
Abstract
Background
Coat protein complex 1 (COPI) is integral in the sorting and retrograde trafficking of proteins and lipids from the Golgi apparatus to the endoplasmic reticulum (ER). In recent years, coat proteins have been implicated in human diseases known collectively as “coatopathies”.
Methods
Whole exome or genome sequencing of two families with a neuro-developmental syndrome, variable microcephaly and cataracts revealed biallelic variants in COPB1, which encodes the beta-subunit of COPI (β-COP). To investigate Family 1’s splice donor site variant, we undertook patient blood RNA studies and CRISPR/Cas9 modelling of this variant in a homologous region of the Xenopus tropicalis genome. To investigate Family 2’s missense variant, we studied cellular phenotypes of human retinal epithelium and embryonic kidney cell lines transfected with a COPB1 expression vector into which we had introduced Family 2’s mutation.
Results
We present a new recessive coatopathy typified by severe developmental delay and cataracts and variable microcephaly. A homozygous splice donor site variant in Family 1 results in two aberrant transcripts, one of which causes skipping of exon 8 in COPB1 pre-mRNA, and a 36 amino acid in-frame deletion, resulting in the loss of a motif at a small interaction interface between β-COP and β’-COP. Xenopus tropicalis animals with a homologous mutation, introduced by CRISPR/Cas9 genome editing, recapitulate features of the human syndrome including microcephaly and cataracts. In vitro modelling of the COPB1 c.1651T>G p.Phe551Val variant in Family 2 identifies defective Golgi to ER recycling of this mutant β-COP, with the mutant protein being retarded in the Golgi.
Conclusions
This adds to the growing body of evidence that COPI subunits are essential in brain development and human health and underlines the utility of exome and genome sequencing coupled with Xenopus tropicalis CRISPR/Cas modelling for the identification and characterisation of novel rare disease genes.
Similar content being viewed by others

Background
The endomembrane system is composed of a group of organelles (including the endoplasmic reticulum (ER), Golgi apparatus and endoplasmic reticulum-Golgi intermediate compartment (ERGIC)). It allows modification, packaging and transport of proteins through the secretory pathway in eukaryotic cells [1]. Proteins move between different organelles in membrane-bound transport vesicles coated with proteins. Protein coat complexes include coatomer protein I (COPI) complex, coatomer protein II (COPII) complex, a number of adaptor protein (AP) complexes including clathrin adaptor protein complexes, and the retromer complex [2]. These protein coats select cargo (proteins and lipids) for transport and facilitate transport vesicle formation. The functioning of protein coats is a fundamental cellular process. Knockout of certain subunits of COPI and COPII, such as COPB2, in mice is embryonic lethal [3, 4]. Depletion of COPI in cancer cells results in decreased cell survival, impaired autophagy and ER stress [5].
COPI consists of seven subunits (α, β (the focus of this study), β’, γ, δ, ε and ζ) (Fig. 1a) [6]. The COPI subunits can be divided into the F- (or Adaptor-like) subcomplex (containing the β, γ, δ and ζ subunits) and the B- (or Cage-like) subcomplex (α, β′ and ε) (Fig. 1a) [7,8,9]. The adaptor-like subcomplex, which contains β-COP, is analogous to the clathrin adaptor AP subcomplexes [10]. Both β-COP and γ-COP contain an α-solenoid “trunk” domain, connected by a linker to a small appendage domain [11].

Structure and function of the COP1 complex. a COPI consists of a scaffold “B-subcomplex” (blue) and an adaptor “F-subcomplex” (green). When GTP-bound, two ARF1 small GTPase molecules associate with the membrane and bind COPI via the β-COP and γ-COP subunits. A number of subunits of this complex have been implicated in human disease as shown. b COPI complexes and their ARF1 molecules associate into triads. Cargo, such as ER-resident proteins which need to be returned from the Golgi, are selected by binding directly with COPI subunits or indirectly through transmembrane receptors which in turn bind with COPI. COPI polymerises on the membrane enabling its deformation/curvature, and eventually budding and scission of the transport vesicle. When released, the vesicle’s coat is shed and ARF1 and COPI dissociate. Adapted from Nickel et al. [12]
ARF1, a small GTPase, catalyses COPI recruitment to the vesicle. When GTP-bound, ARF1 becomes stabilised in the membrane and initiates recruitment of COPI through binding with the trunk domains of the β-COP and γ-COP subunits [13]. COPI causes increased curvature of the membrane, until a vesicle forms and scission occurs [14]. After the vesicle is released, the COPI coat is shed and ARF1 and COPI can dissociate (Fig. 1b).
The primary functions of COPI are returning ER-resident proteins back from the Golgi to the ER, recycling certain transmembrane cargo receptors from the cis-golgi and ERGIC, and intra-Golgi retrograde transport [15]. COPI is also involved in lipid droplet formation and lipolysis [16]. Depletion of β-COP results in defective cargo transport and defective compartmentalisation of the ERGIC trans-Golgi network, Golgi, and recycling endosomes, emphasising its importance in both compartmentalisation and transport [17].
As with other coat protein complexes, several elements of the COPI machinery have been linked to human disease. The term “coatopathies” has been suggested to describe this heterogenous group of conditions (Fig. 1a) [2]. Homozygous variants in COPB2 have been linked to a severe microcephaly syndrome (MIM # 617800) [4]. The COPI complex may also play a role in Alzheimer disease as it is involved in the localisation of amyloid precursor protein and silencing of δ-COP leads to decreased amyloid plaques and improved memory in mice [18]. Interestingly, loss-of-function mutations in ARCN1 (which encodes δ-COP) cause a distinctive syndrome of craniofacial abnormalities, microcephalic dwarfism and mild developmental delay (MIM # 617164) [19]. Loss-of-function variants in COPA cause an autoimmune disorder of arthritis and lung disease (MIM # 616414) [20, 21]. Hypomorphic variants in ARF1 (which though not part of COPI, interacts closely with it, as described above) have been linked to brain malformations including periventricular heterotopia (MIM # 618185) [22]. β-COP has never been associated with Mendelian disease.
This study aimed to identify the molecular cause of a syndromic presentation in two unrelated families and investigate the pathogenicity of putative variants. We describe a new genetic disorder identified in six individuals across two families associated with biallelic variants in COPB1. This syndrome is typified by a severe intellectual disability with variable microcephaly and cataracts.
Methods
Subjects and sequencing
As part of clinical care, Exeter Clinical Laboratory performed whole exome sequencing on lymphocyte-extracted DNA from a duo of two affected sisters from Family 1. This involved exome sequence analysis of the coding region and conserved splice sites of 23,244 genes by next-generation sequencing (Twist Core Human Exome/Illumina NextSeq). The duo analysis utilised a gene-agnostic approach to identify rare homozygous or compound heterozygous variants shared by both sisters. Segregation analysis of a candidate variant was undertaken on lymphocyte-extracted DNA from all family members in keeping with routine clinical practice. As this investigation identified a splicing variant in a non-morbid gene, the participants were recruited to a research study to investigate the functional effect of these variants using lymphocyte-extracted RNA. Patient enrolment took place at University Hospital Southampton UK.
Family 2 were recruited as part of a multicentre clinical genome study in consanguineous Saudi populations [23]. Whole genome sequencing was performed on lymphocyte-extracted DNA and data was analysed with both gene-agnostic and gene panel approaches. The enrolment, data collection and analysis were conducted at King Abdulaziz Medical City, Riyadh, Saudi Arabia. Data collection was performed by analysing results from electronic health records. WGS was carried in clinical CAP/CLIA-accredited laboratories as part of a collaborative project. WGS was performed using HiSeq 4000. This family was identified with use of the GeneMatcher platform [24].
Analysis of splice site variant
In silico analysis
The prediction tools, Human Splicing Finder, Splice Site Prediction by Neural Network, MaxEntScan and SpliceAI, were used to predict the impact of Family 1’s variant on splicing [25,26,27,28].
Reverse transcriptase PCR (RT-PCR)
The effect of Family 1’s variant on splicing was investigated using RT-PCR. Blood from the probands, parents and unaffected siblings was collected in PAXgene® Blood RNA Tubes (PreAnalytiX, Hombrechtikon, Switzerland). RNA was then extracted from blood samples using the PAXgene blood RNA kit (PreAnalytiX, Hombrechtikon, Switzerland) and quality control was performed using a 2100 Bioanalyzer Instrument (Agilent, Santra Clara, USA). RNA extracted from blood samples was converted to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, USA). The primer pairs were designed using Primer 3 and ordered from Integrated DNA Technologies (IDT, Coralville, USA) (COPB1_ c957+1G>T_F_CTGGTGAATGAGAAGGATGCA, COPB1_c957+1G>T_R RV=GGCTTCACGAACAAACTCCA).
PCR experiments were performed using GoTaq G2 Polymerase PCR system (Promega, Madison, USA) and separated by agarose gel electrophoresis. The amplicons were then documented under Chemidoc XRS+ (Bio-Rad, Hercules, USA). The amplicons for further analysis were cloned into plasmids using a TA cloning kit, with pCR 2.1 vector (Thermo Fisher Scientific, Waltham, USA). The plasmids carrying inserts were sent to Source Bioscience (Nottingham, UK) for Sanger sequencing.
Xenopus care
Nigerian strain Xenopus tropicalis were housed and maintained within the European Xenopus Resource Centre, University of Portsmouth, in re-circulating systems (25 °C) with 15% daily water changes on a 13–11-h light-dark cycle. All procedures were conducted in accordance with the Home Office Code of Practice, under PPL 70/8983 with approval from the University of Portsmouth’s Animal Welfare and Ethical Review Body.
Adult female X. tropicalis were primed the evening prior to egg collection with 10 IU hCG (Chorulon, Intervet) and received a boosting dose of 100 IU hCG the following morning. Egg clutches were obtained by gentle abdominal massage and fertilised with cryopreserved sperm. Frozen X. tropicalis spermatozoa were generated within the European Xenopus Resource Centre from 12- to 15-month-old male X. tropicalis exhibiting enhanced nuptial pads. Sperm cryopreservation followed a protocol adapted from Sargent and Mohun [29].
Generating knockout animals using CRISPR-Cas9
The exon structure of the copb1 gene in X. tropicalis and humans is identical despite the difference shown currently on Xenbase’s gene page; we identified an untranslated exon contributing to the 5′ UTR of the gene by using its conservation with X. laevis. The target region within copb1 exon 8 was identified using Xenbase (originally designed against v9.1 of the genome and checked against v10) [30], and two single-stranded oligonucleotides were selected based on the following criteria: high mutagenic activity, minimal predicted off-target events (CRISPRscan) [31] and a high frameshift frequency (indelphi) [32].
sgRNA3-taatacgactcactataGGACTCGCTCATGGGAAGGGgttttagagctagaa
sgRNA4-taatacgactcactataGGAGCACTCGCTCATGGGAAgttttagagctagaa
Single guide RNAs (sgRNA) were synthesised from single-stranded oligonucleotides (Invitrogen, above) containing the T7 promoter as described in the Taq-based method by Nakayama et al., using the T7 megashortscript kit (Invitrogen) [33]. sgRNAs were subsequently purified (SigmaSpin post-reaction clean up columns) and visualised by agarose gel electrophoresis. The quantity and purity were assessed using a NanoDrop 1000 spectrophotometer and sgRNAs were stored at − 80 °C as single use aliquots. Each sgRNA (500 pg) was co-injected with Cas9 protein (2.6 ng, spy Cas9 NLS, NEB) at 1-cell and 2-cell stages into X. tropicalis embryos from both a wild-type strain and a transgenic line ([Xtr.Tg (tubb2b:GFP)Amaya] RRID:EXRC_3001) [34]. Although we show un-injected control tadpoles, these experiments were performed alongside CRISPR-based knockouts of other genes and the phenotypes shown here were specific to the copb1 sgRNA(s).
To determine the efficiency of indel formation, the target region of interest was amplified from genomic DNA (Lysis Buffer: 50 mM Tris (pH 8.5), 1 mM EDTA, 0.5% [v/v] Tween-20, 100 μg/ml Proteinase K) by polymerase chain reaction, using primers designed in Primer3 (http://primer3.ut.ee; Exon8 FWD: TGTGTCCCTAGGCAGCCG, Exon8 REV: CGAGGACACCAGTTCCAGT).
A T7 Endonuclease I (NEB) assay was used to indicate success of indel formation and this was confirmed using Sanger sequencing (Genewiz) [34, 35]. Due to the results seen as a consequence of the Family 1 mutation, we tested whether exon skipping had occurred in these samples. cDNA was prepared using Tristar solution [36] from control and crispant tadpoles; PCR products (amplified using primers spanning Exon6-7:CTACAAGGTTTGCCATGCCAAC and Exon9-10:GATGACCAGCTCTTCCACGTTG) were subcloned into pGEMTeasy and compared by Sanger sequencing (Genewiz).
Phenotypic analysis of knockout tadpoles
Gross morphological differences between un-injected tadpoles and crispant tadpoles were identified using a Zeiss Axio Zoom.V16 Stereomicroscope (with fluorescence for GFP-expressing animals). Detailed structural differences were examined by high-resolution microcomputed tomography (MicroCT). For bright-field/fluorescence microscopy, tadpoles were anaesthetised in 0.025% MS222, whilst tadpoles for MicroCT were fixed in paraformaldehyde following the method described for wholemount in situ hybridisation of Xenopus embryos [37]. These fixed specimens were immediately contrast stained in phosphotungstic acid (PTA) following an adapted protocol from Metscher [38]. Briefly, samples were stained in 1% PTA (in water) for 24 h, washed in methanol and placed in 1% PTA for a further 24 h. Specimens were allowed to rest in methanol for 1 week, before samples were embedded in 0.7% agarose within a 20 μl pipette tip (to prevent movement during imaging). Samples were imaged using the ZEISS Xradia Versa 520 (Carl Zeiss Microscopy, Pleasanton, CA, USA) set to operate at a voltage of 50 kV and a current of 75 mA. A × 4 objective lens was used, resulting in an effective isotropic voxel size of 3.1 μm. In total, 1601 projections were collected over 360° with an exposure time of 2.0 s per projection. Each tomogram was reconstructed to 16-bit grey-level images using the manufacturer’s software (Scout and Scan Reconstructor, Carl Zeiss Microscopy, Pleasanton, CA, USA) which employs a filtered back projection algorithm. The imaged volumes were then visualised using TXM3DViewer (Carl Zeiss Microscopy, Pleasanton, CA, USA).
Analysis of missense variants
In silico analysis
SIFT, PolyPhen 2 and CADD tools were used to predict the impact of Family 2’s variant on β-COP protein structure and function [39,40,41,42]. Conservation at the nucleotide level was assessed using the PhyloP score which attempts to measure deviation from the neutral rate of substitution including conservation and acceleration [43]. ConSurf conservation scores were used to measure the evolutionary rate of the amino acid [44]. PyMOL (Schrodinger) package was used to model the effect of the missense variant on the 3D structure of β-COP in complex with other components of COPI coat complex with the ArfGAP2 uncoating factor (pdb file 5NZS).
Site-directed mutagenesis of COPB1 constructs
Specific patient mutation was introduced into a Myc-DDK-tagged COPB1 transcript 1 (NM_016451) expression vector (Origene, MD, USA) using the Q5 Site-Directed Mutagenesis Kit (New England BioLabs, MA, USA) following manufacturer’s instructions with customised primers designed using NEB Base Changer [45] (COPB1_ c.1651_T>G _F GGATGGAGATgTCTTTGTTGC, COPB1_ c.1651_T>G _R AGAAGGAATCCTCTCAAG).
Plasmids were purified using the QIAprep Spin Miniprep Kit (Qiagen, Hilden, Germany) and successful mutagenesis was confirmed with Sanger sequencing by Source Bioscience (Nottingham, UK) using custom sequencing primers (COPB1_c.1651_T>G TGAGGCACAGGGAAATTCGA).
Transfection-grade plasmid preparation was then performed with the QIAprep Spin Midiprep Kit (Qiagen, Hilden, Germany).
Culture of human cell lines
hTERT RPE-1 cells (immortalised human retinal pigmentary epithelial cells (American Type Culture Collection, Manassas, VA, USA)) were maintained in a 50/50 mix of Dulbecco’s Modified Eagles Medium (DMEM) and Ham’s F12 (DMEM/F12) at 37 °C 5% CO2. Human embryonic kidney 293 cells (HEK 293 cells (American Type Culture Collection, Manassas, VA, USA)) were maintained in high glucose DMEM at 37 °C 5% CO2.
Nucleofection/transfection of cell lines
hTERT-RPE1 cells were nucleofected with mutant plasmid or wild type (WT) plasmid using the Nucleofector 4D (Lonza) and P3 kit (Lonza), following manufacturer’s instructions. HEK293 cells were transfected with mutant or WT plasmid using PEI.
Immunofluorescence staining and confocal imaging
Forty-eight hours after nucleofection, hTERT-RPE1 cells were incubated for 30 min with 5 μM BODIPY-TR-ceramide (Thermo Fisher Scientific, Waltham, MA, USA) complexed with bovine serum albumin (BSA) in phosphate-buffered saline (PBS) prepared following manufacturer’s instructions. After 30 min, cells were rinsed with ice-cold DMEM/F12 and incubated with fresh medium at 37 °C 5% CO2 for a further 30 min. Cells were fixed with 4% paraformaldehyde, blocked with 1% (w/v) non-fat milk powder/PBS and co-stained with mouse anti-c myc antibody (clone 9E10, Sigma Aldrich) and nuclear stain (DAPI (4′,6-diamidino-2-phenylindole) (Thermo Fisher Scientific, Waltham, MA)), and AlexaFluor secondary antibodies (Invitrogen). Alternatively, cells were fixed in ice-cold methanol at − 20 °C for 5 min, permeabilised with 0.1% (v/v) Triton X-100/PBS at room temperature for 5 min, blocked with 1% (w/v) non-fat milk powder/PBS and immunostained with rabbit anti giantin (ab37266, Abcam) and mouse anti-β-COP (ab2899, Abcam) or rabbit anti SERPINH1 (HPA029198, Atlas Antibodies) and mouse anti-c myc antibody (clone 9E10, Sigma Aldrich) and nuclear stain (DAPI (4′,6-diamidino-2-phenylindole) (Thermo Fisher Scientific, Waltham, MA)) and AlexaFluor secondary antibodies (Invitrogen). Cells were imaged using confocal microscopy (Leica TCS SP5 Confocal Laser Scanning Microscope with LASX software). Images were processed in Adobe Photoshop and figures assembled in Adobe Illustrator.
SDS PAGE and western blotting
Forty-eight hours after transfection, HEK293 cells were treated with 10 μg/ml cycloheximide for 16 h. Cells were then lysed with NP40 lysis buffer and total protein levels were assessed using protein assay (DC Protein Assay, Bio-Rad Laboratories, Hercules, CA, USA). Fifteen microgrammes of total protein per sample was loaded onto a 4–12% Bis-Tris polyacrylamide gel and proteins separated by electrophoresis at 200 V for 1 h. Proteins were transferred to PVDF membrane, blocked with 1% non-fat dry milk powder/PBS. COPB1-cmyc was detected using mouse anti-c-myc antibody (clone 9E10, Sigma Aldrich) and beta actin (control) was detected using anti-beta actin antibody (clone AC15, Sigma Aldrich). HRP-conjugated secondary antibodies were used and immunobands visualised with SuperSignal West Femto reagent (Pierce) on a Chemidoc XRS+ (Bio-Rad, Hercules, USA). Densitometry analysis was performed in ImageLab software (Bio-Rad, Hercules, USA).
Results
Clinical information
Family 1
Family 1 are of Roma ethnicity (Fig. 2a). Patient IV2 was the product of an uncomplicated pregnancy and was born at term weighing 3.25 kg (0.2 SD, 50th centile). Antenatal ultrasounds and maternal health were normal. During the neonatal period, she was noted to have torticollis and required physiotherapy. Breastfeeding was normal. She had low tone and came to medical concern at age 8 months as she was not sitting or smiling. Her early milestones were severely delayed (Table 1). She can now go up and down stairs but cannot climb. Her first words were very delayed, occurring at 3 years of age (“Mama”). Her language has subsequently improved and she can speak in short phrases and follow two-step commands. Her speech is unclear but understandable for family members. Her head circumference is − 2.6 standard deviations (SD). Socially she is fully dependant on her parents (Table 1).

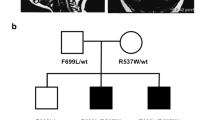
Pedigrees and clinical presentation of individuals with COPB1 mutations. a Family 1 includes two affected individuals IV2 and IV3. Individuals IV5, IV6 and IV7 are also suspected to have been affected with microcephaly and developmental delay. They died in early childhood and no further details were available. IV2 and IV3 have minor dysmorphism with up-slanting palpebral fissures. b Family 2 includes four affected individuals from two nuclear families all from the same Saudi tribe. c Gel electrophoresis of RT PCR amplicons in Family 1 demonstrates 2 bands in the homozygous state (IV2, an affected individual) and in the heterozygous state (III1, an unaffected parent). d Electropherograms of band A and band B from the heterozygous parent and homozygous proband. In the proband’s trace, a G has effectively been deleted due to the creation of a new donor site by the G>T mutation. In band B, Exon 8 has been skipped (proband and parent)
She has a history of recurrent upper respiratory tract infections and tonsillitis. A sleep study was performed for snoring and pauses in breathing during sleep; this was normal. Initial eye exams were normal. A left-sided nuclear sclerotic cataract was first noted at age 13 years and was associated with the development of a dense amblyopia. A divergent squint was also noted at this time. Menarche was early at 9 years of age. Later in childhood, she developed increased appetite, weight gain, acanthosis nigricans (axillary) and facial hypertrichosis. She has no history of seizures, and CT brain was normal in infancy.
Patient IV3 was also the product of an uncomplicated pregnancy with normal antenatal imaging. She had low tone during the neonatal period. Breast feeding was normal. As with her sister, her early milestones were severely delayed. Her gait is described as unsteady and she has difficulty with stairs. She has planovalgus foot deformity of one foot which is managed with orthopaedic footwear. She was noted to have congenital deafness with 65–90-dB hearing loss associated with a homozygous GJB2 c.71G>A variant. She has absent speech. She has difficulty communicating her needs especially pain. She understands some simple commands, such as “give it to me”. Initial eye exams were normal. She was noted to have bilateral cataracts at age 7 years which was treated with lens aspiration and intraocular implants at age 8 years. Her vision is normal. Her microcephaly is more severe than her sister’s and currently measures − 3.2 SD.
She developed focal seizures at 14 years. Her interictal EEG was normal, and seizures are controlled with levetiracetam. Menarche occurred at 11 years. As with her sister, she developed increased appetite, increased weight, facial hypertrichosis and axillary acanthosis nigricans later in childhood.
In addition to IV2 and IV3, three first cousins had microcephaly, developmental delay and died early in childhood (IV5, IV6, IV7). No further phenotypic information was available and genetic testing could not be undertaken.
In terms of dysmorphology, both IV2 and IV3 have up-slanting palpebral fissures, narrow palates and tapering fingers.
Family 2
Family 2 includes four affected individuals from two nuclear families, all from the same Saudi tribe (Fig. 2b). Prenatal and early neonatal information was not available for this family as subjects were born outside of the hospital. All four individuals have severe intellectual disability. Language impairment is variable in this family (individual IV8 is described as verbally fair at 15 years, IV4 has limited verbal output at 11 years whereas IV9 and IV7 have no language at 17 and 11 years of age respectively). All individuals have severe social impairment and are fully dependant for personal care. IV9 and IV4 have severe microcephaly (> − 3 standard deviations). All four affected individuals have developed cataracts and patient IV9 has had surgical treatment for this. Affected individuals in this family also have poor mobility. They have developed spasticity and have delayed motor skills. IV7 and IV8 use a wheelchair whereas IV9 and IV4 cannot sit or stand. IV9 has dystonia.
Immunodeficiency was also identified in Family 2 with severe lymphopenia being present in all four patients. Specifically, T cell lymphopenia was present with very depressed CD3+CD4+ cells. Immunoglobulin levels were normal overall; however, the patients failed to mount antibody responses to any specific antigens tested. Panel-based genetic testing as part of routine clinical care for known causes of severe combined immunodeficiency did not reveal any putative causal variants of this symptom. More detail on this feature is supplied in Additional file 1: Table S1.
Dysmorphology information/clinical photographs were not available for this family.
Genetic findings
Family 1
Family 1 are known to carry a 12;13 insertional translocation of approximately 761 Kb in size. This is present in a balanced form in individual III2 and individual IV2, as a duplication of chromosome 13q13.3 in individuals IV3 and IV1 (who is phenotypically normal) and as a deletion in 13q3.3 in individual IV4 (who is phenotypically normal). The copy number variant includes TRPC4 (MIM * 603651) which has not been linked to monogenic disease and UFM1 (MIM * 610553). Both the deletion and duplication have been classified as likely benign by a clinical laboratory. IV2 was found to have a homozygous GJB2 c.71G>A variant, established as the cause of her deafness. Gene panel testing for microcephaly (PanelApp Cataracts v2.2, PanelApp Severe Microcephaly v1.47) and cataracts performed on a clinical basis did not reveal a cause for the family’s phenotype.
Whole exome sequencing showed no biallelic predicted pathogenic variants in any known disease genes. (In total, 18 homozygous variants across 17 genes were identified. The candidate variants and their interpretation are supplied in Additional file 1: Table S2.) Further review of variants in non-MIM annotated genes identified a homozygous c.957+1G>T variant in COPB1, which affects the + 1 position of the donor site of exon 8 (Chr11(GRCh37):g.14504577C>A). Parents and unaffected siblings were all found to be carriers of this variant, and IV2 and IV3 are homozygous. Four predictive tools were in agreement, all in suggesting donor site loss [25,26,27,28]. SpliceAI additionally predicted a new donor site would be created 1 nucleotide before the WT exon 8 donor site, resulting in a 1 nucleotide frameshift (Table 3).
Modelling of the mutation onto the 3D structure of β-COP protein in complex with other components of the COPI coat leaf in complex with the ArfGAP2 uncoating factor (pdb file 5NZS) was performed [11].
This splice site mutation lies in the trunk domain β-COP but not at the ARF1 binding site. It leads to exon 8 skipping which causes a 108-bp deletion, and an in-frame deletion of 36 amino acids, resulting in the loss of a helix-turn-helix motif from the alpha solenoid structure (trunk) of β-COP (see Fig. 3b). It is noteworthy that this lost motif in the β-COP trunk lies at a small interface between β-COP and β’-COP (COPB2) (Fig. 3c). This is one of the linking points between the adaptor and scaffold coat subcomplexes composing the COPI complex.

Structure and conservation of COPB1 protein, and structural effect of missense variants. a Simple schematic diagram of COPB1 (β-COP) structure showing two main structural domains; trunk domain and appendage domain, and relative location of the c.957+1G>T and p.Phe551Val variants located towards the N terminal of the trunk domain. b COPB1 (β-COP) structure with the location of the 12 amino acids (in red) which are deleted due to exon 8 skipping caused by c957+1G>T. c COPB1 (β-COP) structure in context of COPI complex. Note exon 8 (red) makes up an important link between the scaffold and adaptor subcomplexes. d Alignment of COPB1 amino acid sequence from H. sapiens to yeast showing very high conservation of Phe551 across all species tested. e 3D structural modelling of COPB1 in complex with COPB2 and COPG1 showing the location of Phe551 near the site of interaction with COPG1. f Higher resolution image showing the location of Phe551Val mutation in a turn in the trunk domain
To verify bioinformatic predictions, RT PCR, gel electrophoresis and Sanger sequencing of COPB1 amplicons were performed on cDNA produced from blood-extracted RNA from a homozygote and heterozygote. Both the homozygous proband (IV2) and the heterozygous parent (III1) cDNA amplified 2 bands (A and B) (Fig. 2c). Sequencing of these bands showed that in both the proband and parent, band B was a PCR amplicon from a transcript with in-frame skipping of exon 8. Band A in the parent was a PCR amplicon of the wild-type transcript with exon 8 inclusion, whereas band A in the proband was a PCR amplicon of a mutant transcript in which exon 8 was included with a 1-bp deletion of c.958G. This causes a frameshift, leading to a new in-frame stop codon (TAA) 18 bp downstream of this deletion (and 18 bp downstream of the junction between exon 7 and exon 8). The mechanism through which this mutant transcript escapes nonsense-mediated decay (NMD) in the homozygote proband may be related to the fact that the premature stop codon is only 18 nucleotides downstream of the exon7/exon8 junction, as previous work has shown that premature stop codons less than 50 nucleotides from an exon/exon boundary escape NMD as they are not recognised by the exon junction complex in the NMD pathway [46]. However, why the transcript undergoes NMD in the heterozygote parent (we did not observe this transcript in the parent) is unclear.
Family 2
WGS was undertaken in all affected family members and several unaffected relatives (8 individuals). Only one appropriately segregating homozygous variant was identified, underpinning the power of sequencing multiple family members in large pedigrees [23]. The identified mutation was a homozygous COPB1 missense variant, c.1651T>G in exon 14 causing a Phe551Val amino acid substitution (Chr11(GRCh37):g.14496127A>C), falling in the β-COP-trunk domain (Fig. 3a). This variant was predicted by PolyPhen 2 to be probably damaging, predicted by SIFT to be deleterious and predicted by CADD (PHRED score) to be deleterious, scoring 32 (Table 3) [39,40,41,42].
Phe551 is a highly conserved residue, conserved from human to C. elegans and S. cerevisiae (Fig. 3b), with a high PhyloP score of 5.21(− 14.1;6.4), modest Grantham score at 50 (0–215) and high ConSurf score of 8/9. Allele frequency was assessed using the gnomAD database. The Family 2 variant (c.1651T>G, p.Phe551Val) was absent.
Modelling of this mutation onto the 3D structure of β-COP protein in complex with other components of the COPI was performed [11]. This showed that the mutation falls within the trunk domain, but lies distant to its binding site with ARF1 and COPI subunits (Fig. 3c). Its significance is more likely to be its effect on internal β-COP structural integrity. Phe551Val lies at a turn or a loop connecting two alpha helices which may influence the directionality of the following helix or the folding of the protein (Fig. 3d). Further functional work was undertaken to investigate these predictions, see the “Modelling of Family 2 missense variant” section.
copb1 disruptions in X. tropicalis
In order to investigate the phenotypic effect of β-COP truncation, we used CRISPR/Cas9 to introduce insertions and deletions (indels) into genomic copb1 in the western clawed frog X. tropicalis at a site adjacent to the genomic location of the variant found in participating Family 1. There is now a large body of evidence showing that making targeted indels in the western clawed frog is so efficient that it is possible to analyse the resulting phenotypes in founder animals [47,48,49,50,51,52,53]. This results in robust but more rapid testing of the causality of a gene variant in disease than waiting for F1 or F2 animals to be available. The human and X. tropicalis copb1 genes share exon structures in the updated frog genome (v10, Xenbase), their COPB1 proteins are > 95% identical (Fig. 3d, Additional File 1: Fig. S1a) and both regions containing the patient variants are conserved.
To mimic the Family 1 human genomic change in Xenopus as closely as possible (Family 1; exon 8), sgRNAs were designed to create double-stranded DNA breaks (DSBs) at the end of exon 8 in X. tropicalis copb1 (Fig. 4a, b). Analysis of the target genome region of mosaic, crispant embryos showed both skipping of exon 8 and indels in exon 8 (Fig. 4c, d, Additional file 1: Figure S1) and that both the eyes and the remainder of the tadpole were equally mosaic (Additional file 1: Figure S2). RT-PCR from two separate sets of embryos showed that there was a significant, consistent level of exon skipping across injected embryos (Fig. 4d, Additional file 1: Figure S1). Sanger sequencing of RT-PCR products confirmed the expression of several different transcripts from the targeted region, including transcripts in which exon 8 is skipped, and transcripts in which exon 8 is retained but which contain deletions and insertions (Additional file 1: Figure S1(C,D)). Sequencing of PCR products from genomic DNA from single tadpoles confirmed that the level of mosaicism was high, few cells retained the unaltered DNA sequence at the targeted locus (Additional file 1: Figure S2). PCR products of the CRISPRed locus were subcloned and individually sequenced, showing that a series of deletions had occurred ranging from 5 to 41 bp (Fig. 4c). These large deletions extended well into the intron and were associated with the embryos showing exon 8 skipping.

Targeted CRISPR/Cas9 disruption of copb1 exon 8 generates a range of insertion-deletion changes in vivo. Xenopus tropicalis copb1 has 21 exons, including an untranslated first exon (a). CRISPR/Cas9 directed indel formation disrupting X. tropicalis exon 8 using 2 sgRNAs (red: sgRNA3 and sgRNA4 (b)) results in homozygous frameshift and splice changes akin to those identified within the patient subpopulation. Genotyping analysis of X. tropicalis crispants details the range of indels across three groups of 10 pooled tadpoles (NF41) injected with sgRNA3 following PCR amplification and Sanger sequencing of subclones (c). The 41-bp deletion observed in gDNA subclones extends into the intronic region and is proposed to induce exon 8 skipping in crispant X. tropicalis tadpoles. Total RNA was obtained from 4 groups (un-injected control (1), copb1 sgRNA 3 crispant set 1 (2) and copb1 sgRNA 3 crispant set 2 (3), injected control (4)) of 10 pooled individuals as outlined in Guille 1999 [51] and cDNA synthesised using Primer Design’s Reverse Transcription Premix 2. Amplification across the copb1 region of interest revealed a band at 382 bp (exons: 7, 8 and 9) and an additional band in crispants at 262 bp (exons: 7 and 9, Additional file 1: Fig. S1D)
Two sgRNAs targeted the exon and each produced identical phenotypes in independent experiments, showing that the phenotype was not due to off-target DSBs. Comparing the images from Fig. 5d, g and j with those from Additional file 1: Figure S3 for the crispants makes this clear, with unilateral cataracts, microcephaly and occasional anophthalmia in both sets of tadpoles. The chosen sgRNA(s) were tested for potential off-target sites using CRISPRScan and Cas-OFFinder, neither detected any significant hits. Since these programmes both use previous versions of the genome, we repeated this process manually using BLAST on X. tropicalis genome version 10. In this case, a single potential off-target site was detected, but in an intergenic region. Together with the experimental evidence above, this shows that the observed phenotype is due to cutting of the target locus.

F0 free-feeding wild-type and transgenic [Xtr.Tg (tubb2b:GFP) Amaya, RRID: EXRC_3001] Xenopus tropicalis crispants phenocopy key clinical hallmarks. Images show tadpole head structural morphology under normal conditions (a–c) and across the range of phenotypes observed: mild microcephaly (d–f), microcephaly with cataract(s) (g–i) and microcephaly with absent or missing eye structure(s) (j–l). White arrows on GFP fluorescence images show normal forebrain structures in un-injected control animals (b) and altered forebrain structures in mutants, with an increasing severity of microcephaly (e, h, k). Red arrows demonstrate the same forebrain structural trends in higher resolution MicroCT imaging (1% phosphotungstic acid contrast stain: c, f, i, l). Cataract formation is indicated in bright-field and MicroCT images by yellow arrows (g, i) and can be seen as a loss of GFP expression in fluorescence imaging (h), whilst green arrows highlight absent or missing structures of the eye (j, l). Re-occurring eye abnormalities were documented in 30 un-injected and 30 crispant tadpoles (exon 8, sgRNA3) to show the prevalence of cataract formation (mean, 14 tadpoles) and missing eye structures (mean, 5 tadpoles (m)). Further, 8 transgenic X. tropicalis crispant tadpole brains were imaged and measured (mm) 3 days post-fertilisation (control mean 1 mm, SD 0.02; Crispant mean 0.845 mm, SD 0.12 (t = 3.701; p = 0.007)) and 5 days post-fertilisation (control mean 6.37 mm, SD 0.29; Crispant mean 5.34 mm, SD 0.20 (t = 8.317; p = 0.000)) revealing sustained, significant reduction in brain length. Brain length measured as the distance from the forebrain to the hindbrain (Additional file 1: Fig. S5a) was expressed as a percentage of the mean of the control (N). Kaplan-Meier survival analysis of 50 un-injected control (median survival time 4.4 days) and crispant X. tropicalis tadpoles, injected at the one-cell stage with either sgRNA1 (exon 3 (Additional file 1: Fig. S4, median survival time 2 days) or sgRNA3 (exon 8, median survival time 3 days) show a significant difference in survival by log-rank comparison p = 0.000 (o)
We compared the effects of introducing indels into exon 8 with those of introducing indels into exon 3 (Additional file 1: Figure S4(B,C)), where we hypothesised the effect of truncating the protein would be a more complete knockout, whether by nonsense-mediated decay of the copb1 transcript or by an earlier truncation of the protein. When introduced into exon 3 rather than 8, indels were more damaging to the embryo as evidenced from the survival curve (Fig. 5o) and images of the affected embryos (Additional file 1: Figure S4A). Whilst we cannot formally exclude the possibility that more out of frame indels occur in exon 3 than in exon 8, thus leading to the increased effect observed, the limited sequence data available (Fig. 4c and Additional file 1: Figure S4D) do not support this hypothesis strongly.
All experiments were replicated in embryos from at least three different females and the phenotype data shown are from embryos injected with CRISPR/Cas targeting exon 8 in one cell of a dividing, two-cell embryo. This results in the effect of the protein truncation or internal deletion being focused on one side of the injected embryo along the left-right axis with the other half acting as an internal control [38]. Detailed labelling of structures in tadpoles visualised using MicroCT and in the fluorescent tadpole brain are shown in Additional file 1: Figure S5. The crispants showed a range of deformities, including mild forebrain phenotypes that were only visible in transgenically labelled brains or by MicroCT imaging (Fig. 5): compare the forebrain structures shown by the white arrows in Fig. 5b with Fig. 5e and the red arrows in Fig. 5c with Fig. 5f. More obvious phenotypes include clear microcephaly in 27/30 crispants compared with 2/30 controls (telencephalon to cerebellum length for controls median 0.995 mm ± range of 0.03 mm; crispants median 0.855 mm ± range 0.17 mm; see Fig. 5n). Also, compare the neural structures highlighted by white arrows in Fig. 5b with those in Fig. 5h and Fig. 5k and the red arrows in Fig. 5c with those in Fig. 5i and Fig. 5l. Cataracts or anophthalmia were also observed in 19/30 embryos but were absent in controls (Fig. 5m; highlighted by the yellow arrows in Fig. 5g and i) and revealed by the loss of the fluorescence from the transgenic marker crystallin-GFP in the lens (compare the close-up images of the eyes in Fig. 5b, h). The most extreme phenotypes have a wide range of deformities that include almost complete loss of the eye (see the green arrow in Fig. 5j, l). The few surviving embryos when the gene was disrupted by indels in exon 3 also showed similar, but generally more exaggerated phenotypes (Additional file 1: Figure S4(a,d)). As with the human presentation, the phenotype was specific when taken as a whole, but variable on the individual level.
Modelling of Family 2 missense variant
We hypothesised that the Family 2 variant (c.1651T>G, p.Phe551Val) affects the local structures and folding of β-COP, which could affect its overall affinity of interactions within COPI complex and undertook in vitro analysis to assess this further.
Mutant COPB1 cmyc-DDK-tagged plasmids were generated using site-directed mutagenesis and successful mutagenesis was confirmed using Sanger sequencing. These plasmids were transfected into HEK293 cells, which were then treated with 10 μg/ml cycloheximide to inhibit protein synthesis to provide a more robust test of protein stability. Cells were lysed and protein extracted after 16 h and analysed by SDS PAGE and western blotting. Only a very subtle difference between β-COP-cmyc band intensity was observed between WT and mutant protein (Fig. 6a). Densitometry analysis revealed that relative, normalised levels of β-COP c.1651T>G p.Phe551Val (normalised to beta actin loading control) were lower than β-COP WT levels, suggesting a mild reduction in protein stability of the mutant β-COP (Fig. 6a).

Effect of expression of COPB1 mutation on in vitro localisation and stability of β-COP protein. a Western blot image of protein extracts from HEK293 cells transfected with wild-type, and c.1651T>G (COPB1 tagged with c-myc, immunoblotted with c-myc antibody to detect COPB1, and with beta-actin antibody as loading control). c.1651T>G shows a slightly reduced normalised level. b Immunofluorescence images of hTERT-RPE1 cells nucleofected with wild-type COPB1 (top row), and COPB1 c.1651T>G p. p.Phe551Val (lower row) tagged with c-myc. Cells were stained with Golgi stain Bodipy-TR-ceramide (red), nuclear stain DAPI (blue) and immunostained with anti-myc (green). Mutant expression shows more diffuse β-COP staining throughout the Golgi and outside the Golgi in the wider cytoplasm. Scale bar = 5 μm. c Immunofluorescence images of hTERT-RPE1 cells nucleofected with wild-type COPB1 (top row), and COPB1 c.1651T>G p. p.Phe551Val (lower row) tagged with c-myc. Cells were stained with nuclear stain DAPI (blue) and immunostained with an antibody to resident Golgi protein giantin (green) and anti-β-COP (red). This clarified that wild-type β-COP tended to localise to the edges of the Golgi stacks, whereas mutant β-COP was localised throughout the Golgi. Scale bar = 10 μm
Plasmids were also transfected into hTERT RPE-1 cells and after 48 h the Golgi was stained with BODIPY-TR-ceramide and cells were fixed and β-COP-cmyc was immunostained with anti-cmyc. Confocal microscopy imaging showed a subtle change in localisation of mutant β-COP compared to WT β-COP. Whilst WT β-COP could be seen in a defined pattern at the Golgi, mutant β-COP could be seen in a more diffuse location (Fig. 6b), suggestive of a defect in Golgi to ER recycling of the COPI complex. To investigate this further, we repeated transfections and immunostained the Golgi membrane with an antibody to giantin, which is a protein integral to the Golgi membrane, and co-immunostained with an antibody to β-COP. This showed again that β-COP c.1651T>G p.Phe551Val is more widely distributed throughout the cell, but also more widely distributed throughout the Golgi (Fig. 6c). Whereas wild-type β-COP localised specifically to the edges of the Golgi stacks, mutant β-COP is localised throughout the Golgi, as can be seen by the greater overlap of green (giantin) and red (β-COP) staining, which appears yellow (Fig. 6c). Staining of the ER with anti SERPINH1 showed no difference in localisation of wild-type β-COP and β-COP c.1651T>G p.Phe551Val relative to the ER (Additional file 1: Fig. S6). Together, these data suggest that β-COP c.1651T>G p.Phe551Val is defective in Golgi to ER recycling, with this mutant β-COP getting retarded in the Golgi.
Discussion
In this paper, we present a new syndrome characterised by severe developmental delay and cataracts with the majority of patients also being microcephalic. Family 2 also exhibits spasticity and immunodeficiency, whereas Family 1 demonstrates a “metabolic syndrome” phenotype from late childhood. Our model reflects this highly heterogenous phenotype with some Xenopus showing neither microcephaly nor cataracts, with others being severely affected. Our in silico and in vitro missense modelling work demonstrates a high conservation of the specific residue (c.1651T>G p.Phe551Val) mutated in this family and an impact on localisation of protein with this specific mutant residue observed in cells, with this mutant protein being retarded in the Golgi. Given that the primary role of COPI is to return ER-resident molecules from the Golgi, the localisation abnormality seen associated with the expression of the missense construct is likely to have a significant effect on normal functioning of the endomembrane system. As the splice variant in Family 1 causes skipping of a 108-bp exon, leading to an in-frame deletion of 36 amino acids in the protein, we hypothesise that both the missense and splice variants are hypomorphs which retain some functionality. We further hypothesise that null alleles may be embryonically lethal in mammals, as for other some coatomer subunits [3, 4]. This hypothesis is supported by data from Xenopus in which frameshift indels in exon 3 of copb1 were introduced mosaically using CRISPR/Cas. Survival rates were very low in these knockout animals and survivors’ phenotypes similar to, but generally more severe than those in embryos carrying indels in exon 8.
Microcephaly was a prominent phenotype in this syndrome and clearly reproduced in the Xenopus model. A homozygous variant in the related COPB2 (β’-COP) has been linked to a primary microcephaly syndrome [4]. The authors describe siblings with microcephaly, failure to thrive, spasticity, cortical blindness and severe developmental delay, which share some obvious similarities to, in particular, Family 2 in our study. ARCN1-related syndrome (δ-COP) also includes features of microcephaly and, albeit milder, developmental delay. Interfering RNA screens indicate that COPB1 may play an important role in cell division which could be linked to the microcephaly seen in these patients [54]. Previous studies have indicated that COPI has numerous roles in neuronal function. For example, it may play a role in the transport of RNA in neurons and is also involved in the transport of SNARE complex proteins which traffic neurotransmitters [55, 56]. Interferonopathies are a known cause of microcephaly, and notably a recent study investigating the protein network of interferon-stimulated genes identified β-COP as one of the two most connected proteins in this network of 1400 [57].
All affected individuals in this study had markedly delayed gross motor milestones. Interestingly, COPI (including β-COP) interacts with the protein Staismon which is thought to be important in synaptic transmission in motor circuit neurons and in motor axon outgrowth [58]. Additionally, COPI, through its subunit α-COP, binds with survival motor neuron protein (SMN), which is deficient in spinal muscular atrophy (SMA) and is involved in its axonal transport [59]. Furthermore, SMA patients show abnormal Golgi apparatus morphology which can be corrected by overexpression of α-COP [60].
Six of six individuals identified in this study have developed cataracts. Interestingly, the cataracts in Family 1 developed quite late in childhood, which is a less common presentation for congenital cataracts. Several other coatopathies demonstrate cataracts in their phenotype. For example, cranio-lenticulo-sutural dysplasia has been linked to homozygous variants in the COPII gene SEC23A. These patients had a missense mutation in SEC23A (use italics for this word please) which, when exogenously expressed in cells in culture, displayed a cellular phenotype suggestive of abnormal Golgi to ER trafficking [61]. The pathogenesis of this phenotype is unclear.
Family 2 exhibits the additional phenotype of immunodeficiency; this was absent from Family 1 who have had normal immunology studies. Given the role of coat proteins in the secretory pathway, we hypothesise altered immune function may be, at least in part, due to the high demand of plasma cells on protein (immunoglobulin) expression, modification and secretion. Additionally, β-COP is listed as a secretory granule membrane protein with a role in neutrophil degranulation on reactome database [62]. We also note the role of β-COP in MHC1 and CD4 degradation in HIV-infected cells [63, 64]. It is also possible that the additional feature of immunodeficiency and spasticity may be due to an as yet unidentified second variant in this highly selected family though no appropriate candidates were identified on extensive review.
Conclusions
In conclusion, we have described a novel recessive syndrome caused by biallelic variants in COPB1. Disrupting copb1 in Xenopus clearly shows phenotypic changes in common with the patients, in particular microcephaly and cataracts, strongly supporting the link between the patient variants and their pathology. The utility of these data in making the connection between the gene change, including exon skipping, and phenotype is another demonstration of how successful X. tropicalis is as a tool for the characterisation of novel disease genes. Taken together, our data add to the growing body of evidence that COPI subunits are essential for brain development and human health.
Availability of data and materials
Family 1’s whole exome sequencing data is not publicly available as it was generated in the UK National Health Service as part of the patients’ clinical investigations and cannot be shared outside of this service. Family 2’s whole genome sequencing data cannot be shared as this is not compliant with our IRB approval. The variants identified in both families have been deposited on ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/variation/996016/ [65] and https://www.ncbi.nlm.nih.gov/clinvar/variation/996037/ [66].
Abbreviations
- BSA:
-
Bovine serum albumin
- COPI:
-
Coat protein complex 1
- COPII:
-
Coatomer protein II
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- ER:
-
Endoplasmic reticulum
- ERGIC:
-
Endoplasmic reticulum-Golgi intermediate compartment
- HEK293:
-
Human embryonic kidney 293 cells
- NMD:
-
Nonsense-mediated decay
- MicroCT:
-
Microcomputed tomography
- PBS:
-
Phosphate-buffered saline
- PTA:
-
Phosphotungstic acid
- RT-PCR:
-
Reverse transcriptase PCR
- SD:
-
Standard deviation
- SMA:
-
Spinal muscular atrophy
- SMN:
-
Survival motor neuron protein
- TIDE:
-
Tracking of Indels by Decomposition
- WT:
-
Wild type
References
Vazquez-Martinez R, Diaz-Ruiz A, Almabouada F, Rabanal-Ruiz Y, Gracia-Navarro F, Malagon MM. Revisiting the regulated secretory pathway: from frogs to human. Gen Comp Endocrinol. 2012;175(1):1–9.
Dell'Angelica EC, Bonifacino JS. Coatopathies: genetic disorders of protein coats. Annu Rev Cell Dev Biol. 2019;35:131–68.
Baines AC, Adams EJ, Zhang B, Ginsburg D. Disruption of the Sec24d gene results in early embryonic lethality in the mouse. PLoS One. 2013;8(4):e61114.
DiStasio A, Driver A, Sund K, Donlin M, Muraleedharan RM, Pooya S, et al. Copb2 is essential for embryogenesis and hypomorphic mutations cause human microcephaly. Hum Mol Genet. 2017;26(24):4836–48.
Claerhout S, Dutta B, Bossuyt W, Zhang F, Nguyen-Charles C, Dennison JB, et al. Abortive autophagy induces endoplasmic reticulum stress and cell death in cancer cells. PLoS One. 2012;7(6):e39400.
Waters MG, Serafini T, Rothman JE. ‘Coatomer’: a cytosolic protein complex containing subunits of non-clathrin-coated Golgi transport vesicles. Nature. 1991;349(6306):248–51.
Fiedler K, Veit M, Stamnes MA, Rothman JE. Bimodal interaction of coatomer with the p24 family of putative cargo receptors. Science. 1996;273(5280):1396–9.
Lowe M, Kreis TE. In vitro assembly and disassembly of coatomer. J Biol Chem. 1995;270(52):31364–71.
Lowe M, Kreis TE. In vivo assembly of coatomer, the COP-I coat precursor. J Biol Chem. 1996;271(48):30725–30.
Schledzewski K, Brinkmann H, Mendel RR. Phylogenetic analysis of components of the eukaryotic vesicle transport system reveals a common origin of adaptor protein complexes 1, 2, and 3 and the F subcomplex of the coatomer COPI. J Mol Evol. 1999;48(6):770–8.
Dodonova SO, Aderhold P, Kopp J, Ganeva I, Rohling S, Hagen WJH, et al. 9A structure of the COPI coat reveals that the Arf1 GTPase occupies two contrasting molecular environments. Elife. 2017;6(e26691). https://elifesciences.org/articles/26691.
Nickel W, Brugger B, Wieland FT. Vesicular transport: the core machinery of COPI recruitment and budding. J Cell Sci. 2002;115(Pt 16):3235–40.
Yu X, Breitman M, Goldberg J. A structure-based mechanism for Arf1-dependent recruitment of coatomer to membranes. Cell. 2012;148(3):530–42.
Beck R, Prinz S, Diestelkotter-Bachert P, Rohling S, Adolf F, Hoehner K, et al. Coatomer and dimeric ADP ribosylation factor 1 promote distinct steps in membrane scission. J Cell Biol. 2011;194(5):765–77.
Arakel EC, Schwappach B. Formation of COPI-coated vesicles at a glance. J Cell Sci. 2018;131(5). https://jcs.biologists.org/content/131/5/jcs209890.
Beller M, Sztalryd C, Southall N, Bell M, Jackle H, Auld DS, et al. COPI complex is a regulator of lipid homeostasis. PLoS Biol. 2008;6(11):e292.
Styers ML, O'Connor AK, Grabski R, Cormet-Boyaka E, Sztul E. Depletion of beta-COP reveals a role for COP-I in compartmentalization of secretory compartments and in biosynthetic transport of caveolin-1. Am J Physiol Cell Physiol. 2008;294(6):C1485–98.
Bettayeb K, Hooli BV, Parrado AR, Randolph L, Varotsis D, Aryal S, et al. Relevance of the COPI complex for Alzheimer’s disease progression in vivo. Proc Natl Acad Sci U S A. 2016;113(19):5418–23.
Izumi K, Brett M, Nishi E, Drunat S, Tan ES, Fujiki K, et al. ARCN1 mutations cause a recognizable craniofacial syndrome due to COPI-mediated transport defects. Am J Hum Genet. 2016;99(2):451–9.
Jensson BO, Hansdottir S, Arnadottir GA, Sulem G, Kristjansson RP, Oddsson A, et al. COPA syndrome in an Icelandic family caused by a recurrent missense mutation in COPA. BMC Med Genet. 2017;18(1):129.
Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. 2015;47(6):654–60.
Ge X, Gong H, Dumas K, Litwin J, Phillips JJ, Waisfisz Q, et al. Missense-depleted regions in population exomes implicate ras superfamily nucleotide-binding protein alteration in patients with brain malformation. NPJ Genom Med. 2016;1.
Alfares A, Alsubaie L, Aloraini T, Alaskar A, Althagafi A, Alahmad A, et al. What is the right sequencing approach? Solo VS extended family analysis in consanguineous populations. BMC Med Genet. 2020;13(1):103.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928–30.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67.
Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535–48 e24.
Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4(3):311–23.
Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11(2–3):377–94.
Pearl E, Morrow S, Noble A, Lerebours A, Horb M, Guille M. An optimized method for cryogenic storage of Xenopus sperm to maximise the effectiveness of research using genetically altered frogs. Theriogenology. 2017;92:149–55.
Xenbase Accessed 01/03/2020 [Available from: http://www.xenbase.org/entry. Accessed 19 Feb 2021.
CRISPR Scan Accessed 01/03/2020 [Available from: http://www.crisprscan.org. Accessed 19 Feb 2021.
Indelphi Accessed 01/03/2020 [Available from: https://indelphi.giffordlab.mit.edu. Accessed 19 Feb 2021.
Nakayama T, Blitz IL, Fish MB, Odeleye AO, Manohar S, Cho KW, et al. Cas9-based genome editing in Xenopus tropicalis. Methods Enzymol. 2014;546:355–75.
Vouillot L, Thelie A, Pollet N. Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda). 2015;5(3):407–15.
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822–6.
Steinbach OC, Rupp RA. Quantitative analysis of mRNA levels in Xenopus embryos by reverse transcriptase-polymerase chain reaction (RT-PCR). Methods Mol Biol. 1999;127:41–56.
Guille M. Microinjection into Xenopus oocytes and embryos. Methods Mol Biol. 1999;127:111–23.
Metscher BD. MicroCT for comparative morphology: simple staining methods allow high-contrast 3D imaging of diverse non-mineralized animal tissues. BMC Physiol. 2009;9:11.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–d94.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20(1):110–21.
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–50.
NE Base changer Accessed 01/03/2020 [Available from: http://nebasechanger.neb.com. Accessed 19 Feb 2021.
Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci U S A. 2003;100(1):189–92.
DeLay BD, Corkins ME, Hanania HL, Salanga M, Deng JM, Sudou N, et al. Tissue-specific gene inactivation in Xenopus laevis: knockout of lhx1 in the kidney with CRISPR/Cas9. Genetics. 2018;208(2):673–86.
Deniz E, Mis EK, Lane M, Khokha MK. CRISPR/Cas9 F0 screening of congenital heart disease genes in Xenopus tropicalis. Methods Mol Biol. 1865;2018:163–74.
Hwang WY, Marquez J, Khokha MK. Xenopus: driving the discovery of novel genes in patient disease and their underlying pathological mechanisms relevant for organogenesis. Front Physiol. 2019;10:953.
Kariminejad A, Szenker-Ravi E, Lekszas C, Tajsharghi H, Moslemi AR, Naert T, et al. Homozygous null TBX4 mutations lead to posterior amelia with pelvic and pulmonary hypoplasia. Am J Hum Genet. 2019;105(6):1294–301.
Naert T, Colpaert R, Van Nieuwenhuysen T, Dimitrakopoulou D, Leoen J, Haustraete J, et al. CRISPR/Cas9 mediated knockout of rb1 and rbl1 leads to rapid and penetrant retinoblastoma development in Xenopus tropicalis. Sci Rep. 2016;6:35264.
Naert T, Vleminckx K. CRISPR/Cas9 disease models in zebrafish and Xenopus: the genetic renaissance of fish and frogs. Drug Discov Today Technol. 2018;28:41–52.
Sega AG, Mis EK, Lindstrom K, Mercimek-Andrews S, Ji W, Cho MT, et al. De novo pathogenic variants in neuronal differentiation factor 2 (NEUROD2) cause a form of early infantile epileptic encephalopathy. J Med Genet. 2019;56(2):113–22.
Kittler R, Pelletier L, Heninger AK, Slabicki M, Theis M, Miroslaw L, et al. Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat Cell Biol. 2007;9(12):1401–12.
Todd AG, Lin H, Ebert AD, Liu Y, Androphy EJ. COPI transport complexes bind to specific RNAs in neuronal cells. Hum Mol Genet. 2013;22(4):729–36.
Verrier SE, Willmann M, Wenzel D, Winter U, von Mollard GF, Soling HD. Members of a mammalian SNARE complex interact in the endoplasmic reticulum in vivo and are found in COPI vesicles. Eur J Cell Biol. 2008;87(11):863–78.
Hubel P, Urban C, Bergant V, Schneider WM, Knauer B, Stukalov A, et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat Immunol. 2019;20(4):493–502.
Peter CJ, Evans M, Thayanithy V, Taniguchi-Ishigaki N, Bach I, Kolpak A, et al. The COPI vesicle complex binds and moves with survival motor neuron within axons. Hum Mol Genet. 2011;20(9):1701–11.
Van Alstyne M, Lotti F, Dal Mas A, Area-Gomez E, Pellizzoni L. Stasimon/Tmem41b localizes to mitochondria-associated ER membranes and is essential for mouse embryonic development. Biochem Biophys Res Commun. 2018;506(3):463–70.
Custer SK, Foster JN, Astroski JW, Androphy EJ. Abnormal Golgi morphology and decreased COPI function in cells with low levels of SMN. Brain Res. 2019;1706:135–46.
Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet. 2006;38(10):1192–7.
Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48(D1):D498–d503.
Benichou S, Bomsel M, Bodeus M, Durand H, Doute M, Letourneur F, et al. Physical interaction of the HIV-1 Nef protein with beta-COP, a component of non-clathrin-coated vesicles essential for membrane traffic. J Biol Chem. 1994;269(48):30073–6.
Schaefer MR, Wonderlich ER, Roeth JF, Leonard JA, Collins KL. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog. 2008;4(8):e1000131.
Macken WL GA, Wheyway G, Stals K, Nazlamova L, Ellard S, Alfares A, Aloraini T, Al Subaie L, Alfadhel M, Alajaji S, Wai HA, Self J, Douglas AGL, Kao AP, Guille M, Baralle D. NM_001144061.2(COPB1):c.957+1G>T, Variation ID 996016. ClinVar. https://www.ncbi.nlm.nih.gov/clinvar/variation/996016/. 2021. Accessed 19 Feb 2021.
Macken WL GA, Wheyway G, Stals K, Nazlamova L, Ellard S, Alfares A, Aloraini T, Al Subaie L, Alfadhel M, Alajaji S, Wai HA, Self J, Douglas AGL, Kao AP, Guille M, Baralle D. NM_001144061.2(COPB1):c.1651T>G (p.Phe551Val), Variation ID 996037. ClinVar. https://www.ncbi.nlm.nih.gov/clinvar/variation/996037/. 2021. Accessed 19 Feb 2021.
Acknowledgements
The authors greatly appreciate the participation of the patients and their families in this study. Identification of one of the participating families was made possible by the GeneMatcher portal. We thank the Zeiss Global Centre based at the University of Portsmouth for providing the X-ray MicroCT imaging and data processing facilities. We are grateful to Dr. Colin Sharpe, University of Portsmouth, for his expert feedback on the manuscript. Part of this research was funded by the National Institute for Health Research (NIHR).
Funding
DB is generously supported by a National Institute of Health Research (NIHR) Research Professorship RP-2016-07-011. GW is supported by a Wellcome Trust Seed Award in Science 204378/Z/16/Z. We acknowledge support from the NIHR UK Rare Genetic Disease Research Consortium. The European Xenopus Reference Centre is supported by the Wellcome Trust (101480Z) and BBSRC (BB/K019988/1).
Author information
Authors and Affiliations
Contributions
WM, AG, GW and KS had an equal contribution to the study work (co-first authors). GW, MG and DB designed the study. WM, GW and AG wrote the manuscript. WM, DB and JS phenotyped Family 1. LN, WM, HAW and AGLD were involved in in silico modelling of variants and splicing wet work. WM and GW performed the missense modelling. AG, APK and MG performed Xenopus experiments. KS and SE interpreted the exome data for Family 1. AA, TA, LAS, MA and SA were involved in the clinical and genomic study of Family 2. All authors reviewed the manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Family 1 were investigated on a clinical basis in the UK National Health Service (clinical examination and whole exome sequencing) and were subsequently enrolled in the ‘Splicing and Disease’ study for further functional investigation. This study was approved by South Central – Oxford B Research Ethics Committee (REC Reference 11/SC/0269, IRAS ID 49685). Family 2 were enrolled in a large genome sequencing study for undiagnosed disease. Ethical approval for Family 2’s study was granted by the Institutional Research Board of King Abdullah International Medical Research Center, Riyadh, Saudi Arabia (RC 16/113 and RC16/211/R2). Written informed consent to participate was obtained from the legal guardians for all subjects involved in this study as none had the capacity to consent for themselves. All research conformed with the principles of the Declaration of Helsinki. Xenopus experiments were conducted in accordance with the Home Office Code of Practice, under PPL 70/8983 with approval from the University of Portsmouth’s Animal Welfare and Ethical Review Body.
Consent for publication
Written informed consent for publication of identifiable data was obtained for Family 1 and 2 from the legal guardians of the participants. Consent included the publication of genetic sequence information and clinical information for both families and for clinical photographs for Family 1 only.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1
. Demonstrates how CRISPR/ cas9 genome editing induces exon skipping in X.tropicalis. Fig. S2. Shows the eye (a target organ) and the remainder of the crispant tadpole are equally mosaic. Fig. S3. Confirms that disruption to copb1 exon 8 in X.tropicalis mirrors syndromic hallmarks. Fig. S4. Supplemental figure demonstrates a more exaggerated phenotype in tadpoles injected with CRISPR/cas9 targeting exon 3, copb1. Fig. S5. Illustrates Xenopus anatomy and the significant reduction in brain size seen in transgenic tadpoles. Fig. S6. Shows there is no difference in localisation of wild type beta COP versus the Family 2 variant beta COP. Table S1. Details the immunodeficiency investigations performed in Family 2. Table S2. Details other shared homozygous variants identified in both probands of Family one.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Macken, W.L., Godwin, A., Wheway, G. et al. Biallelic variants in COPB1 cause a novel, severe intellectual disability syndrome with cataracts and variable microcephaly. Genome Med 13, 34 (2021). https://doi.org/10.1186/s13073-021-00850-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-021-00850-w