
Abstract
Genome-wide association studies (GWAS) have uncovered thousands of genetic variants that influence risk for human diseases and traits. Yet understanding the mechanisms by which these genetic variants, mainly noncoding, have an impact on associated diseases and traits remains a significant hurdle. In this review, we discuss emerging experimental approaches that are being applied for functional studies of causal variants and translational advances from GWAS findings to disease prevention and treatment. We highlight the use of genome editing technologies in GWAS functional studies to modify genomic sequences, with proof-of-principle examples. We discuss the challenges in interrogating causal variants, points for consideration in experimental design and interpretation of GWAS locus mechanisms, and the potential for novel therapeutic opportunities. With the accumulation of knowledge of functional genetics, therapeutic genome editing based on GWAS discoveries will become increasingly feasible.
Similar content being viewed by others


Background
Recent genome-wide association studies (GWAS), in which millions of genetic variants across the full allele frequency spectrum are subject to genotype-phenotype association tests, have provided insights into the genetic architecture of complex diseases over the past decades [1, 2]. As of Jan 2021, as many as 246,178 genome-wide significant associations of single-nucleotide polymorphisms (SNPs) with 868 complex traits and diseases (P < 5.0 × 10−8) have been reported (see the National Human Genome Research Institute–European Bioinformatics Institute (NHGRI-EBI) GWAS Catalog) [3]. The majority of variants found by GWAS are common variants (minor allele frequency (MAF) > 5%) in the population and have low to modest effects (OR ~ 1.05–1.20), given that current approaches for association studies are well powered to detect significant effects for such variants [4].
To obtain biological insights from GWAS requires determining the causal variants, identifying their target genes, and importantly, linking the causal variants and target genes to molecular, cellular, and physiological phenotypes [5]. Numerous strategies, including statistical methods and genomic functional annotations, have been extensively applied to prioritize causal variants (termed fine-mapping) and their target genes [6,7,8]; however, laboratory functional studies to validate these causal variants and their targets and to identify molecular mechanisms often lag behind. One of the ultimate goals of genetic research is to inform genomic medicine to enable more effective strategies of disease prevention and treatment. GWAS associations have led to advances in personalized medicine (i.e., individual risk prediction and optimization of therapies based on genotypes), identification of therapeutic targets, and development of novel drugs and gene therapy strategies [1, 2]. Accelerated translation of GWAS to clinical impact is highly anticipated and could alter the future of genomic medicine.
In this review, we will first discuss various experimental approaches, including classic functional experiments and more newly developed genome editing technologies, and their applications in determining the functions of GWAS-identified noncoding variants. Furthermore, we discuss both advantages and disadvantages of each experimental method, which can in turn offer guidance for study designs. We also discuss the therapeutic applications of genome editing which drive translational advances that may enable more effective disease prevention and treatment. Finally, we look ahead and discuss future challenges as our understanding of the genetic basis of complex traits evolves.
General framework for the functional dissection of GWAS loci
After an initial identification of variants, either common or rare (minor allele frequency (MAF) < 5%) variants, associated with a complex trait/disease, several steps may be followed for better visualization and functional dissection of GWAS associations (Fig. 1). As a first step, the GWAS list of SNPs is used to identify disease-associated region, and then each region is visualized, such as with LocusZoom plots, to identify genes within the region and the local LD structure (Fig. 1a) [9].

Flow of a typical process from initial GWAS to functional dissection. a A typical GWAS involves selection of the study populations, either case-control cohorts or general populations; genotyping of variants across the genome by single-nucleotide polymorphism (SNP) array or whole genome sequencing; and statistical analysis of variant-trait/disease associations. Regional Manhattan plots (also termed as LocusZoom plots) are generated to show the P values of all variants in a genomic region, to explore the patterns of linkage disequilibrium (LD) between the sentinel variant and each variant, and to annotate the genes within this region. b Statistical fine-mapping and genomic annotations are used to prioritize candidate causal variants. Normally, a credible set of causal variants are prioritized according to posterior inclusion probability (PIP) of each variant and genomic annotations, including chromatin accessibility, histone markers, and transcription factor binding potential, are summarized to guide the following functional studies. c Target genes are predicted according to enhancer-target gene promoter interaction (chromatin confirmation capture) and correlation between causal variant genotypes and target gene expression. ASE, allele-specific expression. d Various experimental approaches are employed to investigate the functions of causal variants and target genes and to link them back to the original phenotype
As multiple variants in strong LD with the causal variants tend to exhibit similar statistical significance, additional methods are required to discriminate likely functional variants from other nonfunctional correlated variants (Fig. 1b). Publicly available genomic and epigenetic annotations, such as chromatin accessibility, transcription factor (TF) binding, and epigenetic modification [10,11,12], may be utilized to further evaluate the functions of those selected SNPs (Fig. 1b). In addition, several computational approaches have been proposed that build upon the Bayesian frameworks for inferring regulatory hierarchies between genomic regulatory elements, which prioritize critical regulatory regions [13, 14]. One would expect that the genetic variants that fall in these elements are more likely to be disruptive for gene function. Given that the target gene(s) responsible for a GWAS signal are often not clear, identification of the causal variants could thus help determine the target genes. Statistical methods [6], genomic and epigenetic datasets, and bioinformatics tools [5, 15] used for fine-mapping have been reviewed extensively.
Normally, the target gene of a coding variant could be directly inferred according to its genomic location, and the underlying molecular mechanism could be suggested as well based on the mutation type (not further discussed in this review). However, moving from noncoding variants to target genes can be challenging given that cis-regulatory elements (CREs) may affect gene transcription over extended distances by physically interacting with their target promoters through chromatin looping interactions (Fig. 1c) [16]. Multiple lines of evidence have suggested that noncoding variants may exert regulatory effects on target gene expression (known as eQTL) or alternative splicing (known as sQTL) in trait/disease-relevant cell types [17, 18]. Thus, two categories of approaches, collectively termed regulatory target analysis, have been proposed to assign the target genes of noncoding variants: (1) eQTL or sQTL analysis to correlate variant genotypes with candidate gene expression or alternative splicing [5, 8] and (2) proximity ligation methods to delineate interactions between enhancers and target gene promoters [19]. Statistical algorithms to integrate GWAS data (both individual-level data and GWAS summary statistics) and eQTL (or sQTL) [20, 21], and technologies to investigate chromatin conformation and regulatory connections within cells, have been developed [19]. However, a recent study has revealed an inverse relationship between the proportion of h2med (heritability mediated by the cis genetic component of gene expression levels) and expression cis heritability across genes, suggesting that genes with low expression cis heritability may have large effects on complex traits and assayed bulk eQTLs, although disease relevant, may not explain the majority of disease GWAS SNP effects [4]. Finally, laboratory functional experiments are performed in vitro in primary cell culture or in vivo in animal models, to assess the functional consequences of noncoding variants and regulatory effects on their target genes, and to investigate the mechanisms of how dysregulated genes confer risk for complex traits and disease (Fig. 1d). In the following sections, we describe experimental approaches used for functional studies of noncoding variants.
Protein binding assays to determine the molecular functions of noncoding variants
As mentioned above, the vast majority of noncoding GWAS variants are located in CREs that are often occupied by DNA binding proteins, such as TFs [22]. To investigate the binding affinities of noncoding variants with regulatory binding proteins, several approaches have been developed, including ChIP-Seq (or ChIP-qPCR) and electrophoretic mobility shift assays (EMSAs). ChIP-Seq relies on the following hypothesis that normalized sequencing reads covering the variant are expected to be present in equal allelic ratios if the variant does not affect TFs binding, and conversely, deviations from a 50/50 allelic ratio suggest regulatory function of variants [23, 24]. Alternatively, ChIP-qPCR using allele-specific probes or primers can indicate TF binding difference to a variant between the risk and the protective allele [25]. ChIP-Seq (or ChIP-qPCR) should be performed in a cell line or tissue heterozygous for the variant of interest.
In an EMSA experiment, DNA probes surrounding a candidate variant of different alleles (~ 20–100 bp) are incubated with either purified TFs or antibodies raised against candidate TFs in vitro. Difference of electrophoretic mobility shift rate can suggest a difference of TF-variant binding affinity. Sometimes it may be difficult to predict which TFs can bind to the variant; thus, unbiased approaches such as DNA-affinity pulldown followed by mass spectrometry can be advantageous [26]. All DNA-protein complexes are first captured by a probe, and proteins specific to the risk or protective allele are then identified by mass spectrometry. To reduce non-specific binding to the DNA probe observed in conventional DNA pulldown assays, Nigrovic and co-workers developed a novel DNA pulldown method, termed Flanking Restriction Enhanced Pulldown (FREP) which leveraged distinct restriction enzyme sites on either side of the bait sequence [27]. Notably, protein-DNA interactions are regularly reported in a binary on/off manner, which is unsuitable for most noncoding variants identified by GWAS that may not act by critically disrupting the binding motif itself but instead by subtly altering the binding affinity of TFs [28]. Complete characterization of a functional noncoding variant requires knowledge not only of specificity of TF-variant interactions, but also of affinity (i.e., strength in absolute terms for a given interaction). With a semi-quantitative isobaric labeling strategy, several mass spectrometry approaches, such as thermal proteome profiling [29] and chemoproteomic approach [30], have been developed to quantify affinity of biomolecular interactions. More recently, Vermeulen and co-workers reported another quantitative binding assay which uses affinity purifications from nuclear lysates coupled with chemical labeling and mass spectrometry to quantify dissociation constants (KdApp) of nuclear proteins for DNA and nucleosomes [31]. Protein binding assays in the context of nucleosomes may recapitulate the chromatin environment and epigenomic marks associated with a genetic variant [32].
High-throughput protein binding assays
Protein binding assays can also be performed in high throughput which has been reviewed by Stormo et al. [33]. More recently, Li and co-workers developed an unbiased high-throughput screen, termed SNP-seq, to identify functional SNPs that allelically modulate the binding of regulatory proteins [34]. SNP-seq relies on type IIS restriction enzymes, such as BpmI, that can be directed to bind certain variants and cut at a set distance from the binding site; however, pre-binding of regulatory proteins to variants can hinder the binding of type IIS restriction enzymes, thus protecting from cleavage. By incubating a library of these variant-type IIS restriction enzyme constructs with the nuclear extract of disease-related cells or tissues, one can determine which SNPs are bound by regulatory proteins through sequencing the undigested constructs. Then, Flanking Restriction Enhanced DNA Pulldown-Mass Spectrometry (FREP-MS) can be employed to determine the binding proteins of functional variants.
Limitations of protein binding assays
Despite extensive applications of protein binding assays in determining the function of a potential regulatory variant, they might result in false negatives when genetic variants are not supposed to disrupt a well-known TF binding motif but instead are in close proximity to the binding motifs of specific TFs [28, 35]. For instance, a major regulatory modality of red blood cell GWAS functional variants appears to affect GATA1 and co-factor binding by altering the DNA shape in the sequence-flanking core-binding motifs [35]. In addition, protein binding assays are regularly performed in vitro and may lack the appropriate biochemical context in trait/disease-relevant cell types, such as DNA and histone modifications, long-range chromatin interactions, and noncoding RNA binding. Finally, it has become clear that many of the binding sites within the genome do not affect the expression of nearby genes, serving as nonfunctional binding events [36]. These situations can produce both false-negative and false-positive results.
Reporter assays to assess the regulatory activities of noncoding variants
Another approach complementary to the protein binding assay is the reporter assay, widely used for assessing transcriptional regulatory activity of noncoding variants [37]. When an individual variant is analyzed, the region surrounding the variant is cloned into a physiologically relevant position with respect to the reporter gene, usually luciferase or fluorescent proteins, and transiently expressed in a cell line or in a model organism. Variation in regulatory activity can then be measured by comparing reporter activity for each construct.
High-throughput reporter assays
Instead of testing variants individually, researchers can also test tens of thousands of variants in a single experiment using massively parallel reporter assays (MPRAs) [37, 38]. For example, one study used this approach to test 32,373 variants from 3642 cis-eQTL loci for differential allelic effects and found 842 variants with differential expression between alleles, including 53 well-annotated variants linked to diseases and traits in the literature [39]. In addition to focusing on the candidate causal variants, one can use saturation mutagenesis, often by error-prone PCR [40], coupled with either the expression of a reporter gene or a sequencing-based readout to study the function of each nucleotide in a cis-regulatory element. Ahituv and co-workers performed saturation mutagenesis in conjunction with MPRA on 20 cis-regulatory elements associated with rare and common diseases, which enabled functional measurements for over 30,000 single-nucleotide substitutions and deletions [41]. Generally, saturation mutagenesis may facilitate the fine-scale evaluation of effect sizes in regulatory elements, and identification of causal variants not prioritized by fine-mapping due to unavailability of epigenetic annotations. van Steensel and co-workers further developed the survey of regulatory elements (SuRE) reporter technology with much higher throughput (> 100 fold increased scale) and resolution compared with MPRA [42]. Leveraging SuRE reporter technology, the authors survey the effect of 5.9 million SNPs, including 57% of the known common SNPs, on enhancer and promoter activity [42]. These high-throughput technologies enable rapid assessments of numerous alleles associated with a disease or trait of interest.
Limitations of reporter assays
Several limitations and considerations should be kept in mind when interpreting the results of reporter assays. First, reporter assays typically determine the transcriptional regulatory effect of variants in a small segment of plasmid DNA, different from the native chromatin context in which the variants are located. Functional activities of enhancer candidates in a chromosomally integrated context, assessed by lentivirus-based massively parallel reporter assay, showed substantial differences from those assayed on episomes [43]. Even when reporters integrated to the genome are used, they intrinsically lack the relevant genomic context of the native variants and elements. Second, a single GWAS signal might reflect the synergistic actions of multiple co-inherited causal variants. Reporter assays are typically not designed to detect the functions of haplotypes that may include multiple variants within different regulatory elements [44, 45]. Third, both false-negative and false-positive results may occur due to the experimental design, including the size of genomic contexts and the locations of variants relative to the transcriptional start site [8]. Recent improvements in reporter assay, such as longer genomic contexts [46], different promoters or across various cell types/stages, and the ability to detect smaller effect size by the inclusion of more barcodes [47], can partially overcome these shortcomings.
Genome editing technologies
The main limitation of the above experimental approaches is they do not test the function of variants in the native genomic context, which might therefore result in large proportion of false-positive and false-negative results. In light of these considerations, a more physiologically relevant method to investigate the functions of variants might be genome editing, which harnesses DNA repair pathways to yield desired genomic alterations within cells and organisms. Typically genome editing technologies take advantage of double-strand breaks (DSBs) introduced by programmable sequence-specific nucleases (SSNs). DNA DSBs are repaired in two ways: homology-directed repair (HDR) with a donor DNA, or non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ) [48]. NHEJ is the default form of DSB repair, which typically produces short insertions and deletions (indels) of a few bp in length at the cleavage site [49,50,51]. MMEJ, as an alternative NHEJ repair pathway that uses microhomologous sequences flanking the DSB to join the broken ends [52], is thought to be the major contributor to alleles observed after genome editing [49]. By contrast, HDR is often considered to be an error-free pathway which can use a repair template to introduce precise genetic modifications, although partial homology-driven repair events and competing NHEJ/MMEJ repair events mean that typically a range of on-target alleles are usually produced by any genome editing experiment [53]. DNA repair pathways that underlie genome editing and strategies to favor various outcomes have been reviewed extensively [54].
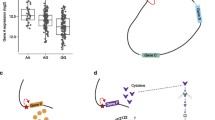
Tremendous effort has been dedicated to developing sequence-specific nucleases (SSNs) that are capable of efficiently introducing targeted DNA breaks [55]. To date, four different types of programmable SSNs have been developed: meganucleases, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR-associated (Cas) nucleases (Fig. 2). Despite their recent discovery, continuous improvements have made CRISPR/Cas systems a widely adopted, low-cost, easy-to-use targeted genetic manipulation tool that has been extensively applied in many organisms.

The genome editing toolbox. A Repair pathways of DNA double-strand breaks (DSBs) induced by nucleases. B CRISPR-mediated gene interference (CRISPRi) or gene activation (CRISPRa) regulates the activity of CREs by sterically blocking or changing epigenetic modifications. C Base editors used a fused deaminase to catalyze the conversion of C to U (C base editor) or A to I (A base editor) on one strand, followed by DNA repair on the non-edited strand. Eventually, the original C:G (or A:T) is converted to T:A (G:C) during DNA replication. D Primer editing leverages nCas9 to nick the genomic target, after which the reverse transcriptase copies the sequence information from the prime editing guide RNA (pegRNA) and replaces the original sequence at the target site
CRISPR/Cas system
The CRISPR/Cas system, comprising CRISPR repeat-spacer arrays and Cas proteins, is an RNA-mediated adaptive immune system in bacteria and archaea [56]. According to their Cas genes and the nature of the interference complex, CRISPR/Cas systems are divided into two classes that have been further subdivided into six types and 33 subtypes [57], and new CRISPR systems are continually being discovered and repurposed. Class 1 CRISPR-Cas systems (types I, III, and IV) have effector modules composed of multiple Cas proteins that form a CRISPR RNA (crRNA)-binding complex, whereas class 2 systems (types II, V, and VI) have a single, multidomain crRNA-binding protein that is functionally analogous to the entire effector complex of class 1. The most well-characterized and widely used Cas effector is SpCas9 from the species Streptococcus pyogenes SF370 [58,59,60]. After repurposing the CRISPR/Cas9 system for gene editing, the CRISPR/Cas9 system has two components: the Cas9 nuclease and a guide RNA, either as separate crRNA and trans-activating crispr RNA (tracrRNA) components or a chimeric single-guide RNA (sgRNA) [61]. DNA binding occurs at a 20-base-pair DNA sequence (called the protospacer) that is complementary to a 20-nucleotide sequence in the guide RNA (spacer sequence) and that can be readily altered for different genome targeting [61, 62] (Fig. 2). The DNA recognition site must be adjacent to a short motif (the protospacer adjacent motif or PAM) that acts as a switch, allowing SpCas9 to bind within the target sequence [61, 62]. The requirement of PAM sequence, like 5′-NGG-3′ for SpCas9, largely restricts the genomic regions targeted by CRISPR/Cas systems. To increase the scope of targetable genomic regions, researchers have engineered Cas proteins to recognize broader PAMs by structure-guided design or directed evolution [58,59,60]. In addition to SpCas9, other natural CRISPR nucleases of diverse PAM sequence requirement have been also engineered for genome editing [63,64,65,66], which greatly expand the scope of target loci amenable to RNA-guided genome editing.
CRISPR-mediated inhibition (CRISPRi) and activation (CRISPRa)
CRISPR-Cas9 has two catalytic domains (HNH and RuvC), and inactivation of both domains by point mutations (D10A and H840A for SpCas9) results in complete loss of DNA cleavage activity (catalytically inactive dead Cas9, dCas9) [67]. Without changing the DNA sequence of a given genome, fusion of dCas enzymes to effector domains enables efficient transcriptional regulation, including CRISPR-mediated inhibition (CRISPRi) and activation (CRISPRa) [67]. Notably, dCas9 itself can strongly bind to the DNA target sequence and the tight binding interferes with the accessibility of other DNA binding proteins (i.e., endogenous TFs and RNA Polymerase II) to target sequence [67, 68]. Fusing a strong repressor, such as Kruppel-associated Box (KRAB) [69] and DNMT3A/3 L [70], to dCas9, results in stronger gene repression than dCas9 alone. On the contrary, dCas9 fused with transcriptional activators can result in robust induction of target gene expression. Various activators (e.g., VP64 [71], P300 [72], ad P65 [73]), and combinations of effector proteins by dCas9 fusions and/or MS2-MCP scaffolding, including the synergistic activation mediator (SAM) system [74], SunTag [75], VP64-p65AD-Rta (VPR) [76], and enCRISPRa [77], have been developed. These technologies have largely enriched the genome editing toolbox, allowing dynamic spatial and temporal control of gene activation.
Base editing
Base editing can generate precise point mutations in genomic DNA or in cellular RNA without generation of DSBs or a DNA donor template [78, 79]. DNA base editors are composed of a base modification enzyme (cytidine deaminase or adenine deaminase) and a catalytically impaired Cas nuclease that operates on single-stranded DNA (ssDNA) but not double-stranded DNA (dsDNA) (Fig. 2). Two types of DNA base editor have been developed: cytosine base editors (CBEs) which convert a C•G base pair into a T•A base pair, and adenine base editors (ABEs) which convert an A•T base pair into a G•C base pair. CBEs have a third fused component, uracil glycosylase inhibitor, which disfavors base excision repair and promotes mismatch repair, substantially increasing the efficiency of C•G to T•A conversion. CBEs and ABEs can collectively achieve four possible transition mutations (C to T, G to A, A to G, and T to C). Before base editing was developed, the introduction of a precise mutation usually required CRISPR/Cas-mediated HDR occurring at a DSB site in a genome via a donor DNA template harboring the desired change [80]. However, due to restriction in the G2 and S phases of cell cycles, inefficient HDR is typically observed in non-dividing cells [81, 82]. Moreover, the majority of edited products will usually contain small insertions or deletions (indels), resulting from competition between NHEJ/MMEJ and HDR [83]. In contrast, base editing does not create a DSB and therefore provides precise genome editing with a high frequency of intended as a fraction of all modified alleles. The development of various base editors and their application in sequence diversification and other areas have been reviewed extensively [84,85,86,87].
Prime editing
Although base editing can efficiently install the four transition mutations without requiring DSBs, base editors (ABE and CBE) cannot yet efficiently perform the eight transversion mutations, as well as small insertions and deletions, although new base editors may generate C>G transitions [88, 89]. Recently, Liu and co-workers developed prime editing, a “search-and-replace” genome editing technology which can precisely install all 12 possible base-to-base conversions, small insertions, small deletions, and their combinations into target DNA sites, without requiring DSBs or donor DNA templates [90]. Prime editors contain two components: a reverse transcriptase (i.e., engineered M-MLV RT) fused to an RNA-programmable nickase (nCas9, H804A) and a prime editing guide RNA (pegRNA) that guides the prime editors to the target site and encodes the desired sequence (Fig. 2).
In comparison with base editors, prime editors induce base substitutions in more extended regions (from 3 bp upstream to 29 bp downstream of a PAM) with fewer bystander mutations at the targeted locus and at predicted off-target sites [90]. Furthermore, prime editors were used to perform insertions even up to 44 bp and deletions up to 80 bp [90]. However, there are a number of variables that need to be optimized for prime editing including the pegRNA and often a second nicking sgRNA (ngRNA) to nick the non-edited strand, which makes the experimental design more complicated than typical CRISPR gene or base editing applications [91]. The factors that affect prime editing efficiency are beginning to be clarified [92].
Applications of genome editing technologies in functional studies of GWAS loci
For a GWAS of interest, genome editing technologies have offered a host of strategies to modify the causal variants and local CREs in physiologically relevant contexts, either in vitro in primary cell culture or in vivo in animal models, making it feasible to investigate their functions and target genes, and more importantly identify their role in determining the original phenotype. Diverse genome editing strategies across the genome editing toolbox have been conceived to modify causal variants or disrupt harboring CREs to dissect GWAS (Table 1).
Introduction of indels nearby causal variants
As mentioned above, DNA DSBs can be patched through NHEJ and MMEJ repair pathways, both of which can yield varied indels without a homologous repair template [48]. The first strategy employs indels to disrupt putative CREs where the causal variants are located [49]. Given the narrow indel spectrum (a typical deletion spectrum of 1–20 base pairs) introduced by NHEJ and MMEJ, this approach requires identification of the candidate causal variants from a relatively broad GWAS risk locus (Fig. 3).

Genome editing strategies for interrogation of GWAS loci. Approaches used to investigate the functions of causal variants included introduction of indels nearby causal variants (to disrupt the core sequence of a putative transcription factor binding motif), deletion of the genomic region surrounding causal variants, CRISPRi/a (to repress or enhance the activity of local CREs), and allele substitution to change causal variants from one allele to the other (to precisely mimic the genotype). Multiplex CRISPR is used to target multiple causal variants where they can function jointly or synergistically. CRISPR/Cas screening can enable high-throughput interrogation of causal variants or target genes. Target gene knockout is typically achieved by introducing frameshifts into the coding sequence
One elegant example that employed indels to disrupt causal variants is regarding an intergenic variant rs35252396, which is associated with renal cancer susceptibility [93]. Given that rs35252396 may alter the activity of hypoxia-inducible transcription factor (HIF) binding to the local enhancer, a sgRNA was designed targeting the center of the SNP-associated HIF-binding signal in 786-O renal cancer cells. After screening 36 clones of cells for indel spectra at the targeting site, the investigators identified 7 clones of cells with mutations that can affect the HIF-binding site. When compared with non-mutant clones of cells, these mutant cells exhibited significantly lower expression of target gene, MYC and PVT1. In another study, CRISPR/Cas9 genome editing was leveraged to generate two heterozygous cell lines with 6-bp or 18-bp genomic deletion surrounding rs558245864 (association with multiple autoimmune diseases) which is located in a CTCF binding motif [13]. ATAC-seq and RNA-seq in the mutant lines revealed a significant downregulation of chromatin accessibility at the focal peak and a concomitant downregulation of BLK expression compared with the parental cell line.
These studies suggested that introduction of indels nearby the causal variants by individual sgRNA targeting is useful for interrogation of causal variants often informed by epigenetic marks or TF binding motifs. However, small indels may be insufficient to disrupt the local TF binding motif given the flexibility of TF binding to target sites (Fig. 3), and in extreme cases, the motif will remain intact after the introduction of small indels. As a result, the consequences of indels nearby the causal variants largely depend on the degree to which local motifs are disrupted. Separation of single cell clones may be required to identify mutant cells with CREs that have disruptive deletions for downstream functional studies but this introduces the problem of clonal variation, in which numerous clones need to be studied to confidently associate gene regulation changes to gene editing, which can be especially challenging when effect sizes are modest.
Genomic deletion surrounding causal variants
Several studies have suggested that the GWAS locus conferring risk for disease can be driven by multiple variants spanning different enhancers that target the same gene [44, 94], in which case modeling an individual variant could fail to exhibit a sufficient genetic or physiological consequence, while deletion of the entire disease risk region with multiple causal variants can be an alternative efficient strategy. Different from indels nearby causal variants, which may not disrupt the local CREs, targeted deletion of the entire genomic regions surrounding causal variants using dual sgRNAs can completely erase the local CREs (Fig. 3).
One study used CRISPR/Cas9-targeted deletion to investigate rs17114036, a common noncoding polymorphism at 1p32.2, which is associated with coronary artery disease (CAD) and ischemic stroke (IS) [95]. To determine the regulatory role of rs17114036 on target gene (PLPP3) expression, Yang and co-workers employed CRISPR/Cas9 to delete an ~ 66-bp genomic region enclosing rs17114036 in human aortic endothelial cells. Compared with non-edited cells, the genome-edited cells showed reduced PLPP3 expression and altered cell behavior in agreement with PLPP3’s roles [96]. In a more recent study, Luo and co-workers identified multiple TFs binding-disruptive SNPs through integrating ChIP-Seq from human brain tissues or neuronal cells and position weight matrix (PWM) data [97]. To verify the regulatory effect of these functional SNPs on target gene expression, Luo and co-workers designed sgRNA pairs to knockout the genomic sequence containing these SNPs. Take the SNP rs3101339 for example, deletion of the genomic region (586 bp) containing rs3101339 led to significant upregulation of NEGR1 expression [97].
Notably, the usage of dual sgRNAs can sometimes result in various outcomes, including targeted genomic deletions, inversions, and more complex genomic rearrangements, which might confound the causal interpretation of target deletion-induced phenotype in bulk cells. Single cell clones with specific editing outcomes (i.e., expected targeted deletion) may help correlate genotypes with phenotypes, although the zygosity of edits with respect to clones, particularly in aneuploid cell lines, needs to be considered carefully.
Epigenetic control of the local CREs with causal variants
Instead of changing genomic sequences by wild type Cas9, modifying the surrounding chromatin by either CRISPRi or CRISPRa equipped by dCas9 may serve as an alternative strategy to investigate the functions of noncoding variants (Fig. 3) [67]. In addition to dCas9, fusing a catalytically inactive ZFN or TALE array to chromatin modifying enzymes can achieve epigenetic control. These non-indel forming approaches may be advantageous to achieve intermediate degrees of gene control in cases where disruption of an enhancer or other regulatory DNA may be cell lethal.
GWAS have reproducibly associated variants within intergenic regions of 1p36.12 locus with osteoporosis [98]. After prioritizing rs6426749 as a potential causal SNP at 1p36.12 through functional genomic and epigenomic analyses, two different genome editing strategies were employed to identify the potential target genes: (1) deletion of a 749-bp enhancer region containing rs6426749 using CRISPR/Cas9 and (2) epigenetically repressing the enhancer activity near rs6426749 locus using dCas9-KRAB in hFOB 1.19 cells [99]. Following both strategies, the authors observed a significant decrease of LINC00339 expression, but not other genes nearby, suggesting that LINC00339 is the target gene responsible for the risk locus.
Despite high efficiency for transcriptional regulation, CRISPRi and CRISPRa have intrinsic shortcomings. First, the regulatory effect by either CRISPRi or CRISPRa may be transient such that the chromatin may revert to its original state after the epigenome modifying machinery is removed, although the duration of epigenetic memory may depend on a number of variables including the specific locus and physiological context [70]. Second, the activity of CRISPRi/a is correlated with the distance of the target site from transcription start sites (TSSs) and core regulatory elements where most of the causal variants lie. For example, strong CRISPRi activity is obtained by targeting a window of DNA from 50 to + 300 bp relative to the TSS of a gene, while strong CRISPRa was observed for sgRNAs targeting 400 to 50 bp upstream from the TSS [71, 100]. For enhancer regions, strong CRISPRi or CRISPRa activity was observed when targeting DNA 100–200 bp away from the accessible chromatin regions [77]. Third, the dynamic range of gene expression control by CRISPRi/CRISPRa may vary depending on genomic and cell-type context, and may exceed or underperform the effects of actual genetic variants, suggesting the potential for false-positive or false-negative effects [101]. Finally, epigenetic modifications induced by CRISPRi repression can encompass a > 1 kb [102], thus limiting the resolution of CRISPRi for variant fine-mapping. Combinational applications of both epigenetic editing and base editing/prime editing (discussed below) might be warranted, to verify the regulatory effect of a GWAS locus on target gene expression and to pinpoint the causal variants responsible for the GWAS locus.
Multiplexed genome editing and epigenetic control
Previous studies have suggested that complex diseases arise from the accumulation of genetic variants that are enriched in genes expressed in molecular networks [103], and individual genes must be understood in the context of molecular networks that define the disease states. More importantly, multiple causal variants may act synergistically, contributing to disease phenotype/trait variance [44, 94, 103]. Multiplexed CRISPR technologies which leverage simultaneous expression of multiple Cas proteins or gRNAs to edit or transcriptionally regulate numerous genetic loci in parallel hold promise for functional study of multiple variants from one GWAS locus (haplotype) or different loci [104] (Fig. 3). For example, in order to investigate the potential synergistic effects of schizophrenia-related genes, Brennand and co-workers used CRISPRa to upregulate SNAP91, TSNARE1, and CLCN4 and RNA interference (RNAi) to repress FURIN in human induced pluripotent stem cells [105]. They observed larger effects of combinatorial perturbation converging on synaptic function, than the additive effects of individually perturbed genes. In another study, Shendure and co-workers used saturation genome editing to assess the pathogenicity of all possible single-nucleotide variants (SNVs) in 13 exons that encode functionally critical domains of BRCA1, a tumor suppressor gene related to both breast and ovarian cancer [106]. To introduce SNVs into haploid human cell line (HAP1), a Cas9/gRNA construct was transfected with a library of plasmids containing all SNVs within approximately 100 bp of genomic sequence (the homology arms) to favor homology-directed repair. Functional scores were systematically derived for 3893 SNVs based on cell survival, independent of prior expectation, which was immediately useful for the clinical interpretation of BRCA1 variants.
Allele substitution of causal variants
While it is crucial to determine the regulatory mechanisms through which a disease-associated variant could affect target gene expression, another question would be how potentially modest variation in a target gene’s expression could result in a disease phenotype, given that the majority of eQTL effects are of relatively small magnitude (< 2-fold change in expression) [107]. Despite high performance in identifying the target gene and determining the function of causal variants, either targeted deletion or epigenetic modification of a CRE may not have the same effect as that of a single-nucleotide change. Therefore, to mutate the causal variant from one allele to the other by HDR, base editing or newly developed prime editing could ensure target gene expression in a physiologically relevant manner (Fig. 3).
Recently, Jaenisch and co-workers leveraged CRISPR-mediated HDR to mutate the candidate causal variants at the SNCA locus, which is associated with Parkinson’s disease (PD) [108]. After prioritizing candidate causal variants based on epigenetic signatures and in silico TF motif predictions, a 500-bp genomic region containing two SNPs in human embryonic stem (ES) cells was first deleted and then the 500-bp region with either the risk or protective alleles of the two SNPs was reinserted using HDR. In comparison with cell clones harboring the protective alleles of the enhancer SNPs, clones bearing the risk-associated alleles showed significantly higher SNCA levels [108]. A more recent study focused on GWAS variants in or near the FAM13A (family with sequence similarity member 13A) associated with chronic obstructive pulmonary disease (COPD) [109]. Following conditional genetic association and MPRAs which together prioritized rs2013701 to be the most promising causal variant responsible for this GWAS locus, CRISPR-based homology-directed repair was applied to generate single clones homozygous for either TT or GG genotype at rs2013701 in 16HBE cell line. Compared with rs2013701 TT clones, GG clones predicted reduced expression of FAM13A and demonstrated an increased rate of cellular proliferation.
The current genome strategies used for allele substitution in functional studies of GWAS loci are restricted to CRISPR/Cas or other nuclease-mediated HDR. However, due to competition between the NHEJ/MMEJ and HDR repair pathway following DSBs, allele substitution using HDR is often inefficient and the occurrence of unintended indels might also cause imprecise editing of the target gene [83], thus limiting its applications in genome editing. Instead, base editing has been on its horizon.
Reiner and co-workers performed whole genome sequencing of over 62,000 ancestrally diverse participants in the TOPMed program and identified 14 single variant-red blood cell associations at 12 unique loci [110]. To further investigate the function of one sentinel variant, rs112097551, underlying red blood cell development, Reiner and co-workers used cytosine base editing to modify the reference G to alternative A allele in HUDEP-2 erythroid precursor cells. Compared to G/G clones, all five G/A heterozygous HUDEP-2 clones showed significantly reduced expression of RUVBL1, but not other nearby genes, suggesting rs112097551-G may exert long-range control of the gene RUVBL1 which is essential for hematopoiesis [110]. Despite potentially higher editing efficiency and product purity, prime editing has yet been employed in GWAS functional studies.
CRISPR/Cas screening
Analogous to the high-throughput protein binding assays and reporter assays, high-throughput CRISPR screens have been also employed in functional studies of GWAS (Fig. 3). One recent study used CRISPRi screens to dissect thousands of noncoding variants at the TNF-α-induced protein 3 (TNFAIP3) locus that is associated with multiple diseases [111]. Hacohen and colleagues first employed either CRISPRi or CRISPRa targeting all regions with accessible chromatin in three cell lines, to identify regions that significantly repress TNFAIP3 expression, and then leveraged MPRAs to test for allele-specific reporter expression induced by individual variants, which finally prioritized 18 causal variants at this locus. Given the relatively broad targeting range by CRISPRi/a, identification of the causal variants required additional tools that can test individual variant, such as EMSA and reporter assay as mentioned above. Instead of CRISPRi/a, CRISPR-mediated varied indels have been also applied for screening of cis-regulatory elements [112, 113]. Previous GWAS and other human genetic studies have highlighted the association between the HBS1L-MYB interval and fetal hemoglobin (HbF) levels [114]. To functionally fine-map the HBS1L-MYB intergenic region, Canver et al. performed variant-aware saturating mutagenesis of this region in HUDEP-2 cells using multiple nucleases with different PAM sequence requirements [112]. Multiple putative functional elements were identified, including the previously known − 84 DNase I-hypersensitive site (DHS) which harbored a potential causal variant rs61028892 [112]. Again, the resolution is a function of the number of sgRNAs available within a given genomic region which in turn depends on the nuclease and its genome targeting range [60].
Therapeutic applications of genome editing
A central objective of genetic research is to translate biological insights into clinical applications that enable effective prevention and treatment of diseases. Human disease genetics has identified thousands of mutations that result in diverse diseases, which provided insights into gene therapy strategies [1, 115, 116]. Therapeutic applications (i.e., accuracy, precision, and safety) of genome editing in monogenic diseases both ex vivo and in vivo, as well as delivery methods of genome editing tools, have been extensively reviewed elsewhere [117, 118]. Here we focused on examples of adult-onset disease to demonstrate the significant advances of translation from genetic variants identified by GWAS to disease gene therapy.
GWAS have identified multiple SNPs associated with increased expression of fetal hemoglobin and a lower severity of both transfusion-dependent β-thalassemia (TDT) and sickle cell disease (SCD) in adults [23, 119]. Some of the SNPs are located in an erythroid-specific enhancer of BCL11A encoding a zinc finger-containing transcription factor that represses γ-globin expression and fetal hemoglobin in erythroid cells [23]. These findings have led to a considerable effort to target BCL11A to increase fetal hemoglobin levels in patients with β-hemoglobin disorders [120, 121]. Disruption of GATA1 binding sequences within the erythroid-specific enhancer of BCL11A in hematopoietic stem and progenitor cells (HSPCs) by either CRISPR/Cas9 or base editors significantly reduced BCL11A expression in erythroid-lineage cells, restored γ-globin synthesis, and reactivated production of fetal hemoglobin, even though the core GATA1 binding sequences are not subject to common genetic variation but rather neighboring sequences [115, 116]. Both TDT and SCD patients infused with autologous CRISPR/Cas9-edited CD34+ HSPCs 9 targeting the GATA1 binding sequences at the erythroid-specific enhancer of BCL11A showed increased in fetal hemoglobin, transfusion independence, and elimination of vaso-occlusive episodes (in the SCD patients) [115]. Therefore, the therapeutic potential of editing a GWAS locus not only depends on finding the causal SNPs per se but rather in understanding the elements and genes impacted which themselves could constitute the ultimate therapeutic target. In contrast to common variants of modest effect size, low-frequency or rare variants (MAF < 5%) uncovered by GWAS, especially those leading to loss-of-function, usually exhibit a relatively large phenotypic impact [122]. For example, carriers with inactivating mutations on PCSK9 were found to have markedly lower LDL cholesterol level and CAD risk, which led to the discovery of two FDA-approved monoclonal antibodies [123]. We argue that the interpretation of genetic variation is presently the rate-limiting step for genomic medicine. With the accumulation of functionally verified genetic variants and continuous advancement of CRISPR/Cas editing technologies, therapeutic genome editing and GWAS-inspired development of small molecules may become feasible for more and more polygenic diseases in the near future.
General considerations for functional genetics
Although genome editing has drastically accelerated the identification of causal variants by linking these variants to target gene expression or the original phenotype, there are several general considerations that are applicable to genome editing approaches in functional studies.
Cell type or cell state
Most of the current functional GWAS studies are performed in human cell models. As mentioned above, the vast majority of disease-associated variants reside in gene distal sequences such as enhancer elements (upstream, downstream, or in introns of target genes). Enhancers control spatiotemporal gene expression programs by engaging in physical contacts with promoters of their cognate genes, often through long-range chromosomal interactions. Since both enhancer repertoires and the enhancer-promoter interactome are highly cell type-specific [124, 125], many disease-associated variants may regulate target gene expression and cellular functions in a cell type-specific manner. Supporting this hypothesis, previous studies have revealed significant enrichment of GWAS SNPs in active regulatory regions in disease or trait-relevant cell or tissue types, compared to random SNPs [126, 127]. For a given disease or trait, several methods have recently been developed that integrate tissue-specific gene expression or genomic annotations with GWAS summary statistics to identify risk loci enrichment in specific cell types. These methods included, but not limited to, SNPsea, DEPICT, RolyPoly, g-chromVAR, and CHEERS [127,128,129,130,131,132]. Such frameworks allow researchers to narrow down potential disease-relevant cell types or states, which is crucial for designing functional follow-up experiments and gaining mechanistic insights. Notably, recapitulating the cell state(s) impacted by GWAS SNPs may include not only cell type but also environmental conditions and transient perturbations, as SNPs may only show a phenotypic difference in response to such cues.
Cellular/physiological function
Determining the causality of a variant ideally requires demonstrating an altered phenotype following allelic replacement. Practically, this may be done in vitro in primary cell culture or in vivo in animal models. Given the potential similarity of conserved physiologic phenotypes between animal models and humans, linking GWAS variants back to the original phenotype might be more directly assessed in animal models. However, for human-specific traits, animal models may have limitations. Reciprocally, the precise functional assays that are disease- or trait-relevant can be challenging to define in human cell cultures. For example, neuronal cell types are thought to be implicated in psychiatric traits [132], but it is not known which specific neuronal functions are compromised in disease. In light of this issue, intermediate phenotypes (measurable cellular functions) might be quite useful for functional assays as long as these phenotypes are truly intermediary to the complex phenotype. A recent study showed that variants associated with susceptibility to infection tend to modulate the secretion of monocyte cytokines (cytokine QTLs) [133]. Thus, it would appear fruitful for functional validation of infection-associated variants to assess monocyte cytokine secretion.
Moreover, global changes in gene expression may be a more general and unbiased phenotype to indicate cellular functions. Recently, several technologies combining CRISPR/Cas-mediated genome editing and single cell RNA-sequencing (scRNA-Seq) have been developed, like CROP-seq [134] and Mosaic-seq [135], which enable matching the transcriptome of single cells with genetic variants introduced by genome editing. In the future, high-throughput phenotyping of human cells will be crucial for identifying the best assays to validate candidate GWAS variants or genes.
Genome editing in primary cells
To investigate the functions of variants, researchers ideally would mimic the exact polymorphisms naturally observed in GWAS by genome editing. Although this approach can be applied in immortalized cell lines, it may be more challenging in primary cells that are not easily expanded in culture. Currently, many strategies are available to deliver Cas proteins and other editors into cells, including plasmid transfection, viral delivery, RNP, and mRNA delivery, which holds promise for high editing efficiency in primary cells [120]. For example, utilizing ts-rSeV and lentivirus to deliver SpCa9 and sgRNAs separately, we have successfully performed CRISPR/Cas9 screening in hematopoietic stem and progenitor cells (HSPCs) to study neutropenia-associated variants, leading to the identification of previously unappreciated mechanisms of neutropenia [136]. Instead of primary cells, human induced pluripotent stem cell (iPSC), which can be differentiated into diverse cell types [137], is an attractive system to study molecular consequences of genetic variants. Several studies have demonstrated that human iPSC-derived cardiomyocytes from long QT syndrome patients can faithfully recapitulate disease phenotypes, allowing scientists to study some of the disease traits in vitro [138]. Moreover, as a result of expansion potential in vitro, iPSCs allow for various types of genome editing followed by selection of clonal cell line with the accurate editing outcome. Multiple studies have integrated genome editing technologies with hiPSC-based studies to study the functions of GWAS variants and genes (Table 1).
Animal models
Model organisms are typically preferable for experimental disease research due to similar anatomy and physiology with human beings. However, there are important differences between model organisms and human beings in terms of genetic architecture (especially noncoding genomic regions), and mutations in humans that result in specific phenotypes that may not be faithfully recapitulated in model organisms [139]. Xenograft models may serve as a compromise allowing the study of human cells in an animal setting, though these may also have some limitations in terms of how reliably the xenograft mimics the physiology of the native human tissue.
Limitations of genome editing
Despite widespread potential applications in functional genetics, CRISPR/Cas genome editing tools still have their limitations. First, genome editing efficiency highly depends on a multitude of factors, including cell type, delivery of genome editing tools, cycling rate of cells, and mutagenesis efficiency. Second, when the CRISPR system recognizes sequences homologous to the target sequence, off-target mutations may occur, especially in mammals given their large genomes, which can lead to undesired functional impacts [140]. In addition to the production of unwanted local mutagenesis, off-target DNA cleavage can potentially give rise to chromosomal rearrangements and disrupt the functionality of otherwise normal genes and regulatory elements [141], which might impact the interpretation of the CRISPR-induced phenotype. Third, several studies have revealed that DSBs introduced by genome editing can select for inactivation of the p53 pathway, which is associated with transformation in numerous cell types [142]. Finally, there is potential for pre-existing immunity against CRISPR components to limit the feasibility and safety of in vivo delivery, given the evidence that pre-existing Cas9 antibodies and reactive T cells have been detected in humans after exposure to pathogenic bacteria of CRISPR systems [117, 143]. Such “side effects” of genome editing can typically be addressed by including appropriate empiric controls, such as performing gene editing in parallel at neutral genomic loci [144, 145] and evaluating the status of the p53 pathway.
Conclusions and future directions
Despite thousands of SNP-trait/disease associations have been identified by GWAS, only a small fraction of them have been functionally investigated. In order to translate these associations to biological insights, one needs to determine the causal variants, target genes, and the underlying mechanisms linking variants and genes to the original phenotype. Moving beyond protein binding assays and reporter assays, which can determine the functions of variants either in vitro or in an ectopic context, genome editing technologies can manipulate variants and their harboring elements in trait-relevant chromatin, genomic, and cellular contexts. Genome editing will undoubtedly spur progress in this field and accelerate the translation of genetic advances to novel therapeutics.
In the next decade, we foresee several important areas where advances are likely to occur. First, functional experimental approaches, especially in high-throughput, performed in trait/disease-relevant cell types will be further developed. Current functional assays, such as SNP-seq [34], largely rely on cell lysates from cell lines, which may lead to both false-positive and false-negative TF-DNA binding as discussed above. These assays could benefit from using either primary cell lysates or three-dimensional organoid cultures. Second, although genome editing technology has been rapidly advanced by fine-tuning the architecture of nucleases, i.e., Cas proteins, to increase the efficiency, specificity, and targetability, there remain many challenges to be overcome before its full potential can be realized. For instance, despite the superiority of human primary cells to model trait/disease phenotype, functional studies of GWAS loci in human primary cells have been scarcely reported, possibly due to low editing efficiency. Third, in addition to single-nucleotide variants, disease-associated structural variations where differences in genomic DNA can range from kilobase to chromosomal magnitude have been discovered through whole genome sequencing. Several genome editing-assisted methods have been developed for targeted insertion, deletion, or replacement of long sequences and genes [146], providing efficient tools to study these structural variations. Finally, despite the inevitable time lag from basic research to clinical implementation, a growing number of examples have highlighted the translational potential of GWAS findings, i.e., to identify individuals at high risk of certain diseases, to inform precision medicine, for drug development, and to design gene therapy strategies [2, 120, 121]. With the cumulative cataloging and understanding of genetic modifiers of common diseases and advancement of genome editing technologies, therapeutics that strive to reproduce, mimic, or augment natural protective genetic variation should flourish in the future.
References
Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5–22.
Tam V, Patel N, Turcotte M, Bosse Y, Pare G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20:467–84.
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–12.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53.
Gallagher MD, Chen-Plotkin AS. The post-GWAS era: from association to function. Am J Hum Genet. 2018;102:717–30.
Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet. 2018;19:491–504.
Broekema RV, Bakker OB, Jonkers IH. A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol. 2020;10:190221.
Cannon ME, Mohlke KL. Deciphering the emerging complexities of molecular mechanisms at GWAS loci. Am J Hum Genet. 2018;103:637–53.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7.
Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5.
Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–59.
de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, et al. The dynamic landscape of open chromatin during human cortical neurogenesis. Cell. 2018;172:289–304 e218.
Kumasaka N, Knights AJ, Gaffney DJ. High-resolution genetic mapping of putative causal interactions between regions of open chromatin. Nat Genet. 2019;51:128–37.
Delaneau O, Zazhytska M, Borel C, Giannuzzi G, Rey G, Howald C, et al. Chromatin three-dimensional interactions mediate genetic effects on gene expression. Science. 2019;364(6439):eaat8266. https://doi.org/10.1126/science.aat8266.
Cano-Gamez E, Trynka G. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front Genet. 2020;11:424.
Pombo A, Dillon N. Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol. 2015;16:245–57.
Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, et al. RNA splicing is a primary link between genetic variation and disease. Science. 2016;352:600–4.
Walker RL, Ramaswami G, Hartl C, Mancuso N, Gandal MJ, de la Torre-Ubieta L, et al. Genetic control of expression and splicing in developing human brain informs disease mechanisms. Cell. 2019;179:750–71 e722.
Kempfer R, Pombo A. Methods for mapping 3D chromosome architecture. Nat Rev Genet. 2020;21:207–26.
Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48:245–52.
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–7.
Deplancke B, Alpern D, Gardeux V. The genetics of transcription factor DNA binding variation. Cell. 2016;166:538–54.
Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–7.
Maurano MT, Haugen E, Sandstrom R, Vierstra J, Shafer A, Kaul R, et al. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat Genet. 2015;47:1393–401.
Huang Q, Whitington T, Gao P, Lindberg JF, Yang Y, Sun J, et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat Genet. 2014;46:126–35.
Xia Q, Deliard S, Yuan CX, Johnson ME, Grant SF. Characterization of the transcriptional machinery bound across the widely presumed type 2 diabetes causal variant, rs7903146, within TCF7L2. Eur J Hum Genet. 2015;23:103–9.
Li G, Cunin P, Wu D, Diogo D, Yang Y, Okada Y, et al. The rheumatoid arthritis risk variant CCR6DNP regulates CCR6 via PARP-1. PLoS Genet. 2016;12:e1006292.
Levo M, Zalckvar E, Sharon E, Dantas Machado AC, Kalma Y, Lotam-Pompan M, et al. Unraveling determinants of transcription factor binding outside the core binding site. Genome Res. 2015;25:1018–29.
Mateus A, Maatta TA, Savitski MM. Thermal proteome profiling: unbiased assessment of protein state through heat-induced stability changes. Proteome Sci. 2016;15:13.
Sharma K, Weber C, Bairlein M, Greff Z, Keri G, Cox J, et al. Proteomics strategy for quantitative protein interaction profiling in cell extracts. Nat Methods. 2009;6:741–4.
Makowski MM, Grawe C, Foster BM, Nguyen NV, Bartke T, Vermeulen M. Global profiling of protein-DNA and protein-nucleosome binding affinities using quantitative mass spectrometry. Nat Commun. 2018;9:1653.
Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010;143:470–84.
Stormo GD, Zhao Y. Determining the specificity of protein-DNA interactions. Nat Rev Genet. 2010;11:751–60.
Li G, Martinez-Bonet M, Wu D, Yang Y, Cui J, Nguyen HN, et al. High-throughput identification of noncoding functional SNPs via type IIS enzyme restriction. Nat Genet. 2018;50:1180–8.
Ulirsch JC, Nandakumar SK, Wang L, Giani FC, Zhang X, Rogov P, et al. Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell. 2016;165:1530–45.
Farnham PJ. Insights from genomic profiling of transcription factors. Nat Rev Genet. 2009;10:605–16.
Trauernicht M, Martinez-Ara M, van Steensel B. Deciphering gene regulation using massively parallel reporter assays. Trends Biochem Sci. 2020;45:90–1.
Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012;30:271–7.
Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell. 2016;165:1519–29.
McCullum EO, Williams BA, Zhang J, Chaput JC. Random mutagenesis by error-prone PCR. Methods Mol Biol. 2010;634:103–9.
Kircher M, Xiong C, Martin B, Schubach M, Inoue F, Bell RJA, et al. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat Commun. 2019;10:3583.
van Arensbergen J, Pagie L, FitzPatrick VD, de Haas M, Baltissen MP, Comoglio F, et al. High-throughput identification of human SNPs affecting regulatory element activity. Nat Genet. 2019;51:1160–9.
Inoue F, Kircher M, Martin B, Cooper GM, Witten DM, McManus MT, et al. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 2017;27:38–52.
Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sal lari R, et al. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res 2014;24:1–13.
Roman TS, Marvelle AF, Fogarty MP, Vadlamudi S, Gonzalez AJ, Buchkovich ML, et al. Multiple hepatic regulatory variants at the GALNT2 GWAS locus associated with high-density lipoprotein cholesterol. Am J Hum Genet. 2015;97:801–15.
Vockley CM, Guo C, Majoros WH, Nodzenski M, Scholtens DM, Hayes MG, et al. Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort. Genome Res. 2015;25:1206–14.
Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell. 2018;172:1132–4.
Ranjha L, Howard SM, Cejka P. Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma. 2018;127:187–214.
Shen MW, Arbab M, Hsu JY, Worstell D, Culbertson SJ, Krabbe O, et al. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature. 2018;563:646–51.
Leenay RT, Aghazadeh A, Hiatt J, Tse D, Roth TL, Apathy R, et al. Large dataset enables prediction of repair after CRISPR-Cas9 editing in primary T cells. Nat Biotechnol. 2019;37:1034–7.
Chen W, McKenna A, Schreiber J, Haeussler M, Yin Y, Agarwal V, et al. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 2019;47:7989–8003.
McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38.
Li S, Li J, Zhang J, Du W, Fu J, Sutar S, et al. Synthesis-dependent repair of Cpf1-induced double strand DNA breaks enables targeted gene replacement in rice. J Exp Bot. 2018;69:4715–21.
Yeh CD, Richardson CD, Corn JE. Advances in genome editing through control of DNA repair pathways. Nat Cell Biol. 2019;21:1468–78.
Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096.
Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71.
Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18:67–83.
Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–62.
Miller SM, Wang T, Randolph PB, Arbab M, Shen MW, Huang TP, et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat Biotechnol. 2020;38:471–81.
Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368:290–6.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579–86.
Knott GJ, Doudna JA. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361:866–9.
Liu JJ, Orlova N, Oakes BL, Ma E, Spinner HB, Baney KLM, et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature. 2019;566:218–23.
Yan WX, Chong S, Zhang H, Makarova KS, Koonin EV, Cheng DR, et al. Cas13d is a compact RNA-targeting type VI CRISPR effector positively modulated by a WYL-domain-containing accessory protein. Mol Cell. 2018;70:327–39 e325.
Komor AC, Badran AH, Liu DR. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168:20–36.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–83.
Shariati SA, Dominguez A, Xie S, Wernig M, Qi LS, Skotheim JM. Reversible disruption of specific transcription factor-DNA interactions using CRISPR/Cas9. Mol Cell. 2019;74:622–33 e624.
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–51.
Amabile A, Migliara A, Capasso P, Biffi M, Cittaro D, Naldini L, et al. Inheritable silencing of endogenous genes by hit-and-run targeted epigenetic editing. Cell. 2016;167:219–32 e214.
Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–61.
Hilton IB, D'Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–7.
Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol. 2015;11:198–200.
Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–8.
Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635–46.
Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer EPR, et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods. 2015;12:326–8.
Li K, Liu Y, Cao H, Zhang Y, Gu Z, Liu X, et al. Interrogation of enhancer function by enhancer-targeting CRISPR epigenetic editing. Nat Commun. 2020;11:485.
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–4.
Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature. 2017;551:464–71.
Song F, Stieger K. Optimizing the DNA donor template for homology-directed repair of double-strand breaks. Mol Ther Nucleic Acids. 2017;7:53–60.
Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510.
Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21:121–31.
Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014;3:e04766.
Hess GT, Tycko J, Yao D, Bassik MC. Methods and applications of CRISPR-mediated base editing in eukaryotic genomes. Mol Cell. 2017;68:26–43.
Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19:770–88.
Molla KA, Yang Y. CRISPR/Cas-mediated base editing: technical considerations and practical applications. Trends Biotechnol. 2019;37:1121–42.
Grunewald J, Zhou R, Lareau CA, Garcia SP, Iyer S, Miller BR, et al. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat Biotechnol. 2020;38:861–4.
Kurt IC, Zhou R, Iyer S, Garcia SP, Miller BR, Langner LM, et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat Biotechnol. 2020;39:41–6.
Zhao D, Li J, Li S, Xin X, Hu M, Price MA, et al. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat Biotechnol. 2021;39:35–40.
Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–57.
Hsu JY, Anzalone AV, Grünewald J, Lam KC, Shen MW, Liu DR, et al. PrimeDesign software for rapid and simplified design of prime editing guide RNAs. bioRxiv Preprint. 2020; https://doi.org/10.1101/2020.05.04.077750.
Kim HK, Yu G, Park J, Min S, Lee S, Yoon S, et al. Predicting the efficiency of prime editing guide RNAs in human cells. Nat Biotechnol. 2021;39:198–206.
Grampp S, Platt JL, Lauer V, Salama R, Kranz F, Neumann VK, et al. Genetic variation at the 8q24.21 renal cancer susceptibility locus affects HIF binding to a MYC enhancer. Nat Commun. 2016;7:13183.
Guo C, Ludvik AE, Arlotto ME, Hayes MG, Armstrong LL, Scholtens DM, et al. Coordinated regulatory variation associated with gestational hyperglycaemia regulates expression of the novel hexokinase HKDC1. Nat Commun. 2015;6:6069.
Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–8.
Krause MD, Huang RT, Wu D, Shentu TP, Harrison DL, Whalen MB, et al. Genetic variant at coronary artery disease and ischemic stroke locus 1p32.2 regulates endothelial responses to hemodynamics. Proc Natl Acad Sci U S A. 2018;115:E11349–58.
Li S, Li Y, Li X, Liu J, Huo Y, Wang J, et al. Regulatory mechanisms of major depressive disorder risk variants. Mol Psychiatry. 2020;25:1926–45.
Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, et al. Multiple genetic loci for bone mineral density and fractures. N Engl J Med. 2008;358:2355–65.
Chen XF, Zhu DL, Yang M, Hu WX, Duan YY, Lu BJ, et al. An osteoporosis risk SNP at 1p36.12 acts as an allele-specific enhancer to modulate LINC00339 expression via long-range loop formation. Am J Hum Genet. 2018;102:776–93.
Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife. 2016;5.
Yeo NC, Chavez A, Lance-Byrne A, Chan Y, Menn D, Milanova D, et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat Methods. 2018;15:611–6.
Thakore PI, D'Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12:1143–9.
Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–23.
McCarty NS, Graham AE, Studena L, Ledesma-Amaro R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat Commun. 2020;11:1281.
Schrode N, Ho SM, Yamamuro K, Dobbyn A, Huckins L, Matos MR, et al. Synergistic effects of common schizophrenia risk variants. Nat Genet. 2019;51:1475–85.
Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562:217–22.
Consortium GT, Laboratory DA, Coordinating Center -Analysis Working Group, Statistical Methods groups-Analysis Working Group, Enhancing Gg, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–13.
Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, et al. Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature. 2016;533:95–9.
Castaldi PJ, Guo F, Qiao D, Du F, Naing ZZC, Li Y, et al. Identification of functional variants in the FAM13A chronic obstructive pulmonary disease genome-wide association study locus by massively parallel reporter assays. Am J Respir Crit Care Med. 2019;199:52–61.
Hu Y, Stilp AM, McHugh CP, Rao SQ, Jain D, Zheng XW, et al. Whole genome sequencing association analysis of quantitative red blood cell phenotypes: the NHLBI TOPMed program. MedRxiv. 2021.
Ray JP, de Boer CG, Fulco CP, Lareau CA, Kanai M, Ulirsch JC, et al. Prioritizing disease and trait causal variants at the TNFAIP3 locus using functional and genomic features. Nat Commun. 2020;11:1237.
Canver MC, Lessard S, Pinello L, Wu Y, Ilboudo Y, Stern EN, et al. Variant-aware saturating mutagenesis using multiple Cas9 nucleases identifies regulatory elements at trait-associated loci. Nat Genet. 2017;49:625–34.
Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–7.
Bae HT, Baldwin CT, Sebastiani P, Telen MJ, Ashley-Koch A, Garrett M, et al. Meta-analysis of 2040 sickle cell anemia patients: BCL11A and HBS1L-MYB are the major modifiers of HbF in African Americans. Blood. 2012;120:1961–2.
Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa203154.
Esrick EB, Lehmann LE, Biffi A, Achebe M, Brendel C, Ciuculescu MF, et al. Williams: Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2029392.
Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578:229–36.
Li B, Niu Y, Ji W, Dong Y. Strategies for the CRISPR-based therapeutics. Trends Pharmacol Sci. 2020;41:55–65.
Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105:1620–5.
Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med. 2019;25:776–83.
Zeng J, Wu Y, Ren C, Bonanno J, Shen AH, Shea D, et al. Therapeutic base editing of human hematopoietic stem cells. Nat Med. 2020;26:535–41.
Vuckovic D, Bao EL, Akbari P, Lareau CA, Mousas A, Jiang T, et al. The polygenic and monogenic basis of blood traits and diseases. Cell. 2020;182:1214–31 e1211.
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Schoenfelder S, Fraser P. Long-range enhancer-promoter contacts in gene expression control. Nat Rev Genet. 2019;20:437–55.
Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 2019;366:1134–9.
Bryois J, Skene NG, Hansen TF, Kogelman LJA, Watson HJ, Liu Z, et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat Genet. 2020;52:482–93.
Ulirsch JC, Lareau CA, Bao EL, Ludwig LS, Guo MH, Benner C, et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat Genet. 2019;51:683–93.
Soskic B, Cano-Gamez E, Smyth DJ, Rowan WC, Nakic N, Esparza-Gordillo J, et al. Chromatin activity at GWAS loci identifies T cell states driving complex immune diseases. Nat Genet. 2019;51:1486–93.
Hu X, Kim H, Stahl E, Plenge R, Daly M, Raychaudhuri S. Integrating autoimmune risk loci with gene-expression data identifies specific pathogenic immune cell subsets. Am J Hum Genet. 2011;89:496–506.
Pers TH, Karjalainen JM, Chan Y, Westra HJ, Wood AR, Yang J, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun. 2015;6:5890.
Slowikowski K, Hu X, Raychaudhuri S. SNPsea: an algorithm to identify cell types, tissues and pathways affected by risk loci. Bioinformatics. 2014;30:2496–7.
Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet. 2018;50:621–9.
Li Y, Oosting M, Smeekens SP, Jaeger M, Aguirre-Gamboa R, Le KTT, et al. A functional genomics approach to understand variation in cytokine production in humans. Cell. 2016;167:1099–110 e1014.
Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods. 2017;14:297–301.
Xie S, Duan J, Li B, Zhou P, Hon GC. Multiplexed engineering and analysis of combinatorial enhancer activity in single cells. Mol Cell. 2017;66:285–99 e285.
Rao S, Yao Y, Soares de Brito J, Yao Q, Shen AH, Watkinson RE, et al. Dissecting ELANE neutropenia pathogenicity by human HSC gene editing. Cell Stem Cell. 2021;28:1–13.
Wilson KD, Wu JC. Induced pluripotent stem cells. JAMA. 2015;313:1613–4.
Sala L, Gnecchi M, Schwartz PJ. Long QT syndrome modelling with cardiomyocytes derived from human-induced pluripotent stem cells. Arrhythm Electrophysiol Rev. 2019;8:105–10.
Sankaran VG, Ghazvinian R, Do R, Thiru P, Vergilio JA, Beggs AH, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012;122:2439–43.
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6.
Kim D, Luk K, Wolfe SA, Kim JS. Evaluating and enhancing target specificity of gene-editing nucleases and deaminases. Annu Rev Biochem. 2019;88:191–220.
Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24:939–46.
Wagner DL, Amini L, Wendering DJ, Burkhardt LM, Akyuz L, Reinke P, et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat Med. 2019;25:242–8.
Morgens DW, Deans RM, Li A, Bassik MC. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat Biotechnol. 2016;34:634–6.
Tycko J, Wainberg M, Marinov GK, Ursu O, Hess GT, Ego BK, et al. Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements. Nat Commun. 2019;10:4063.
Lu Y, Tian Y, Shen R, Yao Q, Wang M, Chen M, et al. Targeted, efficient sequence insertion and replacement in rice. Nat Biotechnol. 2020;38:1402–7.
Acknowledgements
We sincerely thank KaiLong Li (the University of Texas Southwestern Medical Center) for helpful discussions.
Funding
Burroughs Wellcome Fund: DP2HL137300 (D.E.B.), The National Heart, Lung, and Blood Institute: P01HL032262 (D.E.B.), Sichuan Science and Technology Program: 2019YFH0137 (Y.Y.), Chengdu University of Traditional Chinese Medicine: XSGG2019004 (Y.Y.). Funders had no role in the design of the study; in the collection, analysis, and interpretation of the data; or in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
SR, YY, and DEB conducted the literature review. SR and YY drafted the manuscript, and DEB revised it. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rao, S., Yao, Y. & Bauer, D.E. Editing GWAS: experimental approaches to dissect and exploit disease-associated genetic variation. Genome Med 13, 41 (2021). https://doi.org/10.1186/s13073-021-00857-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-021-00857-3