left of the heatmap. Colored columns are depicted at the bottom and to the right and annotate variant type and affected gene as defined at the bottom. b Distribution of similarity scores in each known disease-associated gene group with n = 1 to n = 5 genes. A black line connects the max score of subsequent subsets of groups, e.g., max score of groups with one gene to max score of groups with two genes. c Annotation grid demonstrates individual reported NSD1 duplication proband phenotypes, individual SMARCC2 LoF proband phenotypes, NSD1 DUP and SMARCC2-associated clinical phenotypes summary, and proband phenotypes for BAB9637. From left to right aligned HPO phenotype, blue squares indicate the presence of the phenotype, i.e., HPO term, while gray represents the absence of the term. The clinical phenotype summary was based on the cases used in HPO analysis, with the degree of shading indicating the percent of reported cases shown here for which a particular feature has been observed, as defined in the legend in the top right corner
We evaluated whether the observed phenotypes of BAB9637 are better explained by a combinatorial effect of two or more affected genes, i.e., dual molecular diagnosis or MPV, multilocus pathogenic variation. The combination of NSD1 and SMARCC2 yielded the highest similarity score (0.64) among all pairwise gene combinations (Fig. 5b). Compared with a single gene model, a dual molecular diagnosis model improved the phenotypic similarity score by > 5%. The increase in phenotypic similarity score was not substantial in the 3rd, the 4th, or the 5th gene (~ 1%) compared to the dual molecular diagnosis model (Additional file 1: Table S6). A review of the phenotypes associated with NSD1 and SMARCC2 in grid format reveals that their phenotypic spectrums largely overlap (Fig. 5c). The grid visualization also highlights the presence of phenotypes in the proband that has predominantly or only been observed in NSD1 probands (e.g., absent/small foot bones) or in SMARCC2 probands (e.g., thick eyebrows, inguinal hernia; Fig. 5c). The combination of phenotypic contributions from defects caused by both NSD1 and SMARCC2, therefore, offers the most parsimonious explanation to account for the overall phenotypic presentation in the patient, supporting a multilocus pathogenic variation (MPV) model of blended phenotypic traits caused by perturbation of genes at both loci to constitute the proband’s phenotype.
For the BAB3097 MdnCNV case, two de novo duplication CNV, one (17p12p11.2) with two genes within a contiguous duplication, RAI1 and PMP22 associated with Yuan-Harel-Lupski Syndrome (YUHAL; MIM#616652) [36], and the other (1p36.22p36.13) encompassing established haploinsufficiency gene SPEN, meet ACMG criteria for pathogenicity (pathogenic/likely pathogenic) and appear to be the clinically relevant genes within these CNV intervals (Table 2). RAI1 has the highest similarity score (Lin similarity score 0.52), and PMP22 ranks 11 (Lin similarity score 0.39) among dnCNV encompassed genes (Additional file 1: Table S6). The proband phenotype was analyzed for phenotypic similarity with 17 reported YUHAL cases and OMIM diseases associated with either RAI1 or PMP22. Three clusters were observed, one consisting of 12/17 YUHAL probands, the second of 5/17 of reported YUHAL probands and BAB3097, and the last one containing PMP22 associated neurological diseases (Additional file 1: Figure S7a). The phenotypic grid suggests 7/11 of BAB3097’s observed phenotype matches with YUHAL associated phenotypes reported in Yuan et al. (Additional file 1: Figure S7b) [36]. Additional genes with a high phenotypic similarity score include FLCN (Lin similarity score 0.50, ranked second) located at 17p12p11.2 and SPEN (Lin similarity score 0.45, ranked fourth) at 1p36.22p36.13 (Additional file 1: Table S6). Among all dual molecular diagnosis combinations, 13 have more than 5% improvement of similarity scores over single gene similarity scores. Of those 13 combinations, all have at least one gene within the two regions (17p12p11.2 and 1p36.22p36.13), and 8/13 are made up exclusively of genes within the same regions (Additional file 1: Table S6).
For mCNV3/BAB9484, we classify the de novo duplication at 3q26.32 as likely pathogenic based on ACMG guidelines (Table 2). TBL1XR1 was the only coding gene at the duplication region and had the highest phenotypic similarity score (Lin semantic similarity score = 0.50) among all dnCNV encompassed genes. The second highest gene is EXT2 (Lin similarity score = 0.39). Of all dual molecular diagnosis combinations, three have more than 5% improvement of similarity score over a single gene (Additional file 1: Table S6). The phenotype grid suggests 3/10 exact match of TBL1XR1 associated terms, including hypertelorism, brachycephaly, and wide intermammillary distance (Additional file 1: Figure S8).
Discussion
Our study utilized multimodal genomic approaches to investigate de novo genomic rearrangements in an MdnCNV family, characterize the mutational event that instigated MdnCNV across the genome, identify de novo SNV genome-wide, phase CNV and SNV haplotypes, and explore potential driver genes contributing to the observed clinical phenotype. Accurately capturing structural variants and complexities generated during CNV mutagenesis, including novel DNA rearrangement junctions or DNA break/join-points and other SNV mutational signatures of CNV mutagenesis, is challenging given the length limit of sequencing reads in SR genome sequencing. Genome-wide ascertainment of dnCNVs, breakpoint junction sequence, and dnSNVs were enabled by implementing both LR and SR genome sequencing. LR sequencing, either PacBio [14] or Nanopore as shown here, allows direct phasing of variants multiple kilobases apart, which in turn allows direct observation of dnSNV in cis with breakpoint junctions.
The average size of tandem duplication observed in the newly identified MdnCNV individual is around 1 Mb, consistent with the observation in the five reported MdnCNV individuals [4]. The size is also within a similar size range “window” of replication domains [37] and topologically associating domains (TAD) [38]. Recent study suggests the replication domain boundary is associated with TAD [39]. The size of tandem duplication can be limited within the boundary of the genome organization, e.g., the three-dimensional genome folding and accessibility of DNA during zygotic development. Further analysis on the dynamics of chromatin architecture during zygote development is warranted to address potential influence or constraints on rearrangement size.
We postulate that the CNV generating mechanism could be MMBIR, based on the shared characteristics of microhomology/microhomeology at breakpoint junctions, complexity in the form of associated indels, and the increased occurrence of base substitutions and indels within 4 Mb of junctions. MMBIR, a form of break-induced replication, occurs when a replication fork has collapsed/stalled and restarted. Alternatively, the unrepaired DNA nicks could result in a collapsed fork that is resolved by a mixed NHEJ/MMBIR mechanism, e.g., restart-bypass [40]. Regardless of which mechanism(s) are involved, we suggest that there were multiple broken replication forks in a single zygotic cell and that the MdnCNV event, therefore, may have resulted from cell-wide replication stress, such as energy or substrate unavailability that resulted in multiple broken replication forks.
We found that the mutation rate for dnCNV and substitution was elevated in five MdnCNV families. The post-zygotic mutational events happened at the early development stage, e.g., first cell division has a VAF of 10–35% [41]. The mutation rate during early embryonic development is higher than in germline cells, expecting 2–3 substitutions per generation [41]. The post-zygotic substitution rate in MdnCNV families is three times higher than the rate in control (z-test, p = 0.004, Table 1). The mechanism(s) causing the CNV mutator phenotype is thought to be due to a transient mutagenesis event restricted to the peri-zygotic stage of development [4]. We suspect the mechanism(s) could also lead to SNV hypermutation limited to the peri-zygotic period. However, high-depth genome sequencing is required for the estimation of mosaicism accurately and further supports this hypothesis.
The dnSNV mutational pattern and rare variant analysis suggest multiple mechanisms contributing to the transient peri-zygotic genome instability causing the MdnCNV phenomenon. One of the mechanisms could be that non-inherited variants in the maternal genome affect zygotic genome integrity, whereby maternal mRNA stored in the oocyte impacts the first few cell divisions during embryonic development. Rare variant analyses identified maternal variants in three DNA repair genes POLE, ERCC4, and MSH3 with predicted deleterious effects on the function that could contribute to peri-zygotic genome instability. The gene POLE encodes the catalytic subunit of polymerase ε which plays a major role in the DNA replication [42]. The POLE variant identified in family HOU1209 is located in the C-terminal structure of the subunit which is essential for replisome assembly and checkpoint activation [43, 44]. The maternal variants identified in family HOU3425 (ERCC4, NM_005236:c.476G > A, p.G159D) and HOU3579 (MSH3, NM_002439:c.938C > A, p.A313E) may contribute to replication stress through different mechanisms. ERCC4 (MIM#133520, also known as XPF) encodes the endonuclease catalytic subunit that is involved in nucleotide excision repair (NER) and removes DNA interstrand crosslinking damage [45, 46]. The interstrand crosslink can prevent DNA strand separation and physically block DNA replication and transcription, leading to genomic instability. The mutated residue (ERCC4, NM_005236:c.476G > A, p.G159D) is near two reported functional residues that are relevant to the crosslink repair activity of ERCC4 (Fig. 4e). The nearest one (rs121913050, p.R153P), located 2.2 Å away from the mutated residue, is classified as pathogenic in ClinVar and causes XFE progeroid syndrome (MIM#610965) in a homozygous state [47, 48]. The second variant (rs145402255, p.R150C), located 10.8 Å away from the mutant residue is classified as likely pathogenic in ClinVar. In vitro cell modeling suggests the variant (rs145402255, p.R150C) can mildly disrupt the interstrand crosslink repair activity [49]. MSH3 encodes a protein that forms a heterodimer with MSH2 that is responsible for mismatch repair (MMR) and double-stranded DNA repair [50, 51]. In summary, these variants may represent genetic modifiers rather than causative drivers that may contribute to the MdnCNV phenomenon. Additionally, 4/5 MdnCNV families (Table 1) were of advanced parental age (> 35 years old) at the time of conception, which may have reduced the capacity of DNA repair in oocytes and therefore have a contributory effect on the genomic stability of the zygote.
Mutational signature analysis can be used to decipher the potential mutational processes underlying individual cancer or germline hypermutation [52, 53]. A recent study has revealed genetic and environmental contributions to the germline dnSNV hypermutation [53]. Our study explored the utility of mutational signature analysis to decode potential mutational processes in the context of dnCNV hypermutation. However, further studies are still warranted to understand the mutational process(s) during the peri-zygotic development stage. Mutational signature analysis on embryonic somatic mutations (SNVs and CNVs) may help solve the puzzle. Additionally, further studies are needed to confirm mutational signatures within certain genes or allele-specific effects.
Reciprocal copy number changes in dosage-sensitive gene loci can manifest traits on the opposite ends of a phenotypic spectrum, a phenomenon known as mirror traits [54]. The quantitative trait for head size, or occipital frontal circumference (OFC), illustrates this concept: reciprocal CNVs at multiple loci can drive mirror trait expression manifesting as large (macrocephaly) or small (microcephaly) head size [54,55,56]. Semantic similarity analysis using HPO terms can quantify the similarity of matching phenotype, therefore objectively teasing out the contribution of genes within dnCNV regions. The similarity score correctly ranked the established genes in the dnCNV region and suggested additional genes from the other regions contributing to two of the MdnCNV cases. Quantitative analyses of the clinical phenotypes in BAB9637 versus known disease genes mapping within the duplications implicate NSD1 as a triplosensitivity trait locus that contributes to the clinical phenotype observed. The 5q35 microdeletion causing haploinsufficiency of NSD1 has been associated with Sotos syndrome (MIM#117550). The characteristic clinical features of Sotos syndrome include overgrowth, characteristic facial dysmorphisms, intellectual disability (ID), developmental delay (DD), and macrocephaly. The proband (BAB9637) has overlapping features, including DD and ID. However, regarding the quantitative traits of head circumference and height, the proband (lower-than-average head circumference [Z = − 0.62]; short stature [Z = − 3.01]) with a duplication encompassing NSD1 lies on the opposite end of the phenotypic spectrum compared to individuals with a diagnosis of Sotos syndrome. Furthermore, the clinical presentation of our proband, which includes DD/ID, and short stature, is consistent with that of 31 affected individuals previously reported across multiple studies [57] to have a de novo or familial duplication of the genomic region whose deletion is commonly associated with the Sotos syndrome (Fig. 5, Additional file 1: Figure S9).
The presence of two highly similar clusters for NSD1 duplication and SMARCC2, and their separation from a more dissimilar NSD1 deletion/LoF cluster, highlights the shared phenotypic trait between NSD1 duplication and SMARCC2 associated phenotypes. The contribution of NSD1 does not seem to explain all phenotypes observed in the proband, e.g., scoliosis and craniofacial features. Haploinsufficiency of SMARCC2 has been associated with Coffin-Siris syndrome 8 (MIM#618362). SMARCC2 duplication is ultrarare, less than 0.0001 in one control study [58], and absent in the personal genomes of neurotypical individuals from population databases (gnomAD v2.1.1). SMARCC2 duplication has been reported in three cases (size of duplication < 1 Mb) in the DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources (DECIPHER, https://www.deciphergenomics.org/) [59] (Additional file 1: Figure S9). Two genomes with duplications from unrelated patients have a phenotype described, one (DECIPHER patient ID: 343437) in an individual with growth delay and ID, the second duplication (DECIPHER patient ID: 260552) inherited from a parent who has a similar phenotype, including hypertelorism, hypospadias, broad thumb, delayed cranial suture closure, hypotonia, and ID. Data on segregation of duplication variants in DECIPHER individuals was not available.
The potential molecular diagnoses underlying BAB3097’s phenotype highlight another case where de novo CNV at more than one locus contributes to disease pathobiology. Contiguous gene duplication encompassing RAI1 and PMP22 has been previously described in 17 patients with YUHAL syndrome [36]. Renal phenotypes were observed in 4/17 of patients with Yuan-Harel-Lupski (YUHAL) syndrome, and while a definitive gene within the duplication interval was not associated with renal phenotypes, FLCN was suggested to be a potential contributor. In addition to the duplication at this locus, our analysis suggests duplication at 1p36.22p36.13 may also contribute to the phenotype, with SPEN as the potential driver gene. The gene SPEN is located at the 1p36 deletion syndrome critical region. Furthermore, truncated variants at SPEN suggest the haploinsufficiency of SPEN associated with neurodevelopment phenotype, congenital heart defects, and facial dysmorphism [60]. The whole gene duplication of SPEN was absent from DGV and gnomAD.
Further study of the phenotypic effect of SMARCC2 and SPEN duplication in more patients, rather than deletion, is necessary to understand dosage sensitivity (i.e., triplosensitivity) at these loci; however, these data in aggregate support the contention that the phenotype of the proband is a blended overlapping phenotype driven by multilocus pathogenic variation (MPV) [21, 61], i.e., duplication of the NSD1 and SMARCC2 loci, and their associated traits. Our understanding of emerging concepts, such as which genes or loci are dosage-sensitive, whether haploinsufficiency and triplosensitivity traits will be observed for a given dosage-sensitive gene, and the correlation between dosage-sensitive genes and mirror traits is continuing to evolve with human genetics and genomics studies.
While semantic similarity analysis with patient phenotypes can objectively tease out the contribution of genes within dnCNV regions, there are some caveats to such analysis. First, the variable depth of phenotypic information could affect the clustering, e.g., the unavailability of nerve conduction studies in BAB9484 affecting phenotypic match to previously reported YUHAL probands which were extensively evaluated clinically [36]. Another caveat is that portions of these proband phenotypes may be due to genes encompassed by CNV that are not yet associated with disease in humans or by other yet unrecognized genetic modifiers/pathogenic SNV contributing to the phenotype. Nevertheless, aggregate data of quantitative phenotypic analysis of MdnCNV probands suggests that at least some phenotypic variability is likely explained by dosage changes of genes encompassed by CNV genome-wide in probands. These cases also highlight the possibility of multiple genes, in one case as part of a contiguous gene duplication of PMP22 and RAI1 and in another as duplications of NSD1 and SMARCC2 at separate loci undergoing de novo gains in a single generation to contribute to phenotype manifestation.
Conclusions
Characterization of the MdnCNV phenomenon using a multimodal genomic approach revealed insights that the MdnCNV mutational event likely occurs in the earliest post-zygotic stages of development—potentially in the pronuclear phase or during the first few cell divisions. Moreover, we show the utility of quantitative phenotypic analysis to identify contributory, disease-associated genes within a background of genome-wide dnCNVs and provide evidence for duplications at two genomic loci containing triplosensitive genes that contribute to the patient’s blended phenotype.
Availability of data and materials
The data generated or analyzed during this study are included in this published article. The array data has been submitted to GEO (GSE176427) [62]. The variant data of the study are available with ClinVar accession numbers: SCV002576315—SCV002576337.
Abbreviations
- aCGH:
-
Array comparative genomic hybridization
- dnCNVs:
-
De novo copy number variants
- DNMs:
-
De novo Mutations
- dnSNVs:
-
De novo single-nucleotide variants
- HPO:
-
Human Phenotype Ontology
- MMBIR:
-
Microhomology-mediated break-induced replication
- MPV:
-
Multilocus pathogenic variation
- NHEJ:
-
Non-homologous end joining
- OFC:
-
Occipital frontal circumference
- SNPs:
-
Single-nucleotide polymorphisms
References
Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, et al. A structural variation reference for medical and population genetics. Nature. 2020;581:444–51.
Belyeu JR, Brand H, Wang H, Zhao X, Pedersen BS, Feusier J, et al. De novo structural mutation rates and gamete-of-origin biases revealed through genome sequencing of 2,396 families. Am J Hum Genet. 2021;108:597–607.
Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ, et al. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–81.
Liu P, Yuan B, Carvalho CMB, Wuster A, Walter K, Zhang L, et al. An organismal CNV mutator phenotype restricted to early human development. Cell. 2017;168:830-842.e7.
Köhler S, Gargano M, Matentzoglu N, Carmody LC, Lewis-Smith D, Vasilevsky NA, et al. The human phenotype ontology in 2021. Nucleic Acids Res. 2021;49:D1207–17.
Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med. 2005;7:422–32.
Wiszniewska J, Bi W, Shaw C, Stankiewicz P, Kang S-HL, Pursley AN, et al. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur J Hum Genet. 2014;22:79–87.
Carvalho CMB, Zhang F, Liu P, Patel A, Sahoo T, Bacino CA, et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum Mol Genet. 2009;18:2188–203.
Farek J, Hughes D, Mansfield A, Krasheninina O, Nasser W, Sedlazeck FJ, et al. xAtlas: Scalable small variant calling across heterogeneous next-generation sequencing experiments. bioRxiv. 2018. p. 295071. Available from: https://www.biorxiv.org/content/biorxiv/early/2018/04/05/295071. [cited 2022 Aug 29]
Eldomery MK, Coban-Akdemir Z, Harel T, Rosenfeld JA, Gambin T, Stray-Pedersen A, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:26.
Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10:33.
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101.
Patterson M, Marschall T, Pisanti N, van Iersel L, Stougie L, Klau GW, et al. WhatsHap: weighted haplotype assembly for future-generation sequencing reads. J Comput Biol. 2015;22:498–509.
Beck CR, Carvalho CMB, Akdemir ZC, Sedlazeck FJ, Song X, Meng Q, et al. Megabase length hypermutation accompanies human structural variation at 17p11.2. Cell. 2019;176:1310–132410.
Zarate S, Carroll A, Krashenina O, Sedlazeck FJ, Jun G, Salerno W, et al. Parliament2: fast structural variant calling using optimized combinations of callers. bioRxiv. 2018. p. 424267. Available from: https://www.biorxiv.org/content/biorxiv/early/2018/09/23/424267. [cited 2022 Aug 29]
Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods. 2018;15:461–8.
Cer RZ, Donohue DE, Mudunuri US, Temiz NA, Loss MA, Starner NJ, et al. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res. 2013;41:D94-100.
Halldorsson BV, Palsson G, Stefansson OA, Jonsson H, Hardarson MT, Eggertsson HP, et al. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science. 2019;363. Available from: https://doi.org/10.1126/science.aau1043
Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–63.
Gel B, Serra E. karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics. 2017;33:3088–90.
Herman I, Jolly A, Du H, Dawood M, Abdel-Salam GMH, Marafi D, et al. Quantitative dissection of multilocus pathogenic variation in an Egyptian infant with severe neurodevelopmental disorder resulting from multiple molecular diagnoses. Am J Med Genet A. 2022;188:735–50.
Zhang C, Jolly A, Shayota BJ, Mazzeu JF, Du H, Dawood M, et al. Novel pathogenic variants and quantitative phenotypic analyses of Robinow syndrome: WNT signaling perturbation and phenotypic variability. HGG Adv. 2022;3:100074 (Elsevier BV).
Lima AR, Ferreira BM, Zhang C, Jolly A, Du H, White JJ, et al. Phenotypic and mutational spectrum of ROR2-related Robinow syndrome. Hum Mutat. 2022;43:900–18.
Liu C, Peres Kury FS, Li Z, Ta C, Wang K, Weng C. Doc2Hpo: a web application for efficient and accurate HPO concept curation. Nucleic Acids Res. 2019;47:W566–70 (Oxford Academic).
Köhler S, Carmody L, Vasilevsky N, Jacobsen JOB, Danis D, Gourdine J-P, et al. Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019;47:D1018–27.
Greene D, Richardson S, Turro E. ontologyX: a suite of R packages for working with ontological data. Bioinformatics. 2017;33:1104–6 (Oxford Academic).
Lin D. An information-theoretic definition of similarity. In Proceedings of the 15th International Conference on Machine Learning. 1998. Available from: http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.55.1832. [cited 2022 Aug 29]
Dixon JR, Xu J, Dileep V, Zhan Y, Song F, Le VT, et al. Integrative detection and analysis of structural variation in cancer genomes. Nat Genet. 2018;50:1388–98.
Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–5.
Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33.
Carvalho CMB, Pehlivan D, Ramocki MB, Fang P, Alleva B, Franco LM, et al. Replicative mechanisms for CNV formation are error prone. Nat Genet. 2013;45:1319–26.
Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274:775–80.
Laskowski RA, Stephenson JD, Sillitoe I, Orengo CA, Thornton JM. VarSite: disease variants and protein structure. Protein Sci. 2020;29:111–9.
Ferla MP, Pagnamenta AT, Damerell D, Taylor JC, Marsden BD. MichelaNglo: sculpting protein views on web pages without coding. Bioinformatics. 2020;36:3268–70.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22:245–57.
Yuan B, Harel T, Gu S, Liu P, Burglen L, Chantot-Bastaraud S, et al. Nonrecurrent 17p11.2p12 rearrangement events that result in two concomitant genomic disorders: the PMP22-RAI1 contiguous gene duplication syndrome. Am J Hum Genet. 2015;97:691–707.
Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang C-W, et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245.
McArthur E, Capra JA. Topologically associating domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability. Am J Hum Genet. 2021;108:269–83.
Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515:402–5.
Willis NA, Frock RL, Menghi F, Duffey EE, Panday A, Camacho V, et al. Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature. 2017;551:590–5.
Ju YS, Martincorena I, Gerstung M, Petljak M, Alexandrov LB, Rahbari R, et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017;543:714–8.
Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315.
Handa T, Kanke M, Takahashi TS, Nakagawa T, Masukata H. DNA polymerization-independent functions of DNA polymerase epsilon in assembly and progression of the replisome in fission yeast. Mol Biol Cell. 2012;23:3240–53.
Zhou JC, Janska A, Goswami P, Renault L, Abid Ali F, Kotecha A, et al. CMG-Pol epsilon dynamics suggests a mechanism for the establishment of leading-strand synthesis in the eukaryotic replisome. Proc Natl Acad Sci U S A. 2017;114:4141–6.
Bergstralh DT, Sekelsky J. Interstrand crosslink repair: can XPF-ERCC1 be let off the hook? Trends Genet. 2008;24:70–6.
Wood RD. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ Mol Mutagen. 2010;51:520–6.
Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–43.
Narang A, Uppilli B, Vivekanand A, Naushin S, Yadav A, Singhal K, et al. Frequency spectrum of rare and clinically relevant markers in multiethnic Indian populations (ClinIndb): a resource for genomic medicine in India. Hum Mutat. 2020;41:1833–47.
Osorio A, Bogliolo M, Fernández V, Barroso A, de la Hoya M, Caldés T, et al. Evaluation of rare variants in the new fanconi anemia gene ERCC4 (FANCQ) as familial breast/ovarian cancer susceptibility alleles. Hum Mutat. 2013;34:1615–8.
Kumar C, Williams GM, Havens B, Dinicola MK, Surtees JA. Distinct requirements within the Msh3 nucleotide binding pocket for mismatch and double-strand break repair. J Mol Biol. 2013;425:1881–98.
Reynolds MF, Peterson-Roth EC, Bespalov IA, Johnston T, Gurel VM, Menard HL, et al. Rapid DNA double-strand breaks resulting from processing of Cr-DNA cross-links by both MutS dimers. Cancer Res. 2009;69:1071–9.
Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–59.
Kaplanis J, Ide B, Sanghvi R, Neville M, Danecek P, Coorens T, et al. Genetic and chemotherapeutic influences on germline hypermutation. Nature. 2022;605:503–8 (Nature Publishing Group).
Lupski JR. Structural variation mutagenesis of the human genome: Impact on disease and evolution. Environ Mol Mutagen. 2015;56:419–36.
Ricard G, Molina J, Chrast J, Gu W, Gheldof N, Pradervand S, et al. Phenotypic consequences of copy number variation: insights from Smith-Magenis and Potocki-Lupski syndrome mouse models. PLoS Biol. 2010;8:e1000543.
Heck DH, Gu W, Cao Y, Qi S, Lacaria M, Lupski JR. Opposing phenotypes in mice with Smith-Magenis deletion and Potocki-Lupski duplication syndromes suggest gene dosage effects on fluid consumption behavior. Am J Med Genet A. 2012;158A:2807–14.
Quintero-Rivera F, Eno CC, Sutanto C, Jones KL, Nowaczyk MJM, Wong D, et al. 5q35 duplication presents with psychiatric and undergrowth phenotypes mediated by NSD1 overexpression and mTOR signaling downregulation. Hum Genet. 2021;140:681–90.
Coe BP, Witherspoon K, Rosenfeld JA, van Bon BWM, Vulto-van Silfhout AT, Bosco P, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46:1063–71 (Nature Publishing Group).
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet. 2009;84:524–33.
Radio FC, Pang K, Ciolfi A, Levy MA, Hernández-García A, Pedace L, et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am J Hum Genet. 2021;108:502–16.
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, CobanAkdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21–31 (Massachusetts Medical Society).
Du H, Jolly A, Grochowski CM, Carvalho CMB, Liu P. Multiple de novo copy number variant (MdnCNV) driven mirror traits and blended phenotype. Gene Expression Omnibus GSE176427. Available from: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE176427. [cited 2022 Sep 30]
Acknowledgements
We thank the family and collaborators for their participation in this study.
Funding
Supported in part by US National Institutes of Health, National Human Genome Research Institute (NHGRI)/National Heart, Lung, and Blood Institute (NHLBI) UM1 HG006542 to the Baylor Hopkins Center for Mendelian Genomics, NHGRI U54 HG003273 and NHGRI UM1 HG008898 to RAG, the NHGRI Genomic Research Elucidates Genetics of Rare disease (GREGoR) consortium U01 HG011758 to JEP, JRL, and RAG. NHGRI K08 HG008986 to JEP; the National Institute of General Medical Sciences (NIGMS R01 GM132589 to CMBC and R01 GM106373 to PH and JRL); and the National Institute for Neurological Disorders and Stroke (NINDS R35 NS105078 to JRL); NHGRI R35HG011311 to P.L.
Author information
Authors and Affiliations
Contributions
Conceptualization: H.D., C.M.G., A.J., J.R.L., P.L., and C.C.; Data curation: S.N.J., C.M.G., J.M.F., M.P., S.J., E.H., B.Y., and H.L.; Formal analysis: H.D., C.M.G., and A.J.; Funding acquisition: J.R.L., C.C., P.J.H., R.A.G., P.L., and J.E.P; Visualization: H.D., A.J., C.M.G., and J.M.F.; Methodology: C.C., F.J.S., and Z.C.A.; Resources: M.E.C., A.E.S., and J.E.P.; Supervision: C.C., J.R.L., and P.L.; Writing—original draft: H.D.; Writing—review and editing: H.D., A.J., C.M.G., J.R.L., P.L., C.C., and P.J.H. All author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from the families of BAB9637, BAB3097, and BAB9484 in the research study. This study was approved by the Institutional Review Board (IRB) at Baylor College of Medicine (protocols H-29697 and H-42680). A waiver of consent was approved by the study for investigation of the other families included in this study because the analysis and publication of the human subject genomics data have been de-identified and present minimal risks to the study subjects. The research activities in this study conformed to the principles of the Helsinki Declaration.
Consent for publication
Written consent was obtained to publish detailed clinical information for the families of BAB9637, BAB3097, and BAB9484. A waiver of consent was approved by the study IRB to publish a de-identified synopsis of the clinical summary of the other families using HPO terms.
Competing interests
Baylor College of Medicine (BCM) and Miraca Holdings have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), which performs clinical chromosome microarray analysis (CMA) and other genomic studies (ES, genome sequencing) for patient/family care. J.R.L. serves on the Scientific Advisory Board of BG. J.R.L. has stock ownership in 23andMe, is a paid consultant for the Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. PL and WB are employees of BCM and derive support through a professional service agreement with BG. MP, EH, and SJ are employees of Oxford Nanopore Technologies and are shareholders and/or share option holders. FJS has multiple travels sponsored by Pacbio and ONT. The remaining authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary methods, clinical description of BAB9637, BAB3097, and BAB9484, Table S1, S3, S5, S6, Figure S1-S9.
Additional file 3: Table S4.
dnSNVs in the proband (not include the dnSNV within 150bp to the breakpoint).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Du, H., Jolly, A., Grochowski, C.M. et al. The multiple de novo copy number variant (MdnCNV) phenomenon presents with peri-zygotic DNA mutational signatures and multilocus pathogenic variation. Genome Med 14, 122 (2022). https://doi.org/10.1186/s13073-022-01123-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-022-01123-w