
Abstract
Background
DNA methylation levels change along with age, but few studies have examined the variation in the rate of such changes between individuals.
Methods
We performed a longitudinal analysis to quantify the variation in the rate of change of DNA methylation between individuals using whole blood DNA methylation array profiles collected at 2–4 time points (N = 2894) in 954 individuals (67–90 years).
Results
After stringent quality control, we identified 1507 DNA methylation CpG sites (rsCpGs) with statistically significant variation in the rate of change (random slope) of DNA methylation among individuals in a mixed linear model analysis. Genes in the vicinity of these rsCpGs were found to be enriched in Homeobox transcription factors and the Wnt signalling pathway, both of which are related to ageing processes. Furthermore, we investigated the SNP effect on the random slope. We found that 4 out of 1507 rsCpGs had one significant (P < 5 × 10−8/1507) SNP effect and 343 rsCpGs had at least one SNP effect (436 SNP-probe pairs) reaching genome-wide significance (P < 5 × 10−8). Ninety-five percent of the significant (P < 5 × 10−8) SNPs are on different chromosomes from their corresponding probes.
Conclusions
We identified CpG sites that have variability in the rate of change of DNA methylation between individuals, and our results suggest a genetic basis of this variation. Genes around these CpG sites have been reported to be involved in the ageing process.
Similar content being viewed by others


Background
DNA methylation is a widely studied epigenetic modification with a role in the regulation of gene expression [1]. Local levels of DNA methylation differ within and between individuals. This variation in local methylation is associated with both genetic and environmental factors [2,3,4,5]. The majority of DNA methylation studies in human are based on cross-sectional cohorts. Such studies have reported that methylation levels at many CpG sites in the genome correlate with age [6,7,8,9,10]. Therefore, age is frequently treated as a covariate and adjusted for in a linear regression framework in which differences of DNA methylation between cell types, tissues and diseases are tested [11,12,13]. One implicit assumption behind this correction is that the rate of change at a methylation CpG site across time is constant between individuals, which may not be true. Several studies have revealed that there is a potential change in variability of DNA methylation with age [14, 15], indicating that the rate of change of DNA methylation is different between individuals.
Estimation of the variation in such trajectories of DNA methylation with age between individuals is possible in a longitudinal analysis. Previous longitudinal analyses have investigated the relationship between SNPs and longitudinal DNA methylation [16, 17]. Other studies focused on the association between DNA methylation and a phenotype measured on the same individual [18,19,20]. Differences between individuals in the pattern of change of DNA methylation over time were not considered in these studies. Here, we estimate the variation in trajectories of DNA methylation change among individuals in a longitudinal analysis. This approach may elucidate how epigenetic marks change differently between individuals and whether this variation is associated with genetic factors and biological function.
In this study, we estimated between-individual variation in the rate of change of DNA methylation at 344,000 loci in a longitudinal sample of older people from the Lothian Birth Cohorts 1921 and 1936. For each CpG site, we estimated the variation in the rate of change in each individual. Furthermore, the identification of such probes facilitates the estimation and partitioning of the variation underlying DNA methylation changes, for example, the contribution of genetic factors. We identified genetic loci that are associated with differences in the longitudinal changes in DNA methylation across individuals.
Methods
Methylation data
DNA was extracted from whole blood samples in Lothian Birth Cohort 1921 (LBC1921) at MRC Technology, Western General Hospital, Edinburgh (LBC1921), and the Wellcome Trust Clinical Research Facility (WTCRF), Western General Hospital, Edinburgh (LBC1936), using standard methods. Methylation typing of 485,512 probes was performed at the WTCRF. Bisulphite converted HD Methylation protocol and Tecan robotics (Illumina). Raw intensity data were background-corrected and normalized using internal controls, and methylation M values were generated using the R minfi package [21]. Detailed further quality control steps are given in Additional file 1.
Batch and covariate adjustment
Our analysis was based on the M value of DNA methylation. We regularized the M value by constraining it to be in the interval between − 9.96 and 9.96 (corresponding to the interval 0.001 to 0.999 of the beta-value). Furthermore, for each probe, we removed individuals with DNA methylation three standard deviations above and below the mean M value to exclude outliers. On average, 34 (out of 2894 samples, 1.2%) outliers were removed for each probe. DNA methylation (M value) in most (79.5%) of these outliers is in the range between − 6.6 and 6.6 (corresponding to the interval 0.01 to 0.99 of the beta-value), suggesting the “abnormal” DNA methylation values in the majority of outliers are not extreme values caused by the transformation from beta-value to M value. Covariates including sex, age and cell counts (CC), and batch effects including position in array (PIA), hybridization date (HD), set ID (SI), plate ID (PI) and array ID (AI, both PI and AI were regarded as random effects), were corrected for each probe. We used the residuals after this adjustment for further analysis. If yj is the DNA methylation value for probe j, then we used the residuals from the model
LBC genotype and imputation
Individuals from LBC1921 and LBC1936 were genotyped on Illumina 610-Quad Beadchip arrays. Full details of genotyping procedures are given elsewhere [22]. Standard QC filters were applied, and remaining genotyped SNPs were phased using HAPI-UR [23] and imputed using 1000 Genomes Phase I Version 3 [24] with Impute V2 [25]. Raw imputed SNPs were filtered to remove any SNPs with low imputation quality as defined by an R2 < 0.8. Subsequent quality control removed SNPs with MAF < 0.01, those with HWE P < 1 × 10−6 and a missing rate > 10%. After filtering, 7,760,689 SNPs remained for further analysis.
Estimation of random slope effects
For each probe, we fitted a mean level and a rate of change of DNA methylation for each individual and tested whether the variance due to these random effects was significantly larger than zero, using the mixed model
where yij is the methylation residual after QC steps, i is the ith individual, j represents the jth observation in individual i, u1 is the mean effect, u2 is the mean age effect, ai and bi are the random intercept (mean level of DNA methylation in each individual) and random slope, tij represents standardized age (mean = 0 and variance = 1) and eij is the random error. The random effects of eij, ai and bi are assumed to follow a normal distribution. \( {\sigma}_a^2 \) and \( {\sigma}_b^2 \) are the variances of ai and bi, respectively. σab is the covariance between ai and bi, it was set to be zero under the assumption of independence between ai and bi. The likelihood ratio test (LRT) was used to test if \( {\sigma}_a^2 \) and \( {\sigma}_b^2 \) are equal to zero.
We obtained a P value from the LRT using a χ2(1) distribution and then dividing the P value by two. This can be justified since under the null hypothesis, in 50% of cases, the test statistic is zero (or, follows a χ2(0)), and in 50% of cases it follows a χ2(1) [26]. Probes with a P value smaller than 1.5 × 10−7 (0.05/344,000) for the random slope are defined as rsCpG.
Covariance between a i and b i
We applied an extended model that included the covariance σab between the random intercept and random slope to quantify the effect of covariance on the estimation of random effects. We calculated the Pearson corrections between the estimated random slope effects before and after incorporating the covariance term. All correlations were larger than 0.7 in the 1507 rsCpGs (mean correlation = 0.93). These results indicated that the introduction of the covariance term did not alter the results substantially.
Quadratic effect
We investigated the effect of modelling a quadratic average trajectory by adding a squared term for age (t2) in the model \( {y}_{ij}={u}_1+{u}_2\times {t}_{ij}+{a}_i+{b}_i\times {t}_{ij}+{t}_{ij}^2+{e}_{ij} \). All the correlations of the probes with and without fitting this additional term were found to be larger than 0.98 in 1507 rsCpGs.
Estimating the confidence interval of the correlation based on bootstrapping
Considering the background correlation of DNA methylation between CpG sites, we used bootstrapping to calculate the 95% confidence interval of the correlation (of the estimated variances) between two groups of individuals. We resampled 344,000 pairs of variances with replacement from the original data and estimated the correlation based on these pairs. We repeated this step 30,000 times to calculate the 95% confidence interval of the correlation.
GWAS on random effects
Based on the random effects estimated from the above mixed linear model, we performed a series of genome-wide association studies by using the random effects as the dependent variables with the software PLINK2 [27]. All QC-ed SNPs were used, and P value threshold for the significance was Bonferroni corrected (P < 5 × 10−8/1507).
Permutation analysis of the random slope test statistics
The mean and median test statistic across CpG sites for the effect of the random slope was very large, with a λ inflation value (mean test statistic) of 11.0. To verify if the results are inflated under the null hypothesis, we permuted ages across individuals and waves 500 times. For each round, we re-fitted the full model on the permuted data, and the mean chi-square among the probes was calculated. The mean of this distribution was around 0.73 (SD = 0.32), which shows no significant difference (P = 0.48) with the expected value of 0.5 under the null hypothesis. This indicates the statistical significance of the estimated effects of a random slope is not caused by the violations of the assumptions of the distribution of the test statistic under the null hypothesis.
Mapping CpG Islands and differently methylated region (DMR)
Genomic positions of the CpG island were obtained from the UCSC Genome Browser [28]. Annotation information of the differently methylated region (DMR) was from the Illumina DNA methylation annotation file (GEO ID GPL13534). The significance of enrichment analysis was assessed by permutation.
PANTHER over-representation test
We used all 25,537 genes that are adjacent to the QC-ed 344,000 probes as the background. Eighteen thousand six hundred seven of the genes overlapped with the gene list in the PANTHER database. Ten thousand twenty out of 1235 rsCpG nearby genes were in the PANTHER database. The enrichment test was based on Fisher’s exact test, and protein classes with a false discovery rate (FDR) smaller than 0.05 were selected.
Heritability of probes
We utilized the heritability of the significant probes estimated in the Brisbane Systems Genetics Study (BSGS) cohort [29, 30] to validate the genetic contribution to these probes. The significance of the difference from the null distribution of the mean heritability was estimated based on the average heritability of 1507 randomly selected probes from 30,000 permutation tests.
SNP by age effect on DNA methylation
The SNP effect on random slope can be defined as slopei = βk × dik + ei, i is the ith individual, βk is the effect size of SNP k on random slope, and dik is the dosage of SNP k in individual i. Since the DNA methylation of individual i at time point tij is yij = slopei × tij + eij = (βk × dik + ei) × tij + eij = βk × dik × tij + ei × tij + eij (main effects are ignored here), the SNP effect on random slope can be interpreted as SNP by age effect on DNA methylation. To compare the power in detecting these two effects, we simulated 3000 individuals, each with three age points sampled at round 60, 70 and 80 years old. DNA methylation of individual i at time point tij was simulated by using yij = (βk × dik + ei) × tij + eij. dik was sampled from (0,1,2) assuming Hardy–Weinberg equilibrium, the minor allele frequency of the SNP ranges from 0.05 to 0.5, the random error eij and random slope ei (not explained by SNP) are assumed to follow a standard normal distribution. The effect size βk of the SNP on the random slope was simulated in two ways: (1) βk was sampled from a uniform distribution in the range of − 0.1 to 0.1 and (2) βk was set to zero. For each type of data, we obtained P values in three ways: (1) from association between the SNP and the estimated random slope from a mixed linear model, (2) from association between DNA methylation and a SNP by age effect and (3) from association between DNA methylation and a SNP by age effect with random slope fitted as a covariate. We repeated this simulation 300,000 times and compared P values and median chi-square (λmedian) between these associations. Based on the simulated data with no SNP effect on the random slope (βk = 0), we found the P values from the second analysis method were inflated (λmedian = 2.01). P values from the other two ways were not inflated (λmedian = 1.00), and no difference in the detecting power was identified between these two ways based on the simulated data with SNP effect on the random slope (βk~U(− 0.1,0.1)).
Linkage disequilibrium (LD) clumping
We applied LD clumping on the GWAS significant SNPs using PLINK2 [27], and imputed LBC genotype data was used as the reference. For each significant (P < 5 × 10−8) SNP, LD (R2) between this SNP and other significant (P < 5 × 10−8) SNPs within 1 Mbp distance were calculated and SNPs with LD larger than 0.1 were defined as a clump. Within each clump, only the SNP with smallest P value would be selected during LD clumping.
All analysis was performed by using R package, version 3.2.2 [31]. Figures were generated using ggplot2 [32].
Results
Data
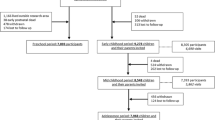
DNA methylation was measured on individuals from Lothian Birth Cohort 1921 (LBC1921) and Lothian Birth Cohort 1936 (LBC1936) [33, 34] using Illumina Infinium HumanMethylation450K BeadChip arrays. There were 3471 samples across all waves of data collection (Additional file 1: Table S1), and 344,000 DNA methylation probes remained after removing probes encompassing SNPs annotated by Illumina (GEO ID GPL13534) and probes identified as potentially cross-hybridizing [35] (see details of quality control steps in Additional file 1). Only individuals with DNA methylation measured at two or more different time points were considered, and samples with inconsistent measurements (match rate < 0.8) of control probes within individuals were removed (Additional file 2: Figure S1), leaving 2894 samples from 954 individuals (Table 1). Among them, 283 individuals had DNA methylation measured at two time points, and 356 and 315 individuals were measured at three and four time points, respectively. The effects of covariates (sex, age and cell counts) and batches (position in the array, hybridization date, set ID, plate ID and array ID) on DNA methylation were removed before further analysis (‘Methods’, Additional file 1: Figures S2 and S3).
Identification of CpG sites with a random slope in methylation
For each CpG site, we estimated the variance of the rate of change (random slope) between individuals in a mixed linear model (‘Methods’). A non-zero variance indicates the existence of individual differences in the rate of change in DNA methylation across time. Forty-two thousand two hundred fifty-three probes were found to have a statistically significant random slope (likelihood ratio test, P < 0.05/344,000, Bonferroni corrected) based on 2894 samples. Permutation test analyses indicated that the statistical significance of the estimated effects of the random slope is not caused by the violations of the assumptions of the test statistic (‘Methods’, Additional file 2: Figures S4 and S5). Moreover, no substantial impact on the estimation of random effects was found by introducing a covariance (‘Methods’, Additional file 2: Figure S6A) between the random slope and the mean level of DNA methylation in each individual into the model, or the inclusion of additional corrections for age effects such as a quadratic term (‘Methods’, Additional file 2: Figure S6B). To obtain a robust set of CpG sites with a statistically significant variation in the rate of change, we divided the individuals into two groups according to the number of time points for which they have a measurement. One group contains individuals with two or three time points, and the other group has individuals with four time points. We applied the mixed linear model on each of these two groups and found that the estimated variances of random slopes for each CpG site were correlated between these independent groups (R = 0.41, 95% bootstrap CI 0.40–0.42, bootstrapping was repeated 30,000 times, ‘Methods’, Fig. 1a). One thousand five hundred seven CpG sites were identified to have a statistically significant (p < 0.05/344,000) variation in the rate of change of DNA methylation in both groups (rsCpGs, Fig. 1b, Additional file 3: Table S2). The overlap is statistically significant (odds ratio = 4.9, P < 3.3 × 10−5, permutation test, 30,000 times), and these 1507 rsCpGs were used for further analysis. A summary of chi-square statistics for the variance of the random slope is presented in Table 2.

a Comparison of estimated variances of random slopes between the group of individuals with four time points and the group of individuals with two or three time points. b Comparison of chi-square test statistics for the variance of random slope between the group of individuals with four time points and the group of individuals with two or three time points. c The change of standard deviation (SD) in 1507 rsCpG across waves (mean age in each wave in parentheses). Each point represents the SD of DNA methylation for one CpG site in each wave, and the SD of each CpG site in different waves are connected by lines. The overall level of SD across all CpG sites in each wave is shown as a boxplot. The red dashed line is the median SD in wave 1 of LBC1936
We observed a larger variation in DNA methylation of rsCpGs compared to randomly selected probes in each wave (Additional file 2: Figure S7). Moreover, an increase in the variability of DNA methylation with age can be identified in most of these rsCpGs (Fig. 1c). These rsCpGs overlapped with probes identified by Slieker and colleagues [14]. Based on a cross-sectional study, Slieker et al. identified 6366 positions that showed changes in variably of methylation with age using 3295 whole blood DNA methylation profiles. Among those positions, 540 probes overlap with the 1507 rsCpGs in our study. This highly significant overlap (odds ratio = 45.9, P < 3.3 × 10−5, permutation test, 30,000 times) provides an independent confirmation of our results.
Genomic locations of CpG sites with random effects
The dynamicity of DNA methylation varies across the human genome [36, 37]. To investigate whether the rsCpGs locate in the more variable genomic regions, we mapped these CpG sites to the genome and applied an enrichment test on these probes (‘Methods’). We observed an enrichment of rsCpGs in the Shore region of CpG islands (regions within 2 kb upstream or downstream of a CpG island are called north shore and south shore, respectively). Genomic positions of the CpG island were obtained from the UCSC Genome Browser [28]), with an odds ratio (OR) of 2.0 for both the north shore (p < 3.3 × 10−5, permutation test, 30,000 times) and the south shore (P < 3.3 × 10−5, permutation test, 30,000 times) (Fig. 2). DNA methylation in the shore region was previously reported to be more dynamic than that in CpG islands [36], and our results indicate that CpG sites with random slopes locate in the regions with more malleable DNA methylation. Similarly, rsCpGs were found to be enriched in reprogramming-specific differently methylated regions (RDMR, regions differentially methylated in the reprogramming process) [37].

Enrichment analysis of rsCpGs in different CpG regions based on the permutation test. For each CpG region, the distribution of odds ratio based on permuted data (30,000 times) and the odds ratio based on the original data (red dashed line) are presented. DMR: differentially methylated region; CDMR: cancer-specific DMR; RDMR: reprogramming-specific DMR; NONE_DMR other CpGs not in DMR. Island: CpG island provided by UCSC [28]; N_Shore: 0–2 Kb upstream of CpG island; S_Shore: 0–2 Kb downstream of CpG island; N_Shelf: 2–4 Kb upstream of CpG island; S_Shelf: 2–4 Kb downstream of CpG island; Sea: 4 Kb away from CpG island. Enhancer: Predicted enhancer elements determined by ENCODE Consortium [46]
Biological enrichment of CpG sites with random effects
To explore the biological function of the rsCpGs, we applied a gene over-representation test on the nearest genes of rsCpGs using PANTHER (version 13.1) [38] (‘Methods’). The result showed that 1235 genes around the 1507 rsCpGs were statistically significantly (Fisher’s exact test, P = 3.7 × 10−10, FDR = 3.9 × 10−8) enriched in Homeodomain (Homeobox) transcription factor (PC00119) protein class (Table 3). We also investigated the significance of these protein classes using a permutation test and found they remained significant (100 repeats). Furthermore, we performed Gene Ontology (GO) analysis on these genes and found that ectoderm development (GO 0007398, P= 5.9 × 10−18, FDR = 1.5 × 10−15) and developmental process (GO 0032502, P = 1.8 × 10−17, FDR = 2.2 × 10−15) were the most significantly over-represented biological processes (Additional file 4: Table S3). Cadherin signalling pathway (P00012), Wnt signalling pathway (P00057) and Heterotrimeric G-protein signalling pathway-Gq alpha and Go alpha-mediated pathway (P00027) were found to be significantly enriched pathways for genes around rsCpGs in a PANTHER pathway analysis (Table 3). Since there are only 177 primarily signalling pathways in PANTHER database [38], we further performed the pathway analysis in an integrated pathway database ConsensusPathDB (version 33) [39]. This analysis on the same gene sets showed that the most significant pathway for rsCpGs was “Neuronal System” (P = 1.4 × 10−9). Full details of significant pathway results are given in the Additional file 5: Table S4.
Genetic effects on the random slope
rsCpGs were found to be enriched in the CpG sites with large heritability (p < 3.3 × 10−5, permutation test, 30,000 times, ‘Methods’, Fig. 3a), indicating a substantial genetic component to their variation. To examine the genetic contribution on the random effects of rsCpGs, we performed genome-wide association studies (GWASs) using PLINK2 [27], fitting the predicted random slope for each person (obtained from the mixed model analysis) as the dependent trait (‘Methods’). Results showed that there were four significant SNP-probe pairs in total (P < 5 × 10−8/1507, after linkage disequilibrium (LD) clumping, ‘Methods’), three of them are cis (in same chromosome) (Table 4, Fig. 3b). In addition, 343 rsCpGs were identified to have at least one genome-wide significant (P < 5 × 10−8, after LD clumping) SNP effect (436 SNP-probe pairs). Ninety-five percent of the SNPs are on different chromosomes from their corresponding probes (Fig. 4, Additional file 6: Table S5). The SNP effect on the random slope can also be interpreted as the SNP by age effect on DNA methylation (‘Methods’, Additional file 2: Figure S8). Van Dongen et al. reported 71,894 CpG sites to have an interaction between genetic effects and age (P < 0.05) on DNA methylation [5], and the 343 rsCpGs with a significant SNP effect on the random slope from our analyses were enriched (P < 3.3 × 10−5, permutation test, 30,000 times) in these probes (Additional file 2: Figure S9). This provides an independent confirmation of our results.

a The distribution of estimated heritability of 1507 rsCpGs and all probes. The heritability of rsCpGs is normally distributed, with a mean of 0.40 (SD = 0.21). It is significantly larger (P < 3.3 × 10−5, permutation test, 30,000 times) than the overall level. No significant correlation (R = − 0.005, P = 0.27) was found between heritability of probes and the distance to their meQTLs [47]. However, there is a small but significant association (R = 0.07, P < 2.2 × 10−16) between the heritability and the mean phenotypic correlation (R2) between a target probe and other probes on the same chromosome. This indicated that CpG sites with substantial heritability could contribute to the estimation of heritability of other CpG sites that they correlate with. b An example to show the significant association between SNP dosage and the random slope of DNA methylation

a The distribution of SNPs with a significant (P < 5 × 10−8) effect on the random slope of DNA methylation. The 14 SNPs associated with the random slope of cg08773226 (with the largest number of associated SNPs) are marked as diamonds. b The Manhattan plot to show the GWAS result on the random slope of cg08773226
Relationship between rate of DNA methylation change and covariates
To detect a possible contribution of the covariates in estimating the random slope of DNA methylation, we investigated two covariates that were previously identified to have a change of variation with ageing: body mass index (BMI) and walking speed (the time to walk 6 m) [20, 40]. We fitted each of the covariates in the full model and re-estimated the significance of the variance of random slope. No significant changes were observed (Additional file 2: Figure S10), implying no contribution of these two covariates to variation of the random slope.
Discussion
We estimated the variation in the rate of change of DNA methylation for each probe by implementing a mixed linear model in a longitudinal analysis. One thousand five hundred seven probes (rsCpGs) were found to have statistically significant variation in the rate of change between individuals. These rsCpGs were enriched in the shore region of CpG island, which is consistent with more dynamicity of DNA methylation in CpG island shore region than the CpG island itself [37].
We found that the closest genes of rsCpGs were enriched in the Homeobox gene cluster, which was also reported in Slieker et al. to be associated with age-related variably methylated positions (aVMP) in cis [14]. The Homeobox gene cluster is involved in the process of cell development [41], and recent evidence showed that it is related to ageing [42, 43]. Pathway analysis on these genes in PANTHER database indicated they were enriched in Wnt signalling pathway (P00057), which was also reported to be related to the ageing process [44, 45]. One of the most significantly over-represented Gene Ontology (GO) category in these genes was the developmental process (GO 0032502), which was discovered to be significant for the probes that consistently drift among twins over time [16]. These results indicate the rsCpGs may be involved in the developmental process (such as ageing) by regulating their nearby genes.
There is a significant higher heritability of the 1507 rsCpGs compared to the overall level. GWAS results on the random slope identified 436 SNP-probe pairs (343 rsCpGs) with a genome-wide significant association (P < 5 × 10−8), suggesting a SNP by age effect on the CpG sites. Among them, 95% of the SNPs were on different chromosomes from their probes, which (in the absence of non-identified confounders) indicated a potential major trans SNP by age effect on DNA methylation of rsCpGs.
Our study has several limitations. Although the permutation test indicates our results will not be inflated by the violations of the assumptions of the distribution of the test statistic under the null hypothesis, our results could be inflated by unknown confounding factors. We adjusted for known possible confounders, including the chronological age at which the samples were taken, but cannot exclude the possibility of unknown confounders that have effects on the mean or variance of the measured DNA methylation. The effects of covariates including age, sex and cell counts were adjusted in our quality control steps, and we further confirmed that BMI and walking speed have no effects on the rate of change in DNA methylation. However, other exposures, like medication, smoking status and disease status may potentially contribute to this variation, which can influence the estimation of random effects. Nevertheless, the 1507 rsCpGs that have a statistically significant random slope in two separate groups of individuals indicate that these results should be robust. There was no gene expression data on the same individuals available, and we simply assume that the expression of a gene can be regulated by its closest DNA methylation CpG site, which may not be true. Finally, our results are based on older individuals and may not apply to different age ranges.
Conclusions
Ageing is strongly correlated with changes in DNA methylation, and the rates of change over time at one CpG site can differ between individuals. We detected CpG sites with different changing rates (random slope) using a mixed linear model and found 1507 CpG sites that have a statistically significant rate of change in methylation between individuals, and that these different rates of change can be partially explained by genetic effect. Genes around rsCpGs were enriched in Homeobox gene clusters and Wnt signalling pathway, both of which have been reported to be involved in the ageing process. Our results imply that the changing rate of DNA methylation varies between individuals at several CpG sites, and this difference is associated with genetic factors. These CpG sites might be useful markers to better understand individual differences in ageing.
Abbreviations
- BMI:
-
Body mass index
- BSGS:
-
Brisbane Systems Genetics Study
- DMR:
-
Differently methylated region
- FDR:
-
False discovery rate
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- GWAS:
-
Genome-wide association study
- LBC:
-
Lothian Birth Cohort
- LD:
-
Linkage disequilibrium
- LRT:
-
Likelihood ratio test
- SNP:
-
Single nucleotide polymorphism
References
Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610.
Feinberg AP, Fallin MD. Epigenetics at the crossroads of genes and the environment. Jama. 2015;314:1129–30.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109.
Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–62.
van Dongen J, Nivard MG, Willemsen G, Hottenga JJ, Helmer Q, Dolan CV, Ehli EA, Davies GE, van Iterson M, Breeze CE, et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat Commun. 2016;7:11115.
Richardson B. Impact of aging on DNA methylation. Ageing Res Rev. 2003;2:245–61.
Hernandez DG, Nalls MA, Gibbs JR, Arepalli S, van der Brug M, Chong S, Moore M, Longo DL, Cookson MR, Traynor BJ. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20:1164–72.
Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76.
Bell JT, Tsai P-C, Yang T-P, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8:e1002629.
Florath I, Butterbach K, Müller H, Bewerunge-Hudler M, Brenner H. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet. 2013;23:1186–201.
Shah S, McRae AF, Marioni RE, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, Pattie A, Corley J. Genetic and environmental exposures constrain epigenetic drift over the human life course. Genome Res. 2014;24:1725–33.
Wockner L, Noble E, Lawford B, Young RM, Morris C, Whitehall V, Voisey J. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl Psychiatry. 2014;4:e339.
Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aïssi D, Wahl S, Meduri E, Morange P-E, Gagnon F, Grallert H. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014;383:1990–8.
Slieker RC, van Iterson M, Luijk R, Beekman M, Zhernakova DV, Moed MH, Mei H, Van Galen M, Deelen P, Bonder MJ. Age-related accrual of methylomic variability is linked to fundamental ageing mechanisms. Genome Biol. 2016;17:191.
Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, Suchiman HED, Slagboom PE, Boomsma DI, Heijmans BT. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11:694–703.
Martino D, Loke YJ, Gordon L, Ollikainen M, Cruickshank MN, Saffery R, Craig JM. Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013;14:R42.
Gaunt TR, Shihab HA, Hemani G, Min JL, Woodward G, Lyttleton O, Zheng J, Duggirala A, McArdle WL, Ho K. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016;17:61.
Simpkin AJ, Suderman M, Gaunt TR, Lyttleton O, McArdle WL, Ring SM, Tilling K, Smith GD, Relton CL. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum Mol Genet. 2015;24:3752–63.
Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, Christensen K. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell. 2016;15:149–54.
Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, Gibson J, Redmond P, Cox SR, Pattie A. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol. 2015;44:1388–96.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Medland SE, Nyholt DR, Painter JN, McEvoy BP, McRae AF, Zhu G, Gordon SD, Ferreira MA, Wright MJ, Henders AK. Common variants in the trichohyalin gene are associated with straight hair in Europeans. Am J Hum Genet. 2009;85:750–5.
Williams AL, Patterson N, Glessner J, Hakonarson H, Reich D. Phasing of many thousands of genotyped samples. Am J Hum Genet. 2012;91:238–51.
Consortium GP. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56.
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955.
Visscher PM. A note on the asymptotic distribution of likelihood ratio tests to test variance components. Twin Res Hum Genet. 2006;9:490–5.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Casper J, Zweig AS, Villarreal C, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Karolchik D, et al. The UCSC genome browser database: 2018 update. Nucleic Acids Res. 2018;46:D762–9.
McRae AF, Powell JE, Henders AK, Bowdler L, Hemani G, Shah S, Painter JN, Martin NG, Visscher PM, Montgomery GW. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 2014;15:R73.
Powell JE, Henders AK, McRae AF, Caracella A, Smith S, Wright MJ, Whitfield JB, Dermitzakis ET, Martin NG, Visscher PM. The Brisbane Systems Genetics Study: genetical genomics meets complex trait genetics. PLoS One. 2012;7:e35430.
Team RC: R: a language and environment for statistical computing. 2013.
Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag; 2016. https://cran.r-project.org/web/packages/ggplot2/citation.html.
Deary IJ, Gow AJ, Pattie A, Starr JM. Cohort profile: the Lothian Birth Cohorts of 1921 and 1936. Int J Epidemiol. 2011;41:1576–84.
Deary IJ, Gow AJ, Taylor MD, Corley J, Brett C, Wilson V, Campbell H, Whalley LJ, Visscher PM, Porteous DJ. The Lothian Birth Cohort 1936: a study to examine influences on cognitive ageing from age 11 to age 70 and beyond. BMC Geriatr. 2007;7:28.
Price EM, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6(1):1756–8935.
Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–81.
Doi A, Park I-H, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S. Differential methylation of tissue-and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–3.
Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2016;45:D183–9.
Kamburov A, Stelzl U, Lehrach H, Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2012;41:D793–800.
Block JP, Subramanian SV, Christakis NA, O’Malley AJ. Population trends and variation in body mass index from 1971 to 2008 in the Framingham Heart Study Offspring Cohort. PLoS One. 2013;8:e63217.
Pearson JC, Lemons D, McGinnis W. Modulating Hox gene functions during animal body patterning. Nat Rev Genet. 2005;6:893–904.
McClay JL, Aberg KA, Clark SL, Nerella S, Kumar G, Xie LY, Hudson AD, Harada A, Hultman CM, Magnusson PKE. A methylome-wide study of aging using massively parallel sequencing of the methyl-CpG-enriched genomic fraction from blood in over 700 subjects. Hum Mol Genet. 2014;23:1175–85.
Bork S, Pfister S, Witt H, Horn P, Korn B, Ho AD, Wagner W. DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell. 2010;9:54–63.
Lezzerini M, Budovskaya Y. A dual role of the Wnt signaling pathway during aging in Caenorhabditis elegans. Aging Cell. 2014;13:8–18.
White BD, Nguyen NK, Moon RT. Wnt signaling: it gets more humorous with age. Curr Biol. 2007;17:R923–5.
Siggens L, Ekwall K. Epigenetics, chromatin and genome organization: recent advances from the ENCODE project. J Intern Med. 2014;276:201–14.
McRae A, Marioni RE, Shah S, Yang J, Powell JE, Harris SE, Gibson J, Henders AK, Bowdler L, Painter JN, et al. Identification of 55,000 replicated DNA methylation QTL. bioRxiv. 2017.
Acknowledgements
Phenotype collection in the Lothian Birth Cohort 1921 was supported by the UK’s Biotechnology and Biological Sciences Research Council (BBSRC), The Royal Society and The Chief Scientist Office of the Scottish Government. Phenotype collection in the Lothian Birth Cohort 1936 was supported by the Age UK (The Disconnected Mind project). Methylation typing was supported by the Centre for Cognitive Ageing and Cognitive Epidemiology (Pilot Fund award), Age UK, The Wellcome Trust Institutional Strategic Support Fund, the Gertrude Winifred Gear Fund, The University of Edinburgh and The University of Queensland.
Funding
This research was supported by the Australian National Health and Medical Research Council (grants 1010374, 1046880 and 1113400) and by the Australian Research Council (DP160102400). PMV, NRW and AFM are supported by the NHMRC Fellowship Scheme (1078037, 1078901 and 1083656). REM and IJD conducted the research in The University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology (CCACE), part of the cross-council Lifelong Health and Wellbeing Initiative (MR/K026992/1); funding from the Biotechnology and Biological Sciences Research Council (BBSRC) and Medical Research Council (MRC) is gratefully acknowledged. D.S. is a Cancer Research UK Career Development Fellow (reference C47648/A20837), and the work in his laboratory is also supported by a Medical Research Council University grant to the MRC Human Genetics Unit.
Availability of data and materials
Lothian Birth Cohort (LBC) data have been submitted to the European Genome-phenome Archive (EGA: https://www.ebi.ac.uk/ega/home) under accession number EGAS00001000910.
Author information
Authors and Affiliations
Contributions
AMcR, PMV and IJD conceived and designed the experiments. QZ performed all statistical analyses. QZ, AMcR and PMV wrote the paper. REM, MRR and NWR advised on statistical methodology, and JH and DS contributed to analyses and interpretation of results. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Following written informed consent, venesected whole blood was collected for DNA extraction in both LBC1921 and LBC1936. Ethics permission for the LBC1921 was obtained from the Lothian Research Ethics Committee (wave 1 LREC/1998/4/183). Ethics permission for the LBC1936 was obtained from the Multi-Centre Research Ethics Committee for Scotland (wave 1 MREC/01/0/56), the Lothian Research Ethics Committee (wave 1 LREC/2003/2/29) and the Scotland A Research Ethics Committee (waves 2 and 3 07/MRE00/58). Research was carried out in compliance with the Declaration of Helsinki.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Quality control steps for DNA methylation. Table S1. Summary information for the age of all samples. Figure S2. Comparison of mean DNA methylation for duplicates between sets. Figure S3. Comparison of mean adjusted DNA methylation for duplicates between sets. (DOCX 614 kb)
Additional file 2:
Figure S1. The distribution of match rates of control probes between and within individuals. Figure S4. Q-Q plot for P values from the detection of the random slope. Figure S5. The distribution of λmean from 500 permutation tests. Figure S6. Quantifying the effect of t2 and covariance between the random slope and random intercept on the estimation of random effects. Figure S7. The comparison of standard deviation (SD) of DNA methylation between 1507 rsCpGs and 1507 randomly selected CpG sites in each wave. Figure S8. Power comparison between two methods in detecting associations between SNP and DNA methylation change. Figure S9. The distribution of P value of the interaction effect between age and genetic effects from Van Dongen et al. [5] of two probe sets. Figure S10. The comparison of the significance of the variation of DNA methylation rate of change before and after fitting BMI and walking in the model. (DOCX 2137 kb)
Additional file 3:
Table S2. Probes with a significant random slope in two groups of different individuals. Probe id, chi-square and P value were provided. (XLS 258 kb)
Additional file 4:
Table S3. Gene Ontology analysis on the genes around the 1507 rsCpGs. (CSV 3 kb)
Additional file 5:
Table S4. Pathway analysis on the genes around the 1507 rsCpGs in ConsensusPathDB database. (CSV 16 kb)
Additional file 6:
Table S5. Four hundred thirty-six SNPs with genome-wide significant (P < 5 × 10−8) effects on the random slope of 343 rsCpGs. (CSV 27 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, Q., Marioni, R.E., Robinson, M.R. et al. Genotype effects contribute to variation in longitudinal methylome patterns in older people. Genome Med 10, 75 (2018). https://doi.org/10.1186/s13073-018-0585-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-018-0585-7