
Abstract
Background
Amyotrophic lateral sclerosis (ALS) is a progressive neurological disease characterised by the degeneration of motor neurons, which are responsible for voluntary movement. There remains limited understanding of disease aetiology, with median survival of ALS of three years and no effective treatment. Identifying genes that contribute to ALS susceptibility is an important step towards understanding aetiology. The vast majority of published human genetic studies, including for ALS, have used samples of European ancestry. The importance of trans-ethnic studies in human genetic studies is widely recognised, yet a dearth of studies of non-European ancestries remains. Here, we report analyses of novel whole-exome sequencing (WES) data from Chinese ALS and control individuals.
Methods
WES data were generated for 610 ALS cases and 460 controls drawn from Chinese populations. We assessed evidence for an excess of rare damaging mutations at the gene level and the gene set level, considering only singleton variants filtered to have allele frequency less than 5 × 10–5 in reference databases. To meta-analyse our results with a published study of European ancestry, we used a Cochran–Mantel–Haenszel test to compare gene-level variant counts in cases vs controls.
Results
No gene passed the genome-wide significance threshold with ALS in Chinese samples alone. Combining rare variant counts in Chinese with those from the largest WES study of European ancestry resulted in three genes surpassing genome-wide significance: TBK1 (p = 8.3 × 10–12), SOD1 (p = 8.9 × 10–9) and NEK1 (p = 1.1 × 10–9). In the Chinese data alone, SOD1 and NEK1 were nominally significantly associated with ALS (p = 0.04 and p = 7 × 10–3, respectively) and the case/control frequencies of rare coding variants in these genes were similar in Chinese and Europeans (SOD1: 1.5%/0.2% vs 0.9%/0.1%, NEK1 1.8%/0.4% vs 1.9%/0.8%). This was also true for TBK1 (1.2%/0.2% vs 1.4%/0.4%), but the association with ALS in Chinese was not significant (p = 0.14).
Conclusions
While SOD1 is already recognised as an ALS-associated gene in Chinese, we provide novel evidence for association of NEK1 with ALS in Chinese, reporting variants in these genes not previously found in Europeans.
Similar content being viewed by others
Background
Amyotrophic lateral sclerosis (ALS) is a progressing motor neuron disease characterised by loss of function (LOF) of motor neurons, which are essential for controlling voluntary muscle activity such as walking, breathing and speaking. This condition leads to premature death with a median survival of about two to three years. Disease likely arises from a combination of genetic susceptibility [1,2,3] and environmental factors [4]. However, our understanding of what these factors are and how they contribute to disease risk, onset and progression remain incomplete.
Likely due to this limited understanding of disease aetiology, there has been limited success in designing any effective treatment for ALS. To date, the most important fundamental insights into the underlying cellular mechanisms have resulted from genetic studies of the known causal mutations [5]. However, highly penetrant identified mutations still only account for up to 10% of cases [6, 7] and thus more work needs to be done. Identification of both causal and risk genes will help build a more complete picture of the underlying mechanisms and pathways for disease and any new ALS molecule is potentially a new therapeutic target [8].
Whole-exome sequencing (WES) studies designed to identify genes enriched for rare variants have been conducted for ALS. Association testing has typically been conducted at the gene level comparing the burden of rare coding variants in cases vs controls. Large sample sizes are needed to detect significant associations due to testing of ~ 20,000 genes and because the multiple testing burden is often increased by considering different genetic models. The largest study to date, comprising 2874 cases and 6405 controls of European ancestry, identified the known ALS gene SOD1 as the only gene passing the multiple-testing corrected threshold for significance of association [9]. A follow-up study of 51 genes in an independent sample of 1318 cases and 2371 controls identified TBK1 as a novel ALS risk gene [9] (discovery association p = 1.13 × 10−5, replication p = 5.78 × 10−7 and combined p = 3.63 × 10−11), with later GWAS support for association of common single nucleotide polymorphisms (SNPs) in the same locus (p = 6.6 × 10–8) [10]. A second gene, NEK1, was highlighted as suggestively significant. Both TBK1 and NEK1 are notable because protein–protein interaction analyses link them with other known ALS genes.
The next largest WES study of ALS, a case-control (1022 cases vs 7315 controls) study with cases selected as index individuals from families with multiple recorded cases of ALS (fALS) [11], identified NEK1 as the only significant gene after correcting for multiple testing (ten known ALS genes had been excluded from the analysis to train modelling parameters). Follow-up analysis in four ALS cases from an isolated Dutch community suggested p.Arg261His as a specific NEK1 candidate variant. An association analysis for this variant in 1022 familial ALS (fALS) plus 6172 sporadic ALS (sALS) cases compared to 11,732 controls found the allele frequency at this locus to be 0.81% in cases compared to 0.35% in controls (odds ratio [OR] = 1.41, p = 1.2 × 10–7), thus confirming NEK1 as an ALS risk gene.
The vast majority of published human genome-wide studies, including for ALS, have used samples of European ancestry. The importance of trans-ethnic studies in human genetic studies is widely recognised [12,13,14], yet a dearth of studies of non-European ancestry remains. In Asians, the lifetime risk of ALS is estimated to be lower (0.1%) [15] than in Europeans (0.3%) [16] and the mean age of onset is estimated to be a few years earlier [17, 18]. This may reflect the different frequencies of many gene variants, including those already identified as risk or causal [19]. For example, SOD1 mutations account for a higher proportion of Asian familial cases compared to European familial cases (30 vs 14.8%) [20], while the reverse is true for the C9orf72 repeat expansion in sALS cases (~5% in Europeans [20] compared to only 0.3% [21] in Asians), likely due to different founder events, and with evidence that it may have arisen on a different haplotype background [21]. Here, we report the largest WES study for ALS in Chinese to date.
Methods
Participants
The samples are a subset of previously published genome-wide association study (GWAS) data of 1324 cases and 3115 controls [22], which were selected for WES based on DNA availability (627 cases and 186 controls). All cases and controls are of Chinese origin from Mainland China. Additional Chinese ancestry controls were provided through collaboration with the Hunan Normal University and the University of Shanghai for Science and Technology (HNU; 86 individuals) and Wenzhou Medical University (WMU; 479 individuals) (Additional file 1: Table S1). The WMU controls are individuals who attended the affiliated hospitals of Wenzhou Medical University with no medical or family history of neurological disorders during the years 2007–2015.
Whole-exome sequencing data
WES data were generated on 611 Chinese sporadic ALS cases (including two C9orf72 carriers), 16 familial cases (those with one or more affected first-degree relatives) and 186 controls. Only the cases were screened for C9orf72 repeat expansion. Samples were indexed and multiplexed in groups of six per lane and sequenced in 101-bp paired-end mode using the Illumina HiSeq 2000 platform, but with a range of capture kits (see Additional file 1: Table S1 for full details). Of note was that HNU samples (n = 86) differed from the other samples in terms of capture kit (NimbleGen SeqCap EZ Exome v2) and in mean on-target coverage (~ 18.0X overall and 13.8X in v3 capture regions compared to ~ 40–50X for other samples).
Since rare variants are less likely to be called if coverage is low, and if differences in coverage are confounded with affected status, as is the case with our HNU controls, then analyses involving case-control comparisons may be biased. To minimise the potential for this problem, we created two sets of samples: one excluded HNU controls (610 cases and 460 controls after quality control [QC]) and the other included HNU controls (610 cases and 545 controls after QC) but was restricted to variants common to the NimbleGen v2 and v3 capture kits (n = 187,512 post-QC SNPs, compared to 446,395 post-QC SNPs for the primary analysis excluding HNU controls; see below for variant calling criteria). QC and analysis of the two sets of samples was performed separately but using the same analytical pipeline. The results of analyses excluding (presented in the main text) and including (Additional file 1: Table S2) HNU controls do not impact the conclusions drawn.
Variant calling
Image processing and sequence extraction were performed using the standard Illumina Genome Analyzer software. The samples were de-multiplexed using CASAVA (v1.8.2) outputting the short reads for each individual sample in ‘fastq’ format. The quality of all raw sequencing reads (also including WMU and HNU controls) was evaluated using the FastQC (v0.10.1) software. We generated ~ 5.94 Tbp of sequence data for a total number of 813 individuals (611 sporadic cases, 16 familial cases and 186 controls), with a mean on-target coverage of 42.42X per individual. In addition, we analysed ~ 3.18 Tbp of sequence data (mean on-target coverage of 45.01X per individual) for 479 WMU controls and ~ 0.16 Tbp of sequence data (mean on-target coverage of 13.83X per individual) for 86 HNU controls.
Sequence alignment and variant calling were performed using the same BWA-Picard-GATK analysis pipeline for all 1378 samples. Briefly, we aligned the paired-end reads to the human reference genome (hg19) using BWA (v0.6.2) [23], performed file conversion from SAM to BAM and generated the sorted and indexed BAM files using SAMtools (v0.1.17) [24], and marked duplicates using the Picard software package (http://broadinstitute.github.io/picard/) (v1.72). We then used GATK (v3.4-0) [25] to perform ‘Indel Realignment’, ‘Base Quality Score Recalibration’, ‘Variant Calling’ (GATK HaplotypeCaller in a gVCF mode), ‘Joint Genotyping’ and ‘Variant Recalibration’ as described in the GATK Best Practices [26] guidelines. Variants tagged as ‘PASS’ by the GATK Variant Quality Score Recalibration (VQSR) module were used for downstream analysis. The GATK resource bundle (v2.5) was used for VQSR, which includes as training data known SNP sites from HapMap v3.3, the Illumina Omni2.5 array, the 1000 Genomes Project phase 1, dbSNP v137, and the Mills [27] and 1000G gold standard indels. The VQSR target sensitivity cut-offs were set to 99.5% for SNPs and 99% for indels. Variants in each individual were required to have a genotype quality score (GQ) of ≥ 20 for further analysis. The analysis-ready variants from the GATK analysis pipeline were annotated using the ANNOVAR software tool (version 2015 June 17) [28].
Quality control
After the variants were called and annotated, we performed QC steps on individuals and variants (Additional file 1: Table S3). Briefly, individual-level QC was based on common SNPs (MAF > 1%) with genotype call rate > 95%. We excluded individuals from the association analysis who: (1) were sex-discordant/ambiguous (20 individuals); (2) had a genotyping call rate < 80% (123 individuals); (3) had an excessive heterozygosity rate (> 3 standard deviations from the mean; 52 individuals; (4) were shown to be ancestry outliers based on the first two principal components (PCs) derived from common SNPs (i.e. > 6 SD from Chinese CHB mean; 34 individuals); and (5) had a genetic relationship matrix value of > 0.1 with another individual (107 individuals from the WMU sample, known relatives). After QC, we had in total 1070 individuals (610 cases and 460 controls; 626 men and 444 women) remaining for the analyses. We performed the same QC steps for the common capture set. The total number of individuals after QC was 1155 (610 cases and 545 controls). After obtaining clean sets of individuals, we excluded genetic variants based on the following criteria: (1) low genotype call rate < 99%; (2) deviation from Hardy–Weinberg Equilibrium in controls (p < 10–6); (3) differential missingness between cases and controls (p < 10–6); and (4) ≥ 3 alleles.
Gene-based burden analysis
We assessed evidence for an excess of rare damaging mutations in ALS cases compared to controls at the gene level using the SKAT-O test [29] implemented in the R SKAT package [30]. We used the SKAT-O test because it optimally combines the burden test, which is most powerful when a high proportion of variants in a gene are causal and have the same direction of effect, with the sequence kernel association test (SKAT), which is best used when only a small proportion of variants in a gene are causal or if both risk and protective variants are present. In order to facilitate meta-analyses of our results with Cirulli et al. [9], we followed their approach for variant filtering and classification of three variant sets under a dominant genetic model. Briefly, we analysed RefSeq genes for each of three variant sets: (1) all non-synonymous variants (‘Dominant coding’); (2) non-synonymous variants excluding those predicted to be benign by PolyPhen-2 [31] (‘Dominant not benign’); and (3) LOF variants, including stop-loss, stop-gain and splicing variants but not frameshift indels due to recognised difficulties calling indels from WES data [32] (‘Dominant LOF’). For consistency with Cirulli et al., we restricted our analyses to variants passing an internal frequency filter of < 5 × 10–4 (corresponding to singleton variants in our sample) and additionally applied a frequency threshold of < 5 × 10–5 in ExAC [33]. RefSeq genes with at least one qualifying variant were analysed for a total of 301,368 tests and a Bonferroni corrected p value of 1.66 × 10–7. SKAT-O tests were corrected for sex and the top ten PCs based on HapMap3 SNPs. We used default settings in the R SKAT package, including for imputation of missing genotypes and re-sampling methods for computing p values.
Gene-set analyses
We performed gene-set burden testing in ALS cases compared to controls, as one means of overcoming study power limitations due to sample size. Briefly, we defined three curated gene-sets: (1) 30 genes robustly associated with risk of ALS; (2) 128 genes associated with risk of ALS (comprising 21 ALS risk genes, 77 ALS candidate genes and the 30 high confidence ALS genes in set 1); and (3) 245 genes associated with risk of ALS (128 genes in set 2) and/or any of five related neuromuscular disorders (fronto-temporal dementia, Charcot–Marie–Tooth disease, hereditary spastic paraplegia, hereditary ataxia, distal myopathy; total of 117 genes) (Additional file 1: Table S4). Qualifying variants were defined as above, for a total of nine gene-set tests (Bonferroni corrected p value for significance = 5.56 × 10–3) (Additional file 1: Table S2). The mean coverage of exonic regions for each gene was 29.16X with individual gene coverage (including 43 that were covered < 10X in cases or controls) provided in Additional file 1: Table S4.
Meta-analysis of European and Chinese variant counts
We used a Cochran–Mantel–Haenszel test to evaluate evidence for association at the gene level in a combined analysis of case-control variant counts in Europeans [9] and our Chinese WES cohort. Each variant set count was separately analysed as described above for gene-based burden testing within our Chinese cohort. Considering genes with at least one qualifying variant in either cohort, we performed a total of 26,214 tests across the three variant classes (Bonferroni corrected p value threshold of 1.91 × 10–6) and we used the Breslow–Day test to assess evidence for homogeneity of ORs for each gene across Chinese and European samples [9].
ALS-variant analysis
To identify known variants previously associated with ALS, cases and controls were screened for any of 1158 ALS variants previously reported in the Human Gene Mutation Database (HGMD, trial professional version, accessed 3rd May 2016) and Amyotrophic Lateral Sclerosis online Database (ALSoD, accessed 1st September 2016) [34] using ANNOVAR [28]. Since variants in these databases may include false positives (benign) or risk variants (i.e. they occur at a population frequency that is inconsistent with the assumed disease prevalence and penetrance), we ignored any known variants identified in our cohort for which the frequency in ExAC populations of any ethnicity (the ‘popmax’ approach [33]) was > 0.01. To identify novel variants in relevant genes we used a previously curated hierarchical gene-set [35] (Additional file 1: Table S4) and restricted the analysis to non-synonymous (missense), stop-gain/loss (nonsense) and splicing (first and last two bases of each intron) variants. To enhance pathogenicity call rates [36], any missense variants classified as ‘tolerated’ by both MetaLR [37] and MetaSVM_pred [37] (integration of 18 current deleteriousness-scoring methods) were excluded. ExAC [33] popmax MAF filters of < 5 × 10–5 and < 0.01 for dominant and recessive genetic architectures, respectively, were applied. These filters for novel variants in known disease genes were more stringent than the filters applied for gene-based testing (described above and adopted from Cirulli et al. to enable meta-analysis of gene-based variant counts) because the objective was to screen for putatively pathogenic variants. Final variant lists were cross-checked with clinical databases (OMIM, Clinvar [38]) and the literature for case reports to assess pathogenicity. In examining the curated set of genes [35] (Additional file 1: Table S4), variants passing all filters present in ≥ 1 individual (case and/or control) were identified.
Putatively pathogenic indels were screened for in a subset of 21 genes, with prior evidence for causative indels and/or LOF variants [35] (Additional file 1: Table S4). These were separated into non-truncating (in-frame) and truncating (frame-shift) insertions and deletions, which were subsequently cross-checked for pathogenicity as above.
Results
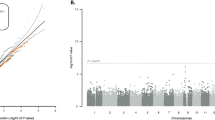
In exome-wide gene-based association testing, no single gene was significantly associated with ALS after multiple testing correction (Additional file 1: Table S5, Additional file 2: Figure S1). This is unsurprising given the size of the sample. Similar to Cirulli et al. [9], we found that many of the top ranked genes, based on burden tests, showed an excess of rare mutations in controls compared to cases. Despite joint calling of variants, this likely reflects ascertainment associated with the additional control samples to increase our control sample size. When we meta-analysed per-gene case-control counts of rare functional mutations in our Chinese sample with those from the largest WES study of European ancestry [9] (Additional file 1: Table S6), three genes surpassed genome-wide significance for association with ALS with smaller p values than in the European ancestry samples alone: TBK1; NEK1;and SOD1 (Table 1; Fig. 1). Both NEK1 and SOD1 were nominally significant in our Chinese sample, while TBK1 was not significant (Table 1), and the case-control frequencies of rare coding variants were similar to Europeans (NEK1 1.8%/0.4% vs 1.9%/0.8%; SOD1: 1.5%/0.2% vs 0.9%/0.1%; TBK1: 1.2%/0.2% vs 1.4%/0.4%). We found no evidence for an excess of rare coding variants in cases in any of three a priori sets of genes associated with risk of ALS or related neuromuscular disorders (Additional file 1: Table S4).

Quantile–quantile plots of the analysis of rare variant counts in combined Chinese and European data (up to 4797 cases and 9236 controls). The Cochran–Mantel–Haenszel test was applied to qualifying variants under three models: (L) dominant coding; (R) dominant not benign; and dominant LOF (Additional file 2: Figure S1). Test statistics are provided for the genes with the top ten associations (blue = increased risk, grey = reduced risk; *no qualifying variants were observed in controls for gene S100A2, so the OR was estimated by adding 0.5 to each cell of the largest cohort). The Bonferroni-corrected significance threshold was p ≤ 1.9 × 10–6, based on 26,214 tests across 18,117 genes. The genomic inflation factor, lambda (λ), was 1.069 for the dominant coding analysis and 1.067 for the dominant not benign analysisrecognised in our Chinese sample
It is well-recognised that many variants reported in databases as ‘pathogenic’ for disease occur at a population frequency too high to be consistent with the reported disease prevalence [33, 39]. With this in mind, WES variants were screened for previously reported ALS variants for which we judged the evidence for pathogenicity was strong. Twenty-one of the Chinese sALS cases, five fALS probands and two of the controls harboured such variants (Additional file 1: Table S7; see Additional file 1: Table S8 for details of variants in NEK1, SOD1 and TBK1 that passed filters for gene-based testing, screening of known ALS variants or both). Considering exome variant results and two C9orf72 carriers jointly, likely pathogenic variants account for 4.6% of ALS cases (28 out of 610) and 0.4% of controls (two out of 460; Fig. 2). This was slightly lower than the proportion of ALS cases with a known causal variant in an Australian clinical ALS cohort (~ 90% European ancestry) which was 10% using an identical filtering technique [35]. For familial probands, 38% (5 out of 13) were carriers of a likely causal variant. This is on the lower end of the range (30–70%) compared to what has previously been reported in European ancestry populations [35, 40]. The lower proportion of identified likely causal variants in both sALS and fALS cases is likely to be explained by a lower prevalence of the C9orf72 repeat expansion which accounts for up to 7% of sALS and 40% of fALS in European populations [2] compared to just 0.3% in sALS cases in this study (as found in other Chinese samples [41, 42]). In contrast, we found a relatively high number of NEK1 variants (nine non-synonymous variants in ten cases) and notably this did not include the recently reported p.Arg261His NEK1 variant identified in a Dutch study [11]. While this can be expected given that ultra-rare variants tend to be highly population-specific [33], it is interesting that this locus has been independently.

Summary of rare variants in Chinese WES sample comprising 597 sporadic (sALS) and 13 familial (fALS) cases. The screening of WES data of Chinese ALS cases identified ~ 5% with previously reported likely causal variants. Variants previously reported for ALS but now found to have population frequency (0.00005 ≤ freq < 0.01) are classified as ‘unlikely causal’. For variants identified in cases only, a number of putatively damaging, rare (MAF < 0.00005 dominant or < 0.01 recessive) variants in a predefined set of known ALS-priority genes (n = 32 cases) and ALS-relevant genes (n = 89 cases) were identified, but these have uncertain significance. Considering only fALS probands (n = 13), WES identified previously reported likely causal variants in five cases (1 DCTN1, 2 FUS, 1 SOD1, 1 TARDBP) with uncertain significance variants (damaging rare in ALS-relevant genes) in four others. Four percent of cases (24/610) and 3% of controls (13/460) were identified to be carrying one or more rare variants in ALS genes (from any category; causal, risk, candidate) and/or similar disease genes (Additional file 1: Table S10), but no individual harboured more than one likely causal variant. The number of cases are defined in the legend and expressed a percentage of total ALS case exomes screened (n = 610)
Discussion
In the largest WES study of ALS in Chinese samples we did not identify any specific gene significantly associated with ALS. Meta-analysing Chinese and European WES data strengthened the evidence for three genes (SOD1, NEK1 and TBK1) reported as significantly associated with ALS in European samples (Table 1, Additional file 1: Table S6). The estimated case-control frequencies of rare coding variants in these genes in Chinese was similar to that reported for Europeans, and thus the nominal statistical associations we report for Chinese (Table 1) are a reflection of the available sample size. While SOD1 is recognised as the most important ALS-associated gene in Chinese [20], evidence that NEK1, recently identified in European samples, may also be associated with ALS in Chinese is novel. Larger Chinese samples with whole exome data will be needed to confirm this result and to establish if TBK1 is also an ALS gene in Chinese. Given the possible differences in genomic architecture of ALS between populations, additional genomic studies of ALS in non-European populations are warranted.
Assessing novel variants in known ALS disease genes revealed > 30 distinct mutations in SOD1, TARDBP, CHMP2B, ERBB4, DCTN1, FIG4, FUS, MATR3, NEK1, SETX, SQSTM1, TBK1 and UBQLN2 that were present in cases but not controls (Additional file 1: Table S9). Characterising the function of these newly identified variants, with respect to other reported variants and disease penetrance, is expected to enhance the ability to understand exactly how gene function and any related genes and/or pathways are impacted to alter ALS risk. Given the size of our cohort, we expect the variants identified to be typical of other clinical cohorts in China (Fig. 2), which will help to provide an evidence-based approach to the design of a targeted genetic screen, and may in future contribute to improved treatment strategies. An important caveat is that the list of identified putatively damaging variants in ALS genes likely contains a proportion of false positives, because our filtering also identified variants in controls (Additional file 1: Table S9). We identified a similar proportion of ‘oligogenic’ individuals (those that harbour two or more rare variants in ALS genes [from any category; causal, risk, candidate] and/or similar disease genes) in cases and controls (4% vs 3%) (Additional file 1: Table S10). Notably, no individual harboured more than one likely causal variant demonstrating that these results cannot yet provide any evidence for an oligogenic, rare variant basis in ALS.
Conclusions
It is well recognised that large sample sizes are needed to detect association of rare variants in complex diseases, such as ALS [43]. Despite being the third largest WES study for ALS and the largest such study in Chinese to date, our study remains limited by sample size. We provide novel evidence for association of NEK1 with ALS in Chinese, reporting variants in these genes not previously found in Europeans. To increase the power for discovery, combining our study with other whole-exome studies (or genome studies) is warranted. To facilitate future meta-analyses, we report per gene counts of all WES variants that pass filtering steps in Chinese (Additional file 1: Tables S5 and S6) and list those variants with ALS-relevant annotation (Additional file 1: Tables S7–S9 and S11),
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- CHB:
-
Han Chinese in Beijing
- ExAC:
-
Exome aggregation consortium
- fALS:
-
Familial amyotrophic lateral sclerosis
- GWAS:
-
Genome-wide association study
- HGMD:
-
Human genome mutation database
- HNU:
-
Hunan Normal University
- QC:
-
Quality control
- sALS:
-
Sporadic ALS
- SKAT:
-
Sequence kernel association test
- SKAT-O:
-
Sequence kernel association test – optimal
- WES:
-
Whole-exome sequencing
- WMU:
-
Wenzhou Medical University
References
Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: New genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 1607;2015:75–93.
Renton AE, Chiò A, Traynor BJ, Cirulli ET, Lasseigne BN, Petrovski S, et al. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23.
He J, Mangelsdorf M, Fan D, Bartlett P, Brown MA. Amyotrophic lateral sclerosis genetic studies. Neurosci. 2015;21:599–615.
Fang F, Quinlan P, Ye W, Barber MK, Umbach DM, Sandler DP, et al. Workplace exposures and the risk of amyotrophic lateral sclerosis. Environ Health Perspect. 2009;117:1387–92.
Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206.
Kenna KP, McLaughlin RL, Byrne S, Elamin M, Heverin M, Kenny EM, et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet. 2013;50:776–83.
Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. 2016;13:96–104.
Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–60.
Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–41.
van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016;48:1043–8.
Kenna KP, van Doormaal PTC, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet. 2016;48:1037–42.
Asimit JL, Hatzikotoulas K, McCarthy M, Morris AP, Zeggini E. Trans-ethnic study design approaches for fine-mapping. Eur J Hum Genet. 2016;24:1330–6.
Zaitlen N, Paşaniuc B, Gur T, Ziv E, Halperin E. Leveraging genetic variability across populations for the identification of causal variants. Am J Hum Genet. 2010;86:23–33.
Morris AP. Transethnic meta-analysis of genomewide association studies. Genet Epidemiol. 2011;35:809–22.
Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41:118–30.
Johnston CA, Stanton BR, Turner MR, Gray R, Blunt AH-M, Butt D, et al. Amyotrophic lateral sclerosis in an urban setting: a population based study of inner city London. J Neurol. 2006;253:1642–3.
Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10:310–23.
Liu MS, Cui LY, Fan DS. Chinese ALS Association. Age at onset of amyotrophic lateral sclerosis in China. Acta Neurol Scand. 2014;129:163–7.
Liu Q, Liu F, Cui B, Lu CX, Guo XN, Wang RR, et al. Mutation spectrum of Chinese patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2016;87:1272–4.
Zou Z-Y, Zhou Z-R, Che C-H, Liu C-Y, He R-L, Huang H-P. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540–9.
He J, Tang L, Benyamin B, Shah S, Hemani G, Liu R, et al. C9orf72 hexanucleotide repeat expansions in Chinese sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2015;36:2660.e1–8.
Benyamin B, He J, Zhao Q, Gratten J, Garton F, Leo PJ, et al. Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis. Nat Commun. 2017;8:611.
Li H, Durbin R. Fast and accurate short read alignment with Burrows – Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
McKenna A, Hanna M, Banks E, Sivachenko AY, Cibulskis K, Kernytsky AM, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Genome Res. 2010;20:491–8.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–33.
Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–90.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13:762–75.
Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet. 2013;92:841–53.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Fang H, Wu Y, Narzisi G, ORawe JA, Barrón LTJ, Rosenbaum J, et al. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014;6:89.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Abel O, Powell JF, Andersen PM, Al-Chalabi A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum Mutat. 2012;33:1345–51.
Garton FC, Benyamin B, Zhao Q, Liu Z, Gratten J, Henders AK, et al. Whole exome sequencing and DNA methylation analysis in a clinical amyotrophic lateral sclerosis cohort. Mol Genet Genomic Med. 2017;5:418–28.
Moreira LGA, Pereira LC, Drummond PR, De Mesquita JF. Structural and functional analysis of human SOD1 in amyotrophic lateral sclerosis. PLoS One. 2013;8:e81979.
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–37.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
McCann EP, Williams KL, Fifita JA, Tarr IS, O’Connor J, Rowe DB, et al. The genotype-phenotype landscape of familial amyotrophic lateral sclerosis in Australia. Clin Genet. 2017;92:259–66.
Jiao B, Tang B, Liu X, Yan X, Zhou L, Yang Y, et al. Identification of C9orf72 repeat expansions in patients with amyotrophic lateral sclerosis and frontotemporal dementia in mainland China. Neurobiol Aging. 2014;35:936. e19-936.e22.
Chen Y, Lin Z, Chen X, Cao B, Wei Q, Ou R, et al. Large C9orf72 repeat expansions are seen in Chinese patients with sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2016;38:217.e15–22.
Agarwala V, Flannick J, Sunyaev S, Altshuler D, Altshuler D. Evaluating empirical bounds on complex disease genetic architecture. Nat Genet. 2013;45:1418–27.
Acknowledgements
We would like to thank all the participants and recruitment staff for their generous contributions to this study.
Funding
This work was funded by the Australian Research Council (ARC) Linkage Grant (MAB, PFB, PMV, HX, RJW, BJM, DCR, MMangelsdorf), the Peter Goodenough Foundation, the National Natural Science Foundation of China (grants to DSF: 81030019, JH: 81601105, ZBJ: 81522014), the National Health and Medical Research Council (NHMRC) (NRW: 1078901, 1083187, BB: 1084417, 1079583, PMV: 1078037, IPB: 1095215, KLW: 1092023, FG: 1121962, JG: 1127440, 1103418), MNDRIA (BB: Mick Roger Benalla Grant, FG: Bill Gole Fellowship), MMangelsdorf and RJW: Ross Maclean Senior Research Fellowships, the Sylvia & Charles Viertel Charitable Foundation.
Availability of data and materials
Additional file 1: Tables S5 and S6 report per gene counts of all WES variants that pass filtering steps. These data as well as the variants contributing to the gene counts have been uploaded to the Project Mine web browser (http://databrowser.projectmine.com/). Our analyses used data from Supplementary Table 6 of Cirulli et al. [9]. Deposit of raw sequence data does not comply with our consent process and ethics approval, but sharing of the primary dataset of 627 cases and 186 controls is possible by emailing the corresponding author.
Author information
Authors and Affiliations
Contributions
Study conception: MAB, DF, RHW, HX, PFB. Funding: MAB, PFB, PMV, HX, RJW, DCR, BJM, MMangelsdorf, RHW, NRW, BB, FG. WES QC and analysis: QZ, JG, BB, PL, FG, Z-HZ, ZLiu, MMarshall, PMV, NRW. Chinese ALS and control samples: DF, JHe, LT, LC, XL, MAB. Wenzhou WES control samples: JW, Z-BJ, ZLi. Project management for Wenzhou WES control samples: AKH, JY. HNU WES controls: H-WD, YL, SR, Y-YH, LJT, X-DC. Genotyping/sequencing: LA, KC, JE, LW, MAB. Manuscript 1st draft: JG, QZ, BB, FG, NRW. All authors contributed to revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
ALS patients were from the ALS specialty clinic at the Department of Neurology of the Peking University Third Hospital, Beijing, China. Control samples were from individuals who attended the same hospital, Shanghai Changzheng Hospital, Hunan Normal University, the University of Shanghai for Science and Technology and Wenzhou Medical University. All cases and controls provided written informed consent to participate in the research. Sample collections were approved by the human research ethics committees of the Peking University Third Hospital, Shanghai Changzheng Hospital, the University of Shanghai for Science and Technology and Wenzhou Medical University Eye Hospital (KYK-2015-18) and by the Hunan University Ethics Committee in their Department of Research Administration. Analyses conducted at the University of Queensland were approved by the University human research ethics committee (Approval no. 2011001173). The study conformed to the principles of the Declaration of Helsinki.
Consent for publication
N/A. Our manuscript does not include any individual person’s data in any form.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Detailed information of samples. Table S2. Analysis of rare coding variants. Table S3. Description and summary of quality control steps of whole-exome sequencing samples. Table S4. Exome sequencing coverage in genes included in the WES analyses. Table S5. Gene-based SKAT-O association test p-values and per gene counts of WES variants that passed filtering steps. Table S6. Gene-based Cochran–Mantel–Haenszel association test results based on WES variant counts in Chinese and Europeans. Table S7. Previously reported variants that are likely causal for ALS identified in individuals in our study. Table S8. NEK1, SOD1, TBK1 variants identified in SKAT-O and/or ALS specific variant/gene testing. Table S9. Not previously reported variants of probable/possible/unknown significance in ALS-related genes identified in at least one individual in our study. Table S10. Individuals identified with two or more variants considered relevant for ALS (oligogenic). Table S11. Previously reported variants that are unlikely causal (due to high minor allele frequency) identified in individuals in our study. (XLSX 7252 kb)
Additional file 2: Figure S1.
Quantile–quantile plots for exome-wide gene-based testing of rare coding variants in the primary analysis of 610 cases and 460 controls. (DOCX 235 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gratten, J., Zhao, Q., Benyamin, B. et al. Whole-exome sequencing in amyotrophic lateral sclerosis suggests NEK1 is a risk gene in Chinese. Genome Med 9, 97 (2017). https://doi.org/10.1186/s13073-017-0487-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-017-0487-0