
Abstract
The histone variant H2A.Z is involved in several processes such as transcriptional control, DNA repair, regulation of centromeric heterochromatin and, not surprisingly, is implicated in diseases such as cancer. Here, we review the recent developments on H2A.Z focusing on its role in transcriptional activation and repression. H2A.Z, as a replication-independent histone, has been studied in several model organisms and inducible mammalian model systems. Its loading machinery and several modifying enzymes have been recently identified, and some of the long-standing discrepancies in transcriptional activation and/or repression are about to be resolved. The buffering functions of H2A.Z, as supported by genome-wide localization and analyzed in several dynamic systems, are an excellent example of transcriptional control. Posttranslational modifications such as acetylation and ubiquitination of H2A.Z, as well as its specific binding partners, are in our view central players in the control of gene expression. Understanding the key-mechanisms in either turnover or stabilization of H2A.Z-containing nucleosomes as well as defining the H2A.Z interactome will pave the way for therapeutic applications in the future.
Similar content being viewed by others
Background
The chromatin structure represents the major modulator of all DNA-based processes such as gene transcription, DNA replication and repair. Chromatin is essentially composed of DNA and histone proteins that together form its basic unit, known as nucleosome. Within the core nucleosome, approximately 146 base pairs (bp) of DNA are wrapped in a left-handed superhelical turn around a protein structure composed of two copies each of the histones H3, H4, H2A and H2B whose crystal structure was solved more than 20 years ago [1]. While histones H3 and H4 form a tetrameric structure known as nucleosome core that is positioned in the inner region of the nucleosome, the histones H2A and H2B are rather located on the nucleosomal surface. In addition, the linker histone H1 can contact the entry and exit sites of the nucleosomal DNA resulting in a more compact structure [2]. Of note, histones are characterized by the presence of a characteristic histone fold domain from which unstructured N- and C-terminal tails protrude [1]. In the process of nucleosome assembly, ATP-dependent remodelers assemble and arrange histone octamers in a highly dynamic fashion [3]. Posttranslational modifications (PTMs) are placed predominantly on flexible histone tails but also within the histone fold domains [4,5,6]. Importantly, replication-independent (hereafter referred to as non-canonical) histone variants can substitute replication-dependent (hereafter referred to as canonical) histones and are specifically positioned within the genome.
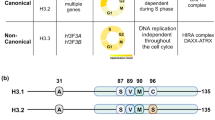
While canonical histones are expressed exclusively during the replication phase of the cell cycle, histone variants are expressed throughout the cell cycle. Canonical histones are encoded by multi-copy genes that lack introns and present a stem loop structure at the 3′-end of their mRNAs. In contrast, genes encoding histone variants are biallelic, sometimes characterized by introns and poly-adenylated at the 3′-end of their mRNAs. As consequence, some histone variants-encoding genes are subjected to alternative splicing. Apart from histone H4, all histone protein families (H2A, H2B, H3 and H1) are characterized by specialized histone variants and among them the most studied family is the H2A, which comprises several members including macroH2A, H2A.X, H2A.Bbd and H2A.Z [7]. As an aside, an H4 variant has been identified in the urochordata Oikopleura dioica [8] and in Trypanosoma brucei [9], suggesting the possibility that H4 variants may be expressed also in other organisms.
The histone variant H2A.Z has been intensively studied over the last three decades elucidating not only the enzymatic activities required for its chromatin deposition but also the interlinked posttranslational regulatory mechanisms as well as its dynamics in response to signaling pathways. The focus of this review is to summarize and discuss the current knowledge on the histone variant H2A.Z. In particular, we will emphasize the mechanisms of its chromatin deposition and removal, its posttranslational regulation and its interaction partners. Further, we will also review the latest developments concerning H2A.Z’s deregulation or mutations in diseases and how newest technologies can be used to manipulate histone variant levels.
Historical perspective and overview
The histone variant H2A.Z was originally identified in 1980 in mouse L1210 cells [10]. Few years later, studies in Tetrahymena thermophila observed the presence of H2A.Z in the transcriptionally active macronucleus but not in the transcriptionally inactive micronucleus [11]. Later, the Drosophila melanogaster homolog H2Av, was identified [12] and shown to be essential [13].
Subsequently, the mammalian H2A.Z gene was cloned in 1990 [14] and similarly to Drosophila, it was found to be essential since the mouse knockout displays an early-lethal embryonic phenotype [15]. Surprisingly, earlier studies revealed that H2A.Z depletion is not lethal in Saccharomyces cerevisiae but “only” leads to reduced growth, a phenotype that can be efficiently rescued via reintroduction of the H2A.Z-encoding gene from Tetrahymena [16], marking the evolutionary conservation of H2A.Z.
Mass-spectrometry (MS) studies identified two different H2A.Z isoforms that differ only in three amino acids (Fig. 1 [17]). These isoforms, known as H2A.Z.1 and H2A.Z.2 [18], are encoded by two separate genes that are well conserved in chordates and known as H2AFZ and H2AFV genes, respectively [19]. Even if these two isoforms differ only in three amino acids, they display specialized functions: For example, H2A.Z.2 is preferentially associated with H3K4me3 [20], while H2A.Z.1 has been shown to better interact with the bromodomain-containing protein 2 (BRD2) [21]. Matsuda and colleagues were able to generate single knockouts of both H2A.Z isoforms in chicken DT40 cells further unveiling the different function of the two isoforms. Compared to H2A.Z.1 knockout cells, H2A.Z.2 knockout cells show a slight reduction in proliferation associated with increased apoptosis that may be the consequence of reduced expression of the anti-apoptotic gene BCL6 [22]. In line with that, H2A.Z.2 depletion in human metastatic melanoma cells leads to downregulation of cell cycle-promoting genes [23] and a recent study further marked the different function of the two isoforms in regulating gene expression in rat neurons [24]. Making use of fluorescence recovery after photobleaching (FRAP) and inverse FRAP (iFRAP), Nishibuchi and colleagues observed that H2A.Z.2 but not H2A.Z.1 is rapidly exchanged at sites of double-strand breaks (DSBs) induced via microirradiation with ultraviolet A (UVA) [25]. Given that RAD51 is required for homologous recombination (HR) at DSBs where it forms foci, the authors investigated the possibility that this mechanism would be differentially influenced by the two H2A.Z isoforms. Surprisingly, they observed reduced RAD51 foci and HR in H2A.Z.2 knockout compared to H2A.Z.1 knockout DT40 cells [25]. However, in the absence of DSBs, another study observed higher mobility of H2A.Z.1 compared to H2A.Z.2 in HeLa cells [26]. Making use of domain swapping experiments, the authors also observed that this difference could be, at least partially, dependent on the substitution in position 38, which corresponds to a serine in H2A.Z.1 and to a threonine in H2A.Z.2 (Fig. 1 [26]).

Schematic representation of the different human H2A.Z isoforms. Alignment of H2A.Z.1, H2A.Z.2.1 and H2A.Z.2.2 protein sequences. Highlighted in gray are residues that differ between H2A.Z.1 and H2A.Z.2.1, while residues highlighted in yellow are the ones not conserved between H2A.Z.2.1 and H2A.Z.2.2. Yellow, red, blue, pink and green balls indicate sites of acetylation, methylation, phosphorylation, SUMOylation and ubiquitination, respectively. Please look at Table 2 for more details about the PTMs of H2A.Z
Recently, this H2A.Z isoform scenario has become more complex due to the identification of an alternatively spliced and primate-specific isoform of H2A.Z.2 (hereafter referred to as H2A.Z.2.1 [18]), known as H2A.Z.2.2 (Fig. 1) that is expressed in a wide range of tissues with maximum transcript expression in human brain tissues [27]. H2A.Z.2.1 and H2A.Z.2.2 differ within their C-terminal region, and H2A.Z.2.2-containing nucleosomes are less stable compared to H2A.Z.2.1-containing ones due to reduced binding to neighboring histones within one octamer [27]. Previously, Adam and colleagues have shown that the C-terminal region of the yeast H2A.Z protein interacts with RNA polymerase II (RNAPII), promoting its recruitment at promoters [28]. Given that the C-terminus of H2A.Z.2.2 significantly differs from the one of H2A.Z.1 and H2A.Z.2.1, it will be interesting to test whether also H2A.Z.2.2 is able to interact with RNAPII and to evaluate whether the different histone variants, present within a different genomic localization, may differentially influence RNAPII recruitment and finally transcription.
H2A.Z has been linked to diverse biological processes such as memory [29,30,31,32] and epithelial-to-mesenchymal transition (EMT, [33]). At molecular level, it has been implicated in heterochromatin regulation [34,35,36,37,38], anti-silencing function at boundaries in yeast [39,40,41,42], DNA repair [25, 43,44,45,46,47,48] and transcriptional regulation [21, 23, 28, 29, 49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88]. How H2A.Z regulates such a wide spectrum of different processes is not fully understood, and it is even more surprising that H2A.Z regulates both transcriptional repression and activation.
Interestingly, the Drosophila H2Av variant, encoded by the His2Av gene, fulfills the functions of both mammalian H2A.Z and H2A.X variants. Similar to mammalian H2A.Z, H2Av regulates heterochromatin formation [89] and gene regulation [90] as also marked by its enrichment in euchromatic regions [91]. On the other hand, the mammalian histone variant H2A.X is a pivotal factor for DNA damage responses. H2A.X is phosphorylated on its unique serine 139 (called γ-H2A.X) upon DSBs and helps recruiting the DNA repair machinery [92]. Similarly, upon DSBs, Drosophila H2Av is specifically phosphorylated on a serine residue conserved in mammalian H2A.X [93]. Subsequently, phosphoH2Av is acetylated by the histone acetyltransferase (HAT) dTip60, leading to its exchange with an unmodified H2Av at DSB sites [94]. In line with that, loss-of-function (LoF) of Tip60 leads to accumulation of phosphoH2Av [95].
In this review, we will discuss the recent literature elucidating the contrasting facets of H2A.Z in gene regulation.
Mechanisms of loading and removal of the histone variant H2A.Z
One major discovery in the histone variant field was the identification of yeast Swr1, a member of the Snf2 family of ATPases, to be the protein complex responsible for loading H2A.Z onto chromatin [62, 63, 98,99,100]. Swr1 loads the H2A.Z-H2B dimer into nucleosomes by a well-defined mechanism: First, one H2A.Z-H2B dimer is loaded generating an H2A-H2A.Z heterotypic nucleosome, and then a second H2A.Z-H2B dimer is loaded generating a homotypic H2A.Z nucleosome [101]. Swr1 itself is part of the multi-subunit SWR1 complex (Table 1, [63, 96,97,98, 100, 102]), whose 3D architecture has been recently solved by electron microscopy [103, 104]. Its function is to partially unwrap the DNA from the histone core, which is dynamically altered by ATP consumption [105]. Furthermore, the crystal structure of the central Swr1 enzyme in complex with the H2A.Z-H2B dimer revealed that Swr1 delivers the H2A.Z-H2B dimer to the DNA-(H3-H4) tetrasome as a histone chaperone [106]. Importantly, six of the SWR1 complex subunits are also found within the NuA4 acetyltransferase complex (Table 1) and/or the Ino80 chromatin remodeling complex [97, 98, 100]. The NuA4 complex is a multi-subunit complex [107] involved in the acetylation of H2A.Z by its specific subunit Esa1 [107, 108]. Esa1 and its mammalian homolog, Tip60, are also required to stimulate H2A.Z loading by acetylation of H4 and H2A histone tails within the nucleosome [100, 109, 110]. Mechanistically, H2A/H4 acetylation is required to recruit the SWR1 complex via its subunit Bdf1, which is able to recognize acetylated histones via its bromodomain [100]. In line with this observation, Bdf1 LoF mutants showed reduced H2A.Z chromatin occupancy in yeast cells [82]. Similarly to the NuA4 complex, also the acetyltransferase activity of the something about silencing (SAS) complex (composed of Sas2, Sas4 and Sas5, [111]) is able to stimulate H2A.Z incorporation in yeast [41], further marking that histone acetylation is a prerequisite for H2A.Z deposition.
Swr1 is evolutionary conserved: The Drosophila homolog is known as Domino, while in mammals there are two homologs called SRCAP (SNF2-related CREBBP activator protein) and Ep400, which are both able to catalyze the incorporation of H2A.Z within chromatin [49, 112]. Biochemical purifications of the human Ep400-containing complex surprisingly unveiled that it is composed of not only homologous subunits of the SWR1 complex but also contains subunits that are exclusively found within the yeast NuA4 complex (Table 1, [109, 113,114,115,116]). The same is true for the Drosophila complex (Table 1, [94]). This suggests that the Drosophila/human complex, known as p400/Tip60 complex, represents a physical merge of the yeast SWR1 and NuA4 complexes. This hypothesis is further supported by the observation that human Ep400 represents a fusion of yeast Swr1 and Eaf1, subunits of SWR1 and NuA4 complexes, respectively [107]. In contrast, biochemical purification of the human SRCAP-containing complex showed that it does not contain any histone acetyltransferase activity (Table 1, [112, 116]).
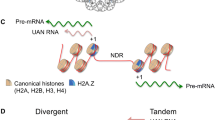
One important question remains: How is the H2A.Z loading and/or acetylation machinery recruited to chromatin? So far, it was shown that a plethora of different transcription factors (TFs) interact with subunits of the H2A.Z loading complexes (Fig. 2a). For example, the p400/Tip60 complex interacts with the Notch/RBPJ coactivator complex [57], Myc [117], ERα (estrogen receptor alpha, [118]), AR (androgen receptor [119]) and PU.1 [120]. Similarly, the GAS41 (also known as YEATS4) subunit of both p400 and SRCAP complexes interacts with the TFIIF subunit of the pre-initiation complex (PIC, Fig. 2b, [121]). In addition to these interactions, some of the subunits of the p400/Tip60 complex contain “reader” domains able to recognize posttranslationally modified histone tails. For example, MRG15 is able to recognize H3K4me1 and H3K4me3 (Fig. 2c, [122]), Tip60 binds to H3K4me1 (Fig. 2c, [118]) and GAS41, via its YEAST domain, recognizes acetylated or succinylated histone tails (Fig. 2d) in both yeast and human [123,124,125,126,127]. In line with this, GAS41 depletion leads to reduced H2A.Z occupancy [124,125,126, 128]. It must be also marked that, at least in yeast, the SWR1 complex recognizes a region devoid of nucleosomes, known as nucleosome-free region (NFR) or nucleosome-depleted region (NDR), which is characteristic of transcriptional starting sites (TSSs) [110, 129]. This further suggests an additional mechanism of SWR1 recruitment that involves its interaction with the NFR and that is not mutually exclusive with the mechanisms involving histone PTMs recognition and/or interactions with TFs and/or components of the PIC. However, the involvement of the NFR in the recruitment of the p400/Tip60 and SRCAP complexes has not yet been investigated.

Mechanisms of recruitment of the Ep400/Tip60 complex. The Ep400/Tip60 complex, involved in loading and acetylation of the histone variant H2A.Z, can be recruited to its target genomic sites via interactions with a transcription factors (TFs) or b subunits of the pre-initiation complex (PIC) composed of general transcription factors (GTF; TFIIA, TFIIB, TFIID, TFIIE, TFIIF and TFIIH) and RNA polymerase II (RNAPII). In addition, the Ep400/Tip60 complex can be recruited to its target genomic sites via interactions with posttranslationally modified histone proteins, for example: c MRG15 binds to H3K4me1 or H3K4me3 [122], while Tip60 binds to H3K4me1 via its chromodomain [118]. d GAS41 binds to H3K14ac, H3K27ac or H3K122suc using its YEAST domain [123,124,125,126,127]. For simplicity reasons, only the Ep400/Tip60 complex is shown; however, similar mechanisms of recruitment can be used by the SRCAP complex. TFBS transcription factor binding site
While the SWR1, p400/Tip60 and SRCAP complexes load H2A.Z within chromatin, there are also mechanisms to evict H2A.Z. ANP32E was recently shown to remove H2A.Z from nucleosomes in human cells during DNA damage [130, 131]. Its depletion leads to increased H2A.Z occupancy, and it co-localizes genome-wide with H2A.Z [130,131,132].
Together, the identification of the protein machineries placing H2A.Z is an important step forward for the better understanding of the dual role of H2A.Z in gene regulation: based on the loading machinery involved in the locus-specific deposition of H2A.Z (SRCAP or p400/Tip60), different PTMs of H2A.Z can be deposited leading to the recruitment of different H2A.Z interactors that finally result in a different transcriptional output (repression or activation).
Posttranslational regulation of H2A.Z determines the transcriptional output
Histone variants, like all the canonical histones, can be dynamically decorated by various PTMs including acetylation, methylation, phosphorylation, SUMOylation and ubiquitination. Genetic data indicate that H2A.Z can serve as a buffer to quench phenotypic noise via modulating transcriptional efficiencies [133]. Looking at gene expression, H2A.Z depletion can either lead to the upregulation of genes, for example ∆Np63α and Notch signaling targets [29, 49, 57, 72, 134], or to downregulation such as estrogen signaling [28, 50, 54]. Thus, H2A.Z is able to modulate, by dampening, either transcriptional repression or activation. In our view, PTMs of H2A.Z play a major role in this transcriptional buffering function.
Historically, H2A.Z acetylation (H2A.Zac) was first described in Tetrahymena [135], while the identification of the exact lysine residues was only fairly recently described (Fig. 1 and Table 2). H2A.Zac is clearly associated with active transcription as first demonstrated in chicken [136, 137]. More recently, these observations have been extended to mammals showing also that H2A.Zac levels positively correlate with transcriptional output [53, 54, 58, 60, 84]. Importantly, H2A.Zac can be dynamically regulated in response to signal transduction [57, 59]. The enzyme responsible for H2A.Zac was initially identified in Saccharomyces cerevisiae as Esa1 (Mst1 in Schizosaccharomyces pombe), a subunit of the NuA4 complex (Table 1, [40, 99, 108, 138,139,140]). Subsequently, its homologs have been linked to H2A.Zac also in Drosophila and mammals (dTip60 and Tip60, respectively [55, 57, 96, 141]). In yeast, acetylation of H2A.Z’s N-terminal lysine residues does not influence its turnover and this PTM is dynamically regulated due to the deacetylase activity of Hda1 [139, 142]. However, how H2A.Z is deacetylated in higher organisms has not been investigated in depth, although global inhibition of histone deacetylases (HDACs) by trichostatin-A (TSA) leads to increased H2A.Zac [30, 53]. Recently, we have shown that the HDAC1/2-containing nucleosome remodeling and deacetylase (NuRD) complex likely participates in H2A.Z deacetylation [143]. Finally, the characterization of H2A.Zac readers is just at the beginning and so far only bromodomain and PHD (plant homeodomain) finger-containing transcription factor (BPTF) protein and Plasmodium falciparum GCN5 (PfGCN5) were shown to recognize a specific pattern of H2A.Zac. BPTF binds to H2A.ZK4acK11ac and H2A.ZK4acK7ac, whereas PfGCN5 interacts with H2A.ZK4acK11ac [144].
Ubiquitination of H2A.Z (H2A.Zub) occurs on different lysine residues as summarized in Fig. 1 and Table 2. However, the function of only few ubiquitinated lysine residues has been described. Sarcinella and colleagues observed ubiquitination of H2A.Z on K120 and K121 and linked these modifications to X-chromosome inactivation (XCI) [145]. K120, K121 and K125 monoubiquitination (K120ub1, K121ub1 and K125ub1, respectively) is mediated by RING1B [58, 145]. Active H2A.Z deubiquitination, mediated by USP10 (Ubiquitin-Specific Protease 10), is required to induce gene expression [51]. Furthermore, RNF168 ubiquitinates H2A.Z, but the exact target lysine is still unknown [146]. Surprisingly, Ku and colleagues observed in mouse embryonic stem cells (mESCs) that a fraction of H2A.Zub1 is also acetylated on its N-terminal tail: This population is more acetylated and contains a differential acetylation profile compared to the non-ubiquitinated H2A.Z [58]. It still needs to be investigated whether such a dually modified H2A.Z is an exclusive feature of mESCs or does also occur in other cell types.
The small ubiquitin-like modifier (SUMO) is another member of the ubiquitin peptide family (Fig. 1 and Table 2). SUMOylation of H2A.Z (H2A.Zsu) in yeast has been linked to DNA repair, as it is required for the recruitment of DSBs to the nuclear periphery [43]. Similarly, in HeLa cells, H2A.Z.2su by the SUMO E3 ligase PIAS4 is involved in DNA repair [147], but the exact site modified by PIAS4 has not yet been identified.
In the last years, also H2A.Z methylation was identified which, based on methylation state and the specific lysine residue to be modified, can have different transcriptional outputs (Fig. 1 and Table 2). Monomethylation of lysine 7 of H2A.Z (H2A.ZK7me1), mediated by SETD6, is associated with gene repression in mESCs [148], while dimethylation of lysine 101 (H2A.ZK101me2) is linked to gene induction in human cells [149].
Together, like the PTMs of canonical histones, there is a complicated network of activating and repressing marks also for H2A.Z. However, there are few valuable marks that will, in our view, pave the way for unraveling the molecular mechanisms of H2A.Z in gene regulation.
The H2A.Z interactome
Assuming that placement of H2A.Z and PTMs of H2A.Z are read and interpreted, it is important to first define the “H2A.Z interactome”. The working hypothesis is interacting factors will give decisive insights about the molecular mechanisms conducting either gene activation or repression.
In the past, several studies examined the H2A.Z interactome using varying methods, such as affinity purification of H2A.Z in nuclear extracts, of either recombinant H2A.Z-containing nucleosomes [150] or H2A.Z mononucleosomes prepared by micrococcal nuclease (MNase) digestion followed by (quantitative) MS and bait protein–protein interaction-sequencing (bPPI-seq) [21, 109, 130, 151, 152]. The results of these studies are summarized in Table 3. Most likely, due to the use of distinctive approaches, different H2A.Z interactors have been identified. Affinity purification of nuclear, not chromatin-bound H2A.Z allowed the identification of H2A.Z-specific chaperone/remodeling complexes (e.g., p400/Tip60, SRCAP complexes, ANP32E and MBTD1) [130]; however, many chromatin-associated proteins remained, most likely, insoluble under the mild conditions used. Consequently, immunoprecipitation of H2A.Z-containing mononucleosomes obtained via MNase digestion of chromatin (MNase-IP) led to the identification of chromatin-bound factors or even large complexes, which stably interact with intact H2A.Z nucleosomes. Nevertheless, this method is entirely restricted to chromatin regions that are accessible for MNase digestion and does not consider strongly compacted and MNase-inaccessible regions. While both of these assays depend on the protein purification quality, bPPi-seq does not. It depends on the formation of a functional eGFP fluorescent protein, when the respective N- and C-terminal parts of eGFP, fused to two proteins, come together in a physical interaction scenario. Hence, bPPi allows the identification of possible direct interactors, meanwhile being limited in identifying large complexes of interactors as the formation of eGFP is dependent on the proximity and the correct steric orientation of the associated factors.
Two of the many found H2AZ interactors (Table 3) were biochemically verified and functionally characterized. The first one is BRD2 that was identified by affinity purification of MNase-digested chromatin as an H2A.Z binder on chromatin level [21]. Further, BRD2 was proposed to be a decisive downstream mediator that couples H2A.Z to AR-induced gene activation [21]. It binds H2A.Z-containing nucleosomes via its bromodomains promoted by H4 hyperacetylation and prefers, mediated by a so far unknown mechanism, binding to the H2A.Z.1 over the H2A.Z.2 isoform [21, 144]. Strikingly, H2A.Z.2 was shown to promote and/or maintain BRD2, E2F1 and histone acetylation levels in malignant melanoma [23]. H2A.Z.2 recruits BRD2 and E2Fs, along with HAT activity, to promoters of E2F target genes in melanoma cells, facilitating expression of cell cycle genes and, ultimately, promoting cell proliferation. The other, recently identified protein is PWWP2A that was shown to tightly bind H2A.Z via a multivalent binding mode [151]. PWWP2A’s direct binding to H2A.Z is predominantly mediated by a C-terminal section of its internal protein region of no known homology or structure. Real-time-lapse microscopy imaging showed halt of PWWP2A-depleted cells in mitosis for up to 10 h. A similar effect has been observed in H2A.Z double knockout vertebrate cells [153]. Hence, PWWP2A might be the mediator of the H2A.Z-dependent cell cycle progression phenotype. Interestingly, PWWP2A, as well as H2A.Z, interacts with an MTA1-specific subcomplex of the NuRD complex that was named “M1HR” [143]. This subcomplex consists exclusively of MTA1, RBBP4/7 and HDAC2 and excludes CHD, GATAD2 and MBD proteins. Depletion of PWWP2A increased acetylation of histones in a subset of H2A.Z-containing enhancers bound by PWWP2A where it presumably regulates histone acetylation levels via M1HR recruitment.
Furthermore, H2A.Z was shown to interact with components of complexes involved in a multitude of biological processes, for example DNA damage repair (e.g., MSH2 and MSH6 of the mismatch repair complex, as well as PIR51, RAD23B and XPC), gene activation (e.g., MLL/KMT complex, PHF2, BRD8, MEAF6, ING3), gene repression (e.g., TIP27/JAZF1, BAHD1, BCORL1, MIER1 and CDYL), various transcription factors (e.g., DIDO1, MYPOP, ZFX/Y), chromatin remodeling (e.g., SMCA1 of the nucleosome remodeling factor (NuRF) complex) and proteins whose function(s) remain yet elusive like the RAI1 complex [154, 155], ZNF512B, MAGEA10, PHF20L1 and ZNF768.
Besides the further need to validate all mentioned putative H2A.Z interaction partners in independent biochemical and functional assays, it is tempting to speculate that these many interactors are one important reason why H2A.Z bears transcriptional activating as well as repressing features. At the same time, it shows that although a lot about H2A.Z’s interactome was resolved, its role in recruiting transcription-regulating complexes to their destinations on chromatin still remains a puzzle.
H2A.Z at enhancers and promoters
In the past, genomic localization of H2A.Z has been mostly reported at the TSS of genes, but more recently it is becoming increasingly clear that H2A.Z is also found at enhancers. In yeast, H2A.Z is strongly enriched at the TSS of both active and inactive genes [156]. Its occupancy at the TSS negatively correlates with gene expression: H2A.Z occupancy is more pronounced at poorly expressed genes compared to induced genes [61,62,63,64]. In contrast, genome-wide studies in human CD4+ T cells observed H2A.Z enrichment mainly at the TSS of active genes [157, 158]. Subsequently, this scenario was further refined with the observation that H2A.Z occupancy at TSS correlates with the level of transcriptional output: While Ku and colleagues observed a negative correlation [58], other studies observed a positive correlation between gene expression and H2A.Z occupancy [60, 67, 84]. Furthermore, usage of dynamic systems showed that gene induction is associated with reduced H2A.Z occupancy at TSS [54, 68, 70,71,72] as well as at enhancers [49, 54, 57, 69, 71, 72]. Similarly, in Drosophila as well as in plants, H2A.Z occupies the promoter in absence of gene expression but it decreases upon gene induction [65, 66]. Notably, H2A.Z occupancy strongly correlates with H3K4 methylation states [58, 74, 157, 159,160,161], further marking its involvement in gene poising and activation. The inverse correlation between H2A.Z occupancy and transcription is also reflected in RNAPII occupancy, [62]: H2A.Z is actively excluded from coding regions by the RNAPII-associated remodelers FACT (facilitates chromatin transcription) and spt6 [162]. Deletion of spt16, a gene encoding a FACT subunit, or of spt6, leads to H2A.Z accumulation at coding regions, a phenotype associated with increased cryptic transcription [162]. This is in line with the observation that H2A.Z-containing nucleosomes are not enriched with H3K36me3, a histone mark associated with transcriptional elongation [143, 159] and provide a mechanistic explanation to the increased H2A.Z occupancy observed at coding regions upon reduced transcription [67, 76, 77]. Additionally, H2A.Z knockdown leads to reduced RNAPII recruitment at TSSs in Saccharomyces cerevisiae and human cells [28, 76] and reduced TBP (TATA-binding protein) occupancy in Saccharomyces cerevisiae [66]. However, it plays a positive function in preventing RNAPII stalling, as its depletion increases this phenomenon [78, 79], further marking the strong relationship between H2A.Z and RNAPII. Increased cryptic transcription observed at coding regions upon H2A.Z accumulation in yeast [162] would suggest the involvement of H2A.Z in promoter usage, but it must be noted that, at least in human cells, H2A.Z is strongly enriched at facultative heterochromatin without leading to cryptic transcription [76]. A further indication that H2A.Z may be involved in promoter usage is represented by the observation that dispersed core promoters (promoters in which the TSS spreads over hundreds of nucleotides) show a stronger H2A.Z enrichment compared to focused core promoters (promoters in which the TSS occurs in a narrow genomic window of few nucleotides [164]). This dispersion in promoter usage may be the consequence of a different stability of H2A.Z-containing nucleosomes [163], an aspect that will be further discussed in the next section. To note, not only H2A.Z enrichment but also the proximity of H2A.Z-containing nucleosomes to the TSSs influences gene expression [80].
As previously marked, the first genome-wide studies of H2A.Z described its strong enrichment near the TSSs in yeast, Drosophila and human [62, 81, 82, 156,157,158, 164, 165]. However, in subsequent studies it became increasingly clear that H2A.Z is not exclusively found at TSSs but it can also be detected at other regulatory elements such as enhancers and insulators in several different species, though to a lesser extent [58, 83,84,85,86, 143, 166]. For example, H2A.Z is enriched at a p53-binding site located approximately 2.2 kb upstream of the TSS of the human p21 gene [49], further suggesting H2A.Z as an enhancer mark. In line with that, H2A.Z is enriched at an ER binding site located just upstream of the TSS of the human TFF1 gene, where it localizes in an estrogen-dependent fashion [50]. Genome-wide studies observed a stronger binding of ERα to its genomic sites associated with H2A.Z compared to those sites depleted of H2A.Z [87]. Additionally, at ER sites enriched for H2A.Z, this histone variant seems to be required for the recruitment of RNAPII, for the induction of enhancer RNAs (eRNAs) and finally for the recruitment of RAD21 that is involved in chromatin looping [87]. Similarly, H2A.Z is enriched at AR responsive enhancer sites such as the ones of the prostate specific antigen (PSA) and kallikrein-like 2 (KLK2) genes [51, 52] as well as at the enhancer site of MyoD [56]. Recently, we have extended these observations also to developmental signaling pathways: H2A.Z localizes at Notch responsive enhancers where it plays a negative role with regards to the expression of Notch target genes [57]. Interestingly, it has been observed that pioneering factors-bound enhancers exist with two different H2A.Z distributions: a) H2A.Z localizes at the center of Ets1- and Oct4-bound enhancer sites, whereas b) it is enriched at nucleosomes flanking forkhead box protein A2 (FoxA2) or C/EBPα-bound enhancer sites [167]. Table 4 provides the complete list of enhancers, which are bound by signal-specific TFs and have been linked to H2A.Z-mediated regulation.
H2A.Z and the nucleosome-free regions (NFRs)
The TSS of active genes was previously known as a nucleosome-free region (NFR) or nucleosome-depleted region (NDR). Interestingly, this has been challenged by Jin and Felsenfeld in 2007 [168]. In this study, the authors observed that nucleosomes containing both H2A.Z and H3.3 histone variants are highly unstable and found at regulatory regions such as promoters and enhancers [168]. When nucleosomes are isolated at low salt concentrations, it is possible to observe occupancy of H3.3/H2A.Z double-containing nucleosomes at NFRs, an occupancy that is lost when high salt concentrations are used [83], further suggesting that NFRs might indeed be not nucleosome-free [169]. Such unstable nucleosomes are also enriched at NFRs in Drosophila and yeast [170, 171]; however, the reason why the H3.3/H2A.Z double-containing nucleosomes are highly unstable remains unclear. While H2A.Z and the canonical H2A differ significantly in their L1 loop [172], cell-free studies observed that nucleosomes composed of both H2A and H2A.Z (defined as heterotypic, [173]) are more stable than H2A.Z only-containing nucleosomes (defined as homotypic), which are less stable than H2A homotypic nucleosomes [174, 175]. However, a highly unstable heterotypic nucleosome occupies the TSS in the G1 phase of the cell cycle [176, 177]. Furthermore, it should be noted that cell-free studies observed that H3.3 does not alter the stability of H2A.Z both homo- and heterotypic nucleosomes [175, 178]. Based on these data, one could think that neither the incorporation of H3.3 into an H2A.Z-containing nucleosome or the presence of an H2A.Z/H2A heterotypic nucleosome could be responsible for the nucleosome instability observed at NFRs; however, the different approaches used, in vitro (cells) versus cell-free assays, may lead to discrepancies and actually the cell-free approaches may lead to underestimate the nucleosome instability as consequence of the lack of PTMs and/or interactors that may contribute to the regulation of nucleosome stability in a physiological context. It is possible that such nucleosomes would be H2A.Z homotypic. However, at least in Drosophila, homotypic H2A.Z nucleosomes are not enriched at the TSS [179], excluding this possibility. In contrast, two more studies observed increased stability of the H2A.Z-containing nucleosomes compared to the H2A-containing nucleosomes in cell-free assays [180, 181]. It seems that this increased stability can be counteracted by histone acetylation, including H2A.Zac [181]. In more detail, it appears that H2A.Zac is the key modification that destabilizes the nucleosome and that acetylation of other histone proteins alone is not sufficient to achieve this destabilization; even more, heterotypic nucleosomes are destabilized by H2A.Zac [182]. While the previous studies focused on H2A.Z.1, another study found structural differences in the L1 loop when comparing this isoform with H2A.Z.2.1 [26]. Furthermore, H2A.Z.2.2-containing nucleosomes seem to be less stable than the H2A.Z.2.1-containing ones [27]. As consequence, at least in vertebrates (or in primates in the case of H2A.Z.2.2), the high instability of the nucleosomes located at NFRs can still be due to the occupancy of the different H2A.Z isoforms that can organize different homo- and heterotypic nucleosomes (also in combination with H3.3) that are regulated by different combinations of PTMs. However, this remains a hypothesis that needs to be tested to identify the mechanism(s) how the nucleosomes occupying the “NFR” become unstable.
The role of H2A.Z in nucleosome positioning
The role of H2A.Z in nucleosome positioning was first described in yeast [81, 183]. Subsequently, Gévry and colleagues extended this observation to mammals when focusing on the promoter of the trefoil factor 1 (TFF1) gene (Fig. 3, [50]). The positioning of the nucleosomes surrounding the ERα-binding element (ERE) at the TFF1 gene promoter is stabilized upon activation of the pathway with estrogen; however, this effect is abolished by depletion of H2A.Z or p400 [50]. Cell-free studies also confirmed the role of H2A.Z in nucleosome positioning; however, the co-occurrence of both H2A.Z and H3.3 variants apparently does not play a major role when compared to the presence of H2A.Z only [178], suggesting that H2A.Z plays an important role in nucleosome positioning. This is also reflected in both the high instability of nucleosomes at the NFR and in the higher H2A.Z occupancy observed at dispersed promoters compared to the focused ones [163]. Additionally, there is a positive correlation between H2A.Z proximity to the TSS and gene expression level with genes highly expressed showing higher TSS proximal H2A.Z enrichment [80]. Together, this strongly supports a role for H2A.Z in nucleosome positioning. In future, it will be interesting to determine mechanistically whether PTMs and/or H2A.Z interactors play a role in the H2A.Z-mediated nucleosome positioning that in turn contributes to the enormous plasticity at promoters.

Regulation of H2A.Z and its involvement in transcription. Two examples are used to explain the function of H2A.Z in gene regulation: a the case of the androgen system focusing on the PSA locus and b the case of the estrogen system focusing on the TFF1 locus. a In a repressed or poised (OFF) state, the H2A.Z-specific loading machineries are recruited to the PSA locus via not well-defined mechanisms that may involve TFs and/or histone modifications. In this scenario, H2A.Z is deposited by SRCAP and/or p400/Tip60 complexes [119, 122] and the deacetylation and ubiquitination machineries are probably recruited via interactions with DNA binding proteins and/or posttranslationally modified histones (not depicted in the figure). The deacetylation machinery removes the acetylation mark from H2A.Z, which is instead ubiquitinated on its C-terminus by E3 ubiquitin ligases (we speculate that RING1B is involved in the AR signaling cascade [58, 145]). Upon gene activation (ON), deubiquitination (for example, USP10 [51]) and loading/acetylation/deubiquitination machineries are recruited/stabilized via interactions with the Androgen Receptor (AR) that binds to its cognate sequences (androgen receptor-binding element, ARE) and/or histone modifications [119, 122]. This leads to H2A.Z deubiquitination [52] and acetylation [59, 122] finally leading to gene activation which is associated with reduced H2A.Z occupancy [51, 52]. b In a repressed (OFF) state, FoxA1 binds to the distal FoxA1-binding site (FBS) of the TFF1 locus where it recruits the p400/Tip60 complex that supports loading of H2A.Z. In this state, H2A.Z is poorly enriched at the TFF1 promoter and, as consequence, nucleosome occupancy is poorly defined [50]. Upon gene induction, the estrogen receptor α (ERα) binds to its cognate sequence (estrogen receptor-binding element, ERE) where it recruits the p400/Tip60 complex leading to loading of H2A.Z at the promoter and as consequence increased nucleosome positioning and finally to gene activation. At the same time, H2A.Z enrichment at the FBS is reduced [50]
H2A.Z and DNA methylation
Studies in Arabidopsis thaliana have shown that H2A.Z is excluded from sites enriched in DNA methylation and H2A.Z-occupied sites display low levels of DNA methylation [184]. This anti-correlation between H2A.Z occupancy and DNA methylation is recapitulated in other organisms [88, 185,186,187,188] and involves acetylation of H2A.Z [60], which is known to be the predominant mechanism in gene activation. Gene reactivation observed upon loss of DNA methylation, obtained via pharmacological inhibition or knockdown of DNA methyltransferases (DNMTs), is associated with a gain in H2A.Z occupancy [184, 189]. Similarly, increased H2A.Z occupancy, obtained via LoF of ANP32E that removes H2A.Z from chromatin [130], leads to reduced DNA methylation [188], while the opposite is observed upon gain-of-function (GoF) of ANP32E, LoF of the H2A.Z loading machinery or depletion of H2A.Z itself [88, 184, 188]. Notably, this mechanism may involve also another histone variant: macroH2A. In fact, it reversely correlates with H2A.Z occupancy but positively correlates with DNA methylation and gene silencing [190]. However, in contrast to the study of Yang and colleagues [189], Barzily-Rokni and colleagues do not observe gain in H2A.Z occupancy upon pharmacological inhibition of DNMTs alone or combined with macroH2A depletion even if gene expression is re-established [190]. One possibility to explain this discrepancy is represented by the different concentration of 5-azacytidine used in these studies [189, 190].
Our current model for H2A.Z in gene regulation
The AR and ER systems represent good examples to explain the function of H2A.Z in gene transcription (Fig. 3). In the AR system, the PSA gene can be considered as the prototype of this pathway (Fig. 3a): In the absence of androgen (OFF state), H2A.Z is loaded by the SRCAP [119] and/or p400/Tip60 [122] complexes. In this repressed configuration, H2A.Z is monoubiquitinated at both enhancers and promoters [51] potentially by RING1B [58, 145]. Upon androgen stimulation (ON), H2A.Z is deubiquitinated by USP10 and its occupancy decreases [51, 52]. Of note, H2A.Zac correlates with AR induction [59, 122] and similarly, the occupancy of the p400/Tip60 complex increases upon AR induction [122]. The recruitment of the p400/Tip60 complex is mediated by its MRG15 subunit which recognizes H3K4 methylation states [122] while SRCAP has been shown to interact with AR [119]. In the case of the estrogen signaling cascade, we focus on the case of the TFF1 locus (Fig. 3b): In the OFF state, forkhead box protein A1 (FoxA1) binds to a distal enhancer (FoxA1-binding site, FBS) of the TFF1 locus where it recruits the p400/Tip60 complex supporting H2A.Z loading [50]. Lack of H2A.Z at the TFF1 promoter, leads to a poorly defined nucleosome occupancy in the repressed/poised state (OFF, [50]). Upon activation of the pathway, the p400/Tip60 complex is recruited at the TFF1 promoter by ERα which binds to its cognate sequences (ERE). At the TFF1 promoter, the p400/Tip60 complex loads H2A.Z leading to a better-defined nucleosome positioning. At the same time, H2A.Z occupancy decreases at the FoxA1-bound distal enhancer [50].
From the above, some general rules for H2A.Z in gene regulation can be postulated: At genes that are poised/repressed (OFF), repressive marks of H2A.Z are found and as consequence its LoF leads to upregulation. At genes that are active, activating PTMs of H2A.Z, such as H2A.Zac, are found and as consequence H2A.Z LoF leads to downregulation.
In a repressed (OFF) or poised state, the H2A.Z deposition machinery is recruited by TFs and/or histone modifications to chromatin. This recruitment can be transient but still allows an exchange of H2A with H2A.Z. In the OFF state, H2A.Z is deacetylated by the deacetylation machinery and ubiquitinated on its C-terminus by RING1B. Upon gene activation (ON), additional TFs and/or histone modifications lead to the recruitment of the loading/acetylation/deubiquitination machinery. This triggers H2A.Z acetylation and deubiquitination, finally leading to transcriptional activation.
Still a number of open questions remain there. For example, the specificity of the p400/Tip60 and SRCAP complexes awaits to be determined in higher eukaryotes. Furthermore, there may be several p400-containing complexes that may act at different stages of gene transcription, the one without the acetyltransferase function and the other with Tip60 present. It is possible that these two complexes act in a stepwise fashion, one after the other. It is also possible that the different complexes are preferentially found at either promoters or enhancer elements (as described above). In addition, the H2A.Z removing factor ANP32E is also found in complex with Ep400 and Tip60 [130] suggesting that deposition, acetylation and removal of H2A.Z could be coupled in a stepwise manner. Chromatin-IP (ChIP) experiments with the relevant antibodies in dynamically regulated systems will answer such questions.
H2A.Z in diseases
The histone variant H2A.Z, its PTMs and interacting proteins have been linked to several diseases, most notably cancer. H2A.Z expression is upregulated in metastatic melanoma [23], breast [191, 192], prostate [52, 59, 193], colorectal [194], liver [195, 196], bladder [74] and lung [126] cancer. Similarly, H2A.Z protein levels are elevated during cardiac hypertrophy [197] but decreased in diseased vascular tissues [161].
In prostate cancer, H2A.Zac has a pro-oncogenic role. It promotes activation of oncogenes and repression of tumor suppressor genes [60]. Regarding genomic occupancy, H2A.Zac increases at the TSS of oncogenes, while it decreases at the TSS of tumor suppressor genes [59, 60] and its genomic redistribution in prostate cancer leads to the activation of AR-associated neo-enhancers [59]. It must be marked that in metastatic melanoma both H2A.Z isoforms, H2A.Z.1 and H2A.Z.2, are upregulated; however, only the depletion of the H2A.Z.2 but not H2A.Z.1 leads to reduced proliferation [22]. In contrast to that, H2AFZ but not H2AFV is overexpressed in liver cancer and its knockdown results in reduced proliferation and inhibits the cancer cells’ metastatic potential [196]. These data support the notion that the different H2A.Z isoforms, which differ in only three amino acids, have distinct roles in the development of different tumor types.
While the data discussed so far highlight the upregulation of H2A.Z in cancer, additional mechanisms of H2A.Z deregulation may involve aberrant expression of the machineries involved in H2A.Z modifications and/or chromatin deposition/removal. For example, the methyltransferase SMYD3, which is upregulated in several cancer types, promotes proliferation of breast cancer cells and tumorigenesis [149]. This is achieved because SMYD3 supports H2A.Z methylation, which is required to activate the expression of the cyclin A1-encoding (CCNA1) gene [149]. Similarly, Tip60 is downregulated in acute myeloid leukemia (AML) samples [198] and present with mono-allelic loss in lymphomas, head-and-neck and mammary carcinoma [199]. In line with that, Tip60 has a tumor suppressor function in colon [200]. However, whether decreased Tip60 expression has any impact on H2A.Z deposition or acetylation is unknown.
Finally, the enzymes involved in the H2A.Z deposition can also be useful therapeutic targets, for example knockdown of SRCAP reduces cell proliferation of prostate cancer cells [119].
The CRISPR/Cas9 system as a tool to deplete H2A.Z
Histone variants emerged as essential regulators of embryonic development as their knockout is frequently lethal. Thus, obtaining even a successful knockdown in tissue culture cells is also a daunting task. We have recently used the CRISPR/Cas9 technology [201] to successfully (and also surprisingly for us) deplete the histone variant H2A.Z in a mouse T cell line [57]. Usually, the CRISPR/Cas9 technology is targeted to the 5′-end of open reading frames (ORF) of coding genes introducing mutations that lead to the generation of premature STOP codons (Fig. 4, [201]). We used this CRISPR/Cas9 system to target the 5′-UTR (untranslated) region of both H2AFZ and H2AFV genes [57]. In the literature, there are few examples of targeting UTRs: The 5′-UTR of the Bcl2 gene in human cells has been targeted [202] and the 3′-UTR of the Tyr gene was targeted to study albinism in rabbits [203]. In another study, the 3′UTR of different chemokine genes was modified [204]. The authors observed differential effects when targeting the 3′-UTRs: while CXCL3, CXCL10, CXCL11, CCL3, CCL4 and CCL7 are upregulated, CXCL1, CXCL6 and CXCL8 are downregulated [204], suggesting that 3′UTRs may contain negative-regulating elements and that the CRISPR/Cas9 system can also be used to increase mRNAs abundance. Similarly, CRISPR/Cas9 targeting of the 3′UTR of the PD-L1 gene leads to its overexpression [205]. Furthermore, the CRISPR/Cas9 system was used to edit the 3′UTR of Cebpg gene to prevent the mTOR (mammalian target of rapamycin)-mediated alternative polyadenylation [206]. Previously, zinc-finger nucleases (ZNFs) [207] were used to target 3′-UTRs in human cells [208]. Of note, most recently, the CRISPR/Cas9 technology was also used to deplete the histone variant H3.3B. However, in this case the authors targeted the coding sequence of the gene [209].

Summary of the different possible strategies that can be employed to deplete a histone gene via the CRISPR/Cas9 technology. A protein-coding gene can be targeted on its 5′-UTR leading to mRNA destabilization or preventing its translation. Alternatively, the ORF can be targeted, leading to the formation of a premature STOP codon. Finally, targeting of the 3′-UTR can lead to mRNA destabilization or to a translational block but it can also increase the mRNA stability or the translation efficiency. UTR untranslated region, ORF open reading frame
In summary (Fig. 4), the CRISPR/Cas9 technology can be used to target the 5′UTR or 3′UTR leading to deregulation of the target by promoting mRNA destabilization or translational block. However, targeting the 3′UTR can also be used to upregulate gene expression by increasing mRNA stability or translation. In our view, it is tempting to speculate that other histone variants or even canonical histones could also be targeted (or tagged/mutated/replaced) in this way as well as, the loading and removal machineries involved in the chromatin regulation of histone variants.
Conclusions and perspective
We propose that the plastic behavior of H2A.Z at regulatory regions is ideally suited to buffer phenotypic noise by modulating transcriptional efficiency, both repressive and activating. Mechanistically, high levels of unmodified H2A.Z and ubiquitination of H2A.Z may serve as a roadblock for transcription; acetylation of H2A.Z and subsequent removal of H2A.Z may enhance transcription rate and/or help recruiting RNAPII or other activating complexes.
In the future, new tools such as highly specific antibodies against single-modified H2A.Z residues are needed to characterize the function of H2A.Z in development and in pathological settings. Since the current antibodies against H2A.Z (unmodified and pan-acetyl-H2A.Z) are exquisitely specific, there is a high chance that more specific reagents will have a major impact on chromatin and transcription research.
Abbreviations
- AhR:
-
aryl hydrocarbon receptor
- AML:
-
acute myeloid leukemia
- AP-1:
-
activator protein-1
- AR:
-
androgen receptor
- ARE:
-
androgen receptor-binding element
- BAH:
-
domain bromo-adjacent homology domain
- bPPI-seq:
-
bait protein–protein interaction-sequencing
- BPTF:
-
bromodomain and PHD finger-containing transcription factor
- BRD2:
-
bromodomain-containing protein 2
- CCNA1 :
-
Cyclin A1
- ChIP:
-
Chromatin-IP
- DNMTs:
-
DNA methyltransferases
- DSBs:
-
double-strand breaks
- ELM2:
-
Egl-27 and MTA1 homology 2 domain
- EMT:
-
epithelial-to-mesenchymal transition
- ER:
-
estrogen receptor
- ERα:
-
estrogen receptor alpha
- ERE:
-
estrogen receptor-binding element
- eRNA:
-
enhancer RNA
- FACT:
-
facilitates chromatin transcription
- FBS:
-
FoxA1-binding site
- FoxA2:
-
forkhead box protein A111
- FoxA2:
-
forkhead box protein A2
- FRAP:
-
fluorescence recovery after photobleaching
- GoF:
-
gain of function
- GR:
-
glucocorticoid receptor
- GTF:
-
general transcription factor
- H2A.Zac:
-
H2A.Z acetylation
- H2A.Zsu:
-
H2A.Z SUMOylation
- H2A.Zub:
-
H2A.Z ubiquitination
- H3K4me1:
-
H3 lysine 4 monomethylation
- H3K4me3:
-
H3 lysine 4 trimethylation
- H3K14ac:
-
H3 lysine 14 acetylation
- H3K27ac:
-
H3 lysine 27 acetylation
- H3K122suc:
-
H3 lysine 122 succinylation
- HAT:
-
histone acetyltransferase
- HDAC:
-
histone deacetylase
- HR:
-
homologous recombination
- iFRAP:
-
inverse fluorescence recovery after photobleaching
- ISGF3:
-
interferon-stimulated gene factor complex 3
- KLK2:
-
kallikrein-like 2
- KMT:
-
lysine methyltransferase
- LoF:
-
loss of function
- MAGE:
-
melanoma antigen-encoding gene
- mESCs:
-
mouse embryonic stem cells
- MNase:
-
Micrococcal nuclease
- MNase-IP:
-
immunoprecipitation of H2A.Z-containing mononucleosomes obtained via MNase digestion of chromatin
- MS:
-
mass spectrometry
- mTOR:
-
mammalian target of rapamycin
- n.d.:
-
not determined
- ND:
-
not defined
- NDR:
-
nucleosome-depleted region
- NFR:
-
nucleosome-free region
- NuRD:
-
nucleosome remodeling and deacetylase
- NuRF:
-
nucleosome remodeling factor
- ORF:
-
open reading frames
- PfGCN5:
-
Plasmodium falciparum GCN5
- PHD:
-
plant homeodomain
- PIC:
-
pre-initiation complex
- PSA:
-
prostate-specific antigen
- PTMs:
-
posttranslational modifications
- RARγ:
-
retinoic acid receptor γ
- RNAPII:
-
RNA polymerase II
- SANT domain:
-
Swi3 Ada2 N-Cor and TFIIIB domain
- SAS:
-
something about silencing
- SRCAP:
-
SNF2-related CREBBP activator protein
- SUMO:
-
small ubiquitin-like modifier
- TBP:
-
TATA-binding protein
- TF:
-
transcription factor
- TFBS:
-
transcription factor binding site
- TFF1:
-
trefoil factor 1
- TSA:
-
trichostatin-A
- TSS:
-
transcription start site
- USP10:
-
ubiquitin-specific protease 10
- UTR:
-
untranslated
- UVA:
-
ultraviolet A
- XCI:
-
X-chromosome inactivation
- ZNFs:
-
zinc-finger nucleases
References
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–60. https://doi.org/10.1038/38444.
Hergeth SP, Schneider R. The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015;16(11):1439–53. https://doi.org/10.15252/embr.201540749.
Hota SK, Bruneau BG. ATP-dependent chromatin remodeling during mammalian development. Development. 2016;143(16):2882–97. https://doi.org/10.1242/dev.128892.8.
Tropberger P, Schneider R. Going global: novel histone modifications in the globular domain of H3. Epigenetics. 2010;5(2):112–7.
Kebede AF, Schneider R, Daujat S. Novel types and sites of histone modifications emerge as players in the transcriptional regulation contest. FEBS J. 2015;282(9):1658–74. https://doi.org/10.1111/febs.13047.
Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications—writers that read. EMBO Rep. 2015;16(11):1467–81. https://doi.org/10.15252/embr.201540945.
Bonisch C, Hake SB. Histone H2A variants in nucleosomes and chromatin: more or less stable? Nucleic Acids Res. 2012;40(21):10719–41. https://doi.org/10.1093/nar/gks865.
Moosmann A, Campsteijn C, Jansen PW, Nasrallah C, Raasholm M, Stunnenberg HG, et al. Histone variant innovation in a rapidly evolving chordate lineage. BMC Evol Biol. 2011;11:208. https://doi.org/10.1186/1471-2148-11-208.
Siegel TN, Hekstra DR, Kemp LE, Figueiredo LM, Lowell JE, Fenyo D, et al. Four histone variants mark the boundaries of polycistronic transcription units in Trypanosoma brucei. Genes Dev. 2009;23(9):1063–76. https://doi.org/10.1101/gad.1790409.
West MH, Bonner WM. Histone 2A, a heteromorphous family of eight protein species. Biochemistry. 1980;19(14):3238–45.
Allis CD, Richman R, Gorovsky MA, Ziegler YS, Touchstone B, Bradley WA, et al. hv1 is an evolutionarily conserved H2A variant that is preferentially associated with active genes. J Biol Chem. 1986;261(4):1941–8.
van Daal A, White EM, Gorovsky MA, Elgin SC. Drosophila has a single copy of the gene encoding a highly conserved histone H2A variant of the H2AF/Z type. Nucl Acids Res. 1988;16(15):7487–97.
van Daal A, Elgin SC. A histone variant, H2AvD, is essential in Drosophila melanogaster. Mol Biol Cell. 1992;3(6):593–602. https://doi.org/10.1091/mbc.3.6.593.
Hatch CL, Bonner WM. The human histone H2A.Z gene sequence and regulation. J Biol Chem. 1990;265(25):15211–8.
Faast R, Thonglairoam V, Schulz TC, Beall J, Wells JR, Taylor H, et al. Histone variant H2A.Z is required for early mammalian development. Curr Biol. 2001;11(15):1183–7.
Jackson JD, Gorovsky MA. Histone H2A.Z has a conserved function that is distinct from that of the major H2A sequence variants. Nucleic Acids Res. 2000;28(19):3811–6.
Coon JJ, Ueberheide B, Syka JE, Dryhurst DD, Ausio J, Shabanowitz J, et al. Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc Natl Acad Sci USA. 2005;102(27):9463–8. https://doi.org/10.1073/pnas.0503189102.
Talbert PB, Ahmad K, Almouzni G, Ausio J, Berger F, Bhalla PL, et al. A unified phylogeny-based nomenclature for histone variants. Epigenet Chromatin. 2012;5:7. https://doi.org/10.1186/1756-8935-5-7.
Eirin-Lopez JM, Gonzalez-Romero R, Dryhurst D, Ishibashi T, Ausio J. The evolutionary differentiation of two histone H2A.Z variants in chordates (H2A.Z-1 and H2A.Z-2) is mediated by a stepwise mutation process that affects three amino acid residues. BMC Evol Biol. 2009;9:31. https://doi.org/10.1186/1471-2148-9-31.
Dryhurst D, Ishibashi T, Rose KL, Eirin-Lopez JM, McDonald D, Silva-Moreno B, et al. Characterization of the histone H2A.Z-1 and H2A.Z-2 isoforms in vertebrates. BMC Biol. 2009;7:86. https://doi.org/10.1186/1741-7007-7-86.
Draker R, Ng MK, Sarcinella E, Ignatchenko V, Kislinger T, Cheung P. CA combination of H2A.Z and H4 acetylation recruits Brd2 to chromatin during transcriptional activation. PLoS Genet. 2012;8(11):e1003047. https://doi.org/10.1371/journal.pgen.1003047.
Matsuda R, Hori T, Kitamura H, Takeuchi K, Fukagawa T, Harata M. Identification and characterization of the two isoforms of the vertebrate H2A.Z histone variant. Nucleic Acids Res. 2010;38(13):4263–73. https://doi.org/10.1093/nar/gkq171.
Vardabasso C, Gaspar-Maia A, Hasson D, Punzeler S, Valle-Garcia D, Straub T, et al. Histone variant H2A.Z.2 mediates proliferation and drug sensitivity of malignant melanoma. Mol Cell. 2015;59(1):75–88. https://doi.org/10.1016/j.molcel.2015.05.009.
Dunn CJ, Sarkar P, Bailey ER, Farris S, Zhao M, Ward JM, et al. Histone hypervariants H2A.Z.1 and H2A.Z.2 play independent and context-specific roles in neuronal activity-induced transcription of Arc/Arg3.1 and other immediate early genes. Neuro. 2017. https://doi.org/10.1523/eneuro.0040-17.2017.
Nishibuchi I, Suzuki H, Kinomura A, Sun J, Liu NA, Horikoshi Y, et al. Reorganization of damaged chromatin by the exchange of histone variant H2A.Z-2. Int J Radiat Oncol Biol Phys. 2014;89(4):736–44. https://doi.org/10.1016/j.ijrobp.2014.03.031.
Horikoshi N, Sato K, Shimada K, Arimura Y, Osakabe A, Tachiwana H, et al. Structural polymorphism in the L1 loop regions of human H2A.Z.1 and H2A.Z.2. Acta Crystallogr Sect D: Biol Crystallogr. 2013;69(Pt 12):2431–9. https://doi.org/10.1107/S090744491302252X.
Bonisch C, Schneider K, Punzeler S, Wiedemann SM, Bielmeier C, Bocola M, et al. H2A.Z.2.2 is an alternatively spliced histone H2A.Z variant that causes severe nucleosome destabilization. Nucleic Acids Res. 2012;40(13):5951–64. https://doi.org/10.1093/nar/gks267.
Adam M, Robert F, Larochelle M, Gaudreau L. H2A.Z is required for global chromatin integrity and for recruitment of RNA polymerase II under specific conditions. Mol Cell Biol. 2001;21(18):6270–9.
Zovkic IB, Paulukaitis BS, Day JJ, Etikala DM, Sweatt JD. Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature. 2014;515(7528):582–6. https://doi.org/10.1038/nature13707.
Narkaj K, Stefanelli G, Wahdan M, Azam AB, Ramzan F, Steininger CFD Jr, et al. Blocking H2A.Z Incorporation via Tip60 inhibition promotes systems consolidation of fear memory in mice. Neuro. 2018. https://doi.org/10.1523/ENEURO.0378-18.2018.
Shen T, Ji F, Wang Y, Lei X, Zhang D, Jiao J. Brain-specific deletion of histone variant H2Az results in cortical neurogenesis defects and neurodevelopmental disorder. Nuclic Acids Res. 2008;46(5):2290–307. https://doi.org/10.1093/nar/gkx1295.
Stefanelli G, Azam AB, Walters BJ, Brimble MA, Gettens CP, Bouchard-Cannon P, et al. Learning and age-related changes in genome-wide H2A.Z binding in the mouse hippocampus. Cell Rep. 2018;22(5):1124–31. https://doi.org/10.1016/j.celrep.2018.01.020.
Domaschenz R, Kurscheid S, Nekrasov M, Han S, Tremethick DJ. The histone variant H2AZ is a master regulator of the Epithelial–Mesenchymal transition. Cell Rep. 2017;21(4):943–52. https://doi.org/10.1016/j.celrep.2017.09.086.
Rangasamy D, Berven L, Ridgway P, Tremethick DJ. Pericentric heterochromatin becomes enriched with H2A.Z during early mammalian development. EMBO J. 2003;22(7):1599–607. https://doi.org/10.1093/emboj/cdg160.
Fan JY, Rangasamy D, Luger K, Tremethick DJ. H2AZ alters the nucleosome surface to promote HP1alpha-mediated chromatin fiber folding. Mol Cell. 2004;16(4):655–61. https://doi.org/10.1016/j.molcel.2004.10.023.
Rangasamy D, Greaves I, Tremethick DJ. RNA interference demonstrates a novel role for H2AZ in chromosome segregation. Nat Struct Mol Biol. 2004;11(7):650–5. https://doi.org/10.1038/nsmb786.
Greaves IK, Rangasamy D, Ridgway P, Tremethick DJ. H2AZ contributes to the unique 3D structure of the centromere. Proc Natl Acad Sci USA. 2007;104(2):525–30. https://doi.org/10.1073/pnas.0607870104.
Ryan DP, Tremethick DJ. The interplay between H2A.Z and H3K9 methylation in regulating HP1alpha binding to linker histone-containing chromatin. Nucleic Acids Res. 2018;46:9353–66. https://doi.org/10.1093/nar/gky632.
Meneghini MD, Wu M, Madhani HD. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell. 2003;112(5):725–36.
Babiarz JE, Halley JE, Rine J. Telomeric heterochromatin boundaries require NuA4-dependent acetylation of histone variant H2A.Z in Saccharomyces cerevisiae. Genes Dev. 2006;20(6):700–10. https://doi.org/10.1101/gad.1386306.
Shia WJ, Li B, Workman JL. SAS-mediated acetylation of histone H4 Lys 16 is required for H2A.Z incorporation at subtelomeric regions in Saccharomyces cerevisiae. Genes Dev. 2006;20(18):2507–12. https://doi.org/10.1101/gad.1439206.
Zhou BO, Wang SS, Xu LX, Meng FL, Xuan YJ, Duan YM, et al. SWR1 complex poises heterochromatin boundaries for antisilencing activity propagation. Mol Cell Biol. 2010;30(10):2391–400. https://doi.org/10.1128/mcb.01106-09.
Kalocsay M, Hiller NJ, Jentsch S. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol Cell. 2009;33(3):335–43. https://doi.org/10.1016/j.molcel.2009.01.016.
Xu Y, Ayrapetov MK, Xu C, Gursoy-Yuzugullu O, Hu Y, Price BD. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol Cell. 2012;48(5):723–33. https://doi.org/10.1016/j.molcel.2012.09.026.
Yu Y, Deng Y, Reed SH, Millar CB, Waters R. Histone variant Htz1 promotes histone H3 acetylation to enhance nucleotide excision repair in Htz1 nucleosomes. Nucleic Acids Res. 2013;41(19):9006–19. https://doi.org/10.1093/nar/gkt688.
Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, et al. SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell. 2014;55(4):626–39. https://doi.org/10.1016/j.molcel.2014.06.027.
Gursoy-Yuzugullu O, Ayrapetov MK, Price BD. Histone chaperone Anp32e removes H2AZ from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair. Proc Natl Acad Sci USA. 2015;112(24):7507–12. https://doi.org/10.1073/pnas.1504868112.
Rona G, Roberti D, Yin Y, Pagan JK, Homer H, Sassani E, et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. Elife. 2018;7:38771. https://doi.org/10.7554/eLife.38771.
Gevry N, Chan HM, Laflamme L, Livingston DM, Gaudreau L. p21 transcription is regulated by differential localization of histone H2AZ. Genes Dev. 2007;21(15):1869–81. https://doi.org/10.1101/gad.1545707.
Gevry N, Hardy S, Jacques PE, Laflamme L, Svotelis A, Robert F, et al. Histone H2A.Z is essential for estrogen receptor signaling. Genes Dev. 2009;23(13):1522–33. https://doi.org/10.1101/gad.1787109.
Draker R, Sarcinella E, Cheung P. USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res. 2011;39(9):3529–42. https://doi.org/10.1093/nar/gkq1352.
Dryhurst D, McMullen B, Fazli L, Rennie PS, Ausio J. Histone H2A.Z prepares the prostate specific antigen (PSA) gene for androgen receptor-mediated transcription and is upregulated in a model of prostate cancer progression. Cancer Lett. 2012;315(1):38–47. https://doi.org/10.1016/j.canlet.2011.10.003.
Bellucci L, Dalvai M, Kocanova S, Moutahir F, Bystricky K. Activation of p21 by HDAC inhibitors requires acetylation of H2A.Z. PLoS ONE. 2013;8(1):e54102. https://doi.org/10.1371/journal.pone.0054102.
Dalvai M, Fleury L, Bellucci L, Kocanova S, Bystricky K. TIP48/Reptin and H2A.Z requirement for initiating chromatin remodeling in estrogen-activated transcription. PLoS Genet. 2013;9(4):e1003387. https://doi.org/10.1371/journal.pgen.1003387.
Dalvai M, Bellucci L, Fleury L, Lavigne AC, Moutahir F, Bystricky K. H2A.Z-dependent crosstalk between enhancer and promoter regulates cyclin D1 expression. Oncogene. 2013;32(36):4243–51. https://doi.org/10.1038/onc.2012.442.
Law C, Cheung P. Expression of non-acetylatable H2A.Z in myoblast cells blocks myoblast differentiation through disruption of MyoD expression. J Biol Chem. 2015;290(21):13234–49. https://doi.org/10.1074/jbc.M114.595462.
Giaimo BD, Ferrante F, Vallejo DM, Hein K, Gutierrez-Perez I, Nist A, et al. Histone variant H2A.Z deposition and acetylation directs the canonical Notch signaling response. Nucleic Acids Res. 2018;46(16):8197–215. https://doi.org/10.1093/nar/gky551.
Ku M, Jaffe JD, Koche RP, Rheinbay E, Endoh M, Koseki H, et al. H2A.Z landscapes and dual modifications in pluripotent and multipotent stem cells underlie complex genome regulatory functions. Genome Biol. 2012;13(10):R85. https://doi.org/10.1186/gb-2012-13-10-r85.
Valdes-Mora F, Gould CM, Colino-Sanguino Y, Qu W, Song JZ, Taylor KM, et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat Commun. 2017;8(1):1346. https://doi.org/10.1038/s41467-017-01393-8.
Valdes-Mora F, Song JZ, Statham AL, Strbenac D, Robinson MD, Nair SS, et al. Acetylation of H2AZ is a key epigenetic modification associated with gene deregulation and epigenetic remodeling in cancer. Genome Res. 2012;22(2):307–21. https://doi.org/10.1101/gr.118919.110.
Santisteban MS, Kalashnikova T, Smith MM. Histone H2A.Z regulates transcription and is partially redundant with nucleosome remodeling complexes. Cell. 2000;103(3):411–22.
Li B, Pattenden SG, Lee D, Gutierrez J, Chen J, Seidel C, et al. Preferential occupancy of histone variant H2AZ at inactive promoters influences local histone modifications and chromatin remodeling. Proc Natl Acad Sci USA. 2005;102(51):18385–90. https://doi.org/10.1073/pnas.0507975102.
Buchanan L, Durand-Dubief M, Roguev A, Sakalar C, Wilhelm B, Stralfors A, et al. The Schizosaccharomyces pombe JmjC-protein, Msc1, prevents H2A.Z localization in centromeric and subtelomeric chromatin domains. PLoS Genet. 2009;5(11):e1000726. https://doi.org/10.1371/journal.pgen.1000726.
Wan Y, Saleem RA, Ratushny AV, Roda O, Smith JJ, Lin CH, et al. Role of the histone variant H2A.Z/Htz1p in TBP recruitment, chromatin dynamics, and regulated expression of oleate-responsive genes. Mol Cell Biol. 2009;29(9):2346–58. https://doi.org/10.1128/mcb.01233-08.
Kusch T, Mei A, Nguyen C. Histone H3 lysine 4 trimethylation regulates cotranscriptional H2A variant exchange by Tip60 complexes to maximize gene expression. Proc Natl Acad Sci USA. 2014;111(13):4850–5. https://doi.org/10.1073/pnas.1320337111.
Hu Y, Shen Y, Conde ESN, Zhou DX. The role of histone methylation and H2A.Z occupancy during rapid activation of ethylene responsive genes. PLoS ONE. 2011;6(11):e28224. https://doi.org/10.1371/journal.pone.0028224.
Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4(1):80–93. https://doi.org/10.1016/j.stem.2008.11.011.
Farris SD, Rubio ED, Moon JJ, Gombert WM, Nelson BH, Krumm A. Transcription-induced chromatin remodeling at the c-myc gene involves the local exchange of histone H2A.Z. J Biol Chem. 2005;280(26):25298–303. https://doi.org/10.1074/jbc.m501784200.
John S, Sabo PJ, Johnson TA, Sung MH, Biddie SC, Lightman SL, et al. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol Cell. 2008;29(5):611–24. https://doi.org/10.1016/j.molcel.2008.02.010.
Sutcliffe EL, Parish IA, He YQ, Juelich T, Tierney ML, Rangasamy D, et al. Dynamic histone variant exchange accompanies gene induction in T cells. Mol Cell Biol. 2009;29(7):1972–86. https://doi.org/10.1128/MCB.01590-08.
Amat R, Gudas LJ. RARgamma is required for correct deposition and removal of Suz12 and H2A.Z in embryonic stem cells. J Cell Physiol. 2011;226(2):293–8. https://doi.org/10.1002/jcp.22420.
Chauhan S, Boyd DD. Regulation of u-PAR gene expression by H2A.Z is modulated by the MEK-ERK/AP-1 pathway. Nucleic Acids Res. 2012;40(2):600–13. https://doi.org/10.1093/nar/gkr725.
Au-Yeung N, Horvath CM. Histone H2AZ suppression of interferon-stimulated transcription and antiviral immunity is modulated by GCN5 and BRD2. iScience. 2018;6:68–82. https://doi.org/10.1016/j.isci.2018.07.013.
Kim K, Punj V, Choi J, Heo K, Kim JM, Laird PW, et al. Gene dysregulation by histone variant H2A.Z in bladder cancer. Epigenetics Chromatin. 2013;6(1):34. https://doi.org/10.1186/1756-8935-6-34.
Martinato F, Cesaroni M, Amati B, Guccione E. Analysis of Myc-induced histone modifications on target chromatin. PLoS ONE. 2008;3(11):e3650. https://doi.org/10.1371/journal.pone.0003650.
Hardy S, Jacques PE, Gevry N, Forest A, Fortin ME, Laflamme L, et al. The euchromatic and heterochromatic landscapes are shaped by antagonizing effects of transcription on H2A.Z deposition. PLoS Genet. 2009;5(10):e1000687. https://doi.org/10.1371/journal.pgen.1000687.
Lashgari A, Millau JF, Jacques PE, Gaudreau L. Global inhibition of transcription causes an increase in histone H2A.Z incorporation within gene bodies. Nucleic Acids Res. 2017;45(22):12715–22. https://doi.org/10.1093/nar/gkx879.
Weber CM, Ramachandran S, Henikoff S. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol Cell. 2014;53(5):819–30. https://doi.org/10.1016/j.molcel.2014.02.014.
Day DS, Zhang B, Stevens SM, Ferrari F, Larschan EN, Park PJ, et al. Comprehensive analysis of promoter-proximal RNA polymerase II pausing across mammalian cell types. Genome Biol. 2016;17(1):120. https://doi.org/10.1186/s13059-016-0984-2.
Bargaje R, Alam MP, Patowary A, Sarkar M, Ali T, Gupta S, et al. Proximity of H2A.Z containing nucleosome to the transcription start site influences gene expression levels in the mammalian liver and brain. Nucleic Acids Res. 2012;40(18):8965–78. https://doi.org/10.1093/nar/gks665.
Guillemette B, Bataille AR, Gevry N, Adam M, Blanchette M, Robert F, et al. Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol. 2005;3(12):e384. https://doi.org/10.1371/journal.pbio.0030384.
Zhang H, Roberts DN, Cairns BR. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell. 2005;123(2):219–31. https://doi.org/10.1016/j.cell.2005.08.036.
Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, et al. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet. 2009;41(8):941–5. https://doi.org/10.1038/ng.409.
Hu G, Cui K, Northrup D, Liu C, Wang C, Tang Q, et al. H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2013;12(2):180–92. https://doi.org/10.1016/j.stem.2012.11.003.
Dai X, Bai Y, Zhao L, Dou X, Liu Y, Wang L, et al. H2A.Z Represses gene expression by modulating promoter nucleosome structure and enhancer histone modifications in arabidopsis. Mol Plant. 2017;10(10):1274–92. https://doi.org/10.1016/j.molp.2017.09.007.
Johnson TA, Chereji RV, Stavreva DA, Morris SA, Hager GL, Clark DJ. Conventional and pioneer modes of glucocorticoid receptor interaction with enhancer chromatin in vivo. Nucleic Acids Res. 2018;46(1):203–14. https://doi.org/10.1093/nar/gkx1044.
Brunelle M, Nordell Markovits A, Rodrigue S, Lupien M, Jacques PE, Gevry N. The histone variant H2A.Z is an important regulator of enhancer activity. Nucleic Acids Res. 2015;43(20):9742–56. https://doi.org/10.1093/nar/gkv825.
Marques M, Laflamme L, Gaudreau L. Estrogen receptor alpha can selectively repress dioxin receptor-mediated gene expression by targeting DNA methylation. Nucleic Acids Res. 2013;41(17):8094–106. https://doi.org/10.1093/nar/gkt595.
Swaminathan J, Baxter EM, Corces VG. The role of histone H2Av variant replacement and histone H4 acetylation in the establishment of Drosophila heterochromatin. Genes Dev. 2005;19(1):65–76. https://doi.org/10.1101/gad.1259105.
Kotova E, Lodhi N, Jarnik M, Pinnola AD, Ji Y, Tulin AV. Drosophila histone H2A variant (H2Av) controls poly(ADP-ribose) polymerase 1 (PARP1) activation in chromatin. Proc Natl Acad Sci USA. 2011;108(15):6205–10. https://doi.org/10.1073/pnas.1019644108.
Leach TJ, Mazzeo M, Chotkowski HL, Madigan JP, Wotring MG, Glaser RL. Histone H2A.Z is widely but nonrandomly distributed in chromosomes of Drosophila melanogaster. J Biol Chem. 2000;275(30):23267–72. https://doi.org/10.1074/jbc.m910206199.
Ismail IH, Hendzel MJ. The gamma-H2A.X: is it just a surrogate marker of double-strand breaks or much more? Environ Mol Mutagen. 2008;49(1):73–82. https://doi.org/10.1002/em.20358.
Madigan JP, Chotkowski HL, Glaser RL. DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic Acids Res. 2002;30(17):3698–705.
Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR 3rd, et al. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306(5704):2084–7. https://doi.org/10.1126/science.1103455.
Flegel K, Grushko O, Bolin K, Griggs E, Buttitta L. Roles for the histone modifying and exchange complex NuA4 in cell cycle progression in drosophila melanogaster. Genetics. 2016;203(3):1265–81. https://doi.org/10.1534/genetics.116.188581.
Krogan NJ, Keogh MC, Datta N, Sawa C, Ryan OW, Ding H, et al. A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell. 2003;12(6):1565–76.
Kobor MS, Venkatasubrahmanyam S, Meneghini MD, Gin JW, Jennings JL, Link AJ, et al. A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2AZ into euchromatin. PLoS Biol. 2004;2(5):E131. https://doi.org/10.1371/journal.pbio.0020131.
Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science. 2004;303(5656):343–8. https://doi.org/10.1126/science.1090701.
Kim HS, Vanoosthuyse V, Fillingham J, Roguev A, Watt S, Kislinger T, et al. An acetylated form of histone H2AZ regulates chromosome architecture in Schizosaccharomyces pombe. Nat Struct Mol Biol. 2009;16(12):1286–93. https://doi.org/10.1038/nsmb.1688.
Altaf M, Auger A, Monnet-Saksouk J, Brodeur J, Piquet S, Cramet M, et al. NuA4-dependent acetylation of nucleosomal histones H4 and H2A directly stimulates incorporation of H2AZ by the SWR1 complex. J Biol Chem. 2010;285(21):15966–77. https://doi.org/10.1074/jbc.m110.117069.
Luk E, Ranjan A, Fitzgerald PC, Mizuguchi G, Huang Y, Wei D, et al. Stepwise histone replacement by SWR1 requires dual activation with histone H2A.Z and canonical nucleosome. Cell. 2010;143(5):725–36. https://doi.org/10.1016/j.cell.2010.10.019.
Wu WH, Alami S, Luk E, Wu CH, Sen S, Mizuguchi G, et al. Swc2 is a widely conserved H2AZ-binding module essential for ATP-dependent histone exchange. Nat Struct Mol Biol. 2005;12(12):1064–71. https://doi.org/10.1038/nsmb1023.
Nguyen VQ, Ranjan A, Stengel F, Wei D, Aebersold R, Wu C, et al. Molecular architecture of the ATP-dependent chromatin-remodeling complex SWR1. Cell. 2013;154(6):1220–31. https://doi.org/10.1016/j.cell.2013.08.018.
Watanabe S, Tan D, Lakshminarasimhan M, Washburn MP, Hong EJ, Walz T, et al. Structural analyses of the chromatin remodelling enzymes INO80-C and SWR-C. Nat Commun. 2015;6:7108. https://doi.org/10.1038/ncomms8108.
Willhoft O, Ghoneim M, Lin CL, Chua EYD, Wilkinson M, Chaban Y, et al. Structure and dynamics of the yeast SWR1-nucleosome complex. Science. 2018. https://doi.org/10.1126/science.aat7716.
Hong J, Feng H, Wang F, Ranjan A, Chen J, Jiang J, et al. The catalytic subunit of the SWR1 remodeler is a histone chaperone for the H2A.Z-H2B dimer. Mol Cell. 2014;53(3):498–505. https://doi.org/10.1016/j.molcel.2014.01.010.
Auger A, Galarneau L, Altaf M, Nourani A, Doyon Y, Utley RT, et al. Eaf1 is the platform for NuA4 molecular assembly that evolutionarily links chromatin acetylation to ATP-dependent exchange of histone H2A variants. Mol Cell Biol. 2008;28(7):2257–70. https://doi.org/10.1128/MCB.01755-07.
Keogh MC, Mennella TA, Sawa C, Berthelet S, Krogan NJ, Wolek A, et al. The Saccharomyces cerevisiae histone H2A variant Htz1 is acetylated by NuA4. Genes Dev. 2006;20(6):660–5. https://doi.org/10.1101/gad.1388106.
Choi J, Heo K, An W. Cooperative action of TIP48 and TIP49 in H2A.Z exchange catalyzed by acetylation of nucleosomal H2A. Nucleic Acids Res. 2009;37(18):5993–6007. https://doi.org/10.1093/nar/gkp660.
Ranjan A, Mizuguchi G, FitzGerald PC, Wei D, Wang F, Huang Y, et al. Nucleosome-free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell. 2013;154(6):1232–45. https://doi.org/10.1016/j.cell.2013.08.005.
Osada S, Sutton A, Muster N, Brown CE, Yates JR 3rd, Sternglanz R, et al. The yeast SAS (something about silencing) protein complex contains a MYST-type putative acetyltransferase and functions with chromatin assembly factor ASF1. Genes Dev. 2001;15(23):3155–68. https://doi.org/10.1101/gad.907201.
Ruhl DD, Jin J, Cai Y, Swanson S, Florens L, Washburn MP, et al. Purification of a human SRCAP complex that remodels chromatin by incorporating the histone variant H2A.Z into nucleosomes. Biochemistry. 2006;45(17):5671–7. https://doi.org/10.1021/bi060043d.
Cai Y, Jin J, Tomomori-Sato C, Sato S, Sorokina I, Parmely TJ, et al. Identification of new subunits of the multiprotein mammalian TRRAP/TIP60-containing histone acetyltransferase complex. J Biol Chem. 2003;278(44):42733–6. https://doi.org/10.1074/jbc.C300389200.
Doyon Y, Selleck W, Lane WS, Tan S, Cote J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol. 2004;24(5):1884–96.
Cai Y, Jin J, Florens L, Swanson SK, Kusch T, Li B, et al. The mammalian YL1 protein is a shared subunit of the TRRAP/TIP60 histone acetyltransferase and SRCAP complexes. J Biol Chem. 2005;280(14):13665–70. https://doi.org/10.1074/jbc.M500001200.
Robert F, Hardy S, Nagy Z, Baldeyron C, Murr R, Dery U, et al. The transcriptional histone acetyltransferase cofactor TRRAP associates with the MRN repair complex and plays a role in DNA double-strand break repair. Mol Cell Biol. 2006;26(2):402–12. https://doi.org/10.1128/MCB.26.2.402-412.2006.
Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003;4(6):575–80. https://doi.org/10.1038/sj.embor.embor861.
Jeong KW, Kim K, Situ AJ, Ulmer TS, An W, Stallcup MR. Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat Struct Mol Biol. 2011;18(12):1358–65. https://doi.org/10.1038/nsmb.2153.
Slupianek A, Yerrum S, Safadi FF, Monroy MA. The chromatin remodeling factor SRCAP modulates expression of prostate specific antigen and cellular proliferation in prostate cancer cells. J Cell Physiol. 2010;224(2):369–75. https://doi.org/10.1002/jcp.22132.
Ye B, Liu B, Yang L, Huang G, Hao L, Xia P, et al. Suppression of SRCAP chromatin remodelling complex and restriction of lymphoid lineage commitment by Pcid2. Nat Commun. 2017;8(1):1518. https://doi.org/10.1038/s41467-017-01788-7.
Heisel S, Habel NC, Schuetz N, Ruggieri A, Meese E. The YEATS family member GAS41 interacts with the general transcription factor TFIIF. BMC Mol Biol. 2010;11:53. https://doi.org/10.1186/1471-2199-11-53.
Ito S, Kayukawa N, Ueda T, Taniguchi H, Morioka Y, Hongo F, et al. MRGBP promotes AR-mediated transactivation of KLK3 and TMPRSS2 via acetylation of histone H2A.Z in prostate cancer cells. Biochim Biophys Acta Gene Regul Mech. 2018. https://doi.org/10.1016/j.bbagrm.2018.07.014.
Cho HJ, Li H, Linhares BM, Kim E, Ndoj J, Miao H, et al. GAS41 recognizes diacetylated histone H3 through a bivalent binding mode. ACS Chem Biol. 2018;13(9):2739–46. https://doi.org/10.1021/acschembio.8b00674.
Klein BJ, Ahmad S, Vann KR, Andrews FH, Mayo ZA, Bourriquen G, et al. Yaf9 subunit of the NuA4 and SWR1 complexes targets histone H3K27ac through its YEATS domain. Nucleic Acids Res. 2018;46(1):421–30. https://doi.org/10.1093/nar/gkx1151.
Hsu CC, Zhao D, Shi J, Peng D, Guan H, Li Y, et al. Gas41 links histone acetylation to H2AZ deposition and maintenance of embryonic stem cell identity. Cell Discov. 2018;4:28. https://doi.org/10.1038/s41421-018-0027-0.
Hsu CC, Shi J, Yuan C, Zhao D, Jiang S, Lyu J, et al. Recognition of histone acetylation by the GAS41 YEATS domain promotes H2A.Z deposition in non-small cell lung cancer. Genes Dev. 2018;32(1):58–69. https://doi.org/10.1101/gad.303784.117.
Wang Y, Jin J, Chung MWH, Feng L, Sun H, Hao Q. Identification of the YEATS domain of GAS41 as a pH-dependent reader of histone succinylation. Proc Natl Acad Sci USA. 2018;115(10):2365–70. https://doi.org/10.1073/pnas.1717664115.
Wang AY, Schulze JM, Skordalakes E, Gin JW, Berger JM, Rine J, et al. Asf1-like structure of the conserved Yaf9 YEATS domain and role in H2A.Z deposition and acetylation. Proc Natl Acad Sci U S A. 2009;106(51):21573–8. https://doi.org/10.1073/pnas.0906539106.
Yen K, Vinayachandran V, Pugh BF. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell. 2013;154(6):1246–56. https://doi.org/10.1016/j.cell.2013.08.043.
Obri A, Ouararhni K, Papin C, Diebold ML, Padmanabhan K, Marek M, et al. ANP32E is a histone chaperone that removes H2AZ from chromatin. Nature. 2014;505(7485):648–53. https://doi.org/10.1038/nature12922.
Mao Z, Pan L, Wang W, Sun J, Shan S, Dong Q, et al. Anp32e, a higher eukaryotic histone chaperone directs preferential recognition for H2A.Z. Cell Res. 2014;24(4):389–99. https://doi.org/10.1038/cr.2014.30.
Shin H, He M, Yang Z, Jeon YH, Pfleger J, Sayed D, et al. Transcriptional regulation mediated by H2A.Z via ANP32e-dependent inhibition of protein phosphatase 2A. Biochim Biophys Acta Gene Regul Mech. 2018;1861(5):481–96. https://doi.org/10.1016/j.bbagrm.2018.03.002.
Richard M, Yvert G. How does evolution tune biological noise? Front Genet. 2014;5:374. https://doi.org/10.3389/fgene.2014.00374.
Gallant-Behm CL, Ramsey MR, Bensard CL, Nojek I, Tran J, Liu M, et al. DeltaNp63alpha represses anti-proliferative genes via H2A.Z deposition. Genes Dev. 2012;26(20):2325–36. https://doi.org/10.1101/gad.198069.112.
Vavra KJ, Allis CD, Gorovsky MA. Regulation of histone acetylation in Tetrahymena macro- and micronuclei. J Biol Chem. 1982;257(5):2591–8.
Bruce K, Myers FA, Mantouvalou E, Lefevre P, Greaves I, Bonifer C, et al. The replacement histone H2AZ in a hyperacetylated form is a feature of active genes in the chicken. Nucleic Acids Res. 2005;33(17):5633–9. https://doi.org/10.1093/nar/gki874.
Myers FA, Lefevre P, Mantouvalou E, Bruce K, Lacroix C, Bonifer C, et al. Developmental activation of the lysozyme gene in chicken macrophage cells is linked to core histone acetylation at its enhancer elements. Nucleic Acids Res. 2006;34(14):4025–35. https://doi.org/10.1093/nar/gkl543.
Cheng X, Auger A, Altaf M, Drouin S, Paquet E, Utley RT, et al. Eaf1 links the NuA4 histone acetyltransferase complex to Htz1 incorporation and regulation of purine biosynthesis. Eukaryot Cell. 2015;14(6):535–44. https://doi.org/10.1128/EC.00004-15.
Mehta M, Braberg H, Wang S, Lozsa A, Shales M, Solache A, et al. Individual lysine acetylations on the N terminus of Saccharomyces cerevisiae H2A.Z are highly but not differentially regulated. J Biol Chem. 2010;285(51):39855–65. https://doi.org/10.1074/jbc.m110.185967.
Chittuluru JR, Chaban Y, Monnet-Saksouk J, Carrozza MJ, Sapountzi V, Selleck W, et al. Structure and nucleosome interaction of the yeast NuA4 and Piccolo-NuA4 histone acetyltransferase complexes. Nat Struct Mol Biol. 2011;18(11):1196–203. https://doi.org/10.1038/nsmb.2128.
Rajagopalan D, Tirado-Magallanes R, Bhatia SS, Teo WS, Sian S, Hora S, et al. TIP60 represses activation of endogenous retroviral elements. Nucleic Acids Res. 2018;46(18):9456–70. https://doi.org/10.1093/nar/gky659.
Lin YY, Qi Y, Lu JY, Pan X, Yuan DS, Zhao Y, et al. A comprehensive synthetic genetic interaction network governing yeast histone acetylation and deacetylation. Genes Dev. 2008;22(15):2062–74. https://doi.org/10.1101/gad.1679508.
Link S, Spitzer RMM, Sana M, Torrado M, Volker-Albert MC, Keilhauer EC, et al. PWWP2A binds distinct chromatin moieties and interacts with an MTA1-specific core NuRD complex. Nat Commun. 2018;9(1):4300. https://doi.org/10.1038/s41467-018-06665-5.
Perell GT, Mishra NK, Sudhamalla B, Ycas PD, Islam K, Pomerantz WCK. Specific acetylation patterns of H2AZ form transient interactions with the BPTF bromodomain. Biochemistry. 2017;56(35):4607–15. https://doi.org/10.1021/acs.biochem.7b00648.
Sarcinella E, Zuzarte PC, Lau PN, Draker R, Cheung P. Monoubiquitylation of H2AZ distinguishes its association with euchromatin or facultative heterochromatin. Mol Cell Biol. 2007;27(18):6457–68. https://doi.org/10.1128/mcb.00241-07.
O’Connor HF, Lyon N, Leung JW, Agarwal P, Swaim CD, Miller KM, et al. Ubiquitin-activated interaction traps (UBAITs) identify E3 ligase binding partners. EMBO Rep. 2015;16(12):1699–712. https://doi.org/10.15252/embr.201540620.
Fukuto A, Ikura M, Ikura T, Sun J, Horikoshi Y, Shima H, et al. SUMO modification system facilitates the exchange of histone variant H2A.Z-2 at DNA damage sites. Nucleus. 2018;9(1):87–94. https://doi.org/10.1080/19491034.2017.1395543.
Binda O, Sevilla A, LeRoy G, Lemischka IR, Garcia BA, Richard S. SETD6 monomethylates H2AZ on lysine 7 and is required for the maintenance of embryonic stem cell self-renewal. Epigenetics. 2013;8(2):177–83. https://doi.org/10.4161/epi.23416.
Tsai CH, Chen YJ, Yu CJ, Tzeng SR, Wu IC, Kuo WH, et al. SMYD3-mediated H2A.Z.1 methylation promotes cell cycle and cancer proliferation. Cancer Res. 2016;76(20):6043–53. https://doi.org/10.1158/0008-5472.can-16-0500.
Fujimoto S, Seebart C, Guastafierro T, Prenni J, Caiafa P, Zlatanova J. Proteome analysis of protein partners to nucleosomes containing canonical H2A or the variant histones H2A.Z or H2A.X. Biol Chem. 2012;393(1–2):47–61. https://doi.org/10.1515/bc-2011-216.
Punzeler S, Link S, Wagner G, Keilhauer EC, Kronbeck N, Spitzer RM, et al. Multivalent binding of PWWP2A to H2A.Z regulates mitosis and neural crest differentiation. EMBO J. 2017;36(15):2263–79. https://doi.org/10.15252/embj.201695757.
Zhang Y, Ku WL, Liu S, Cui K, Jin W, Tang Q, et al. Genome-wide identification of histone H2A and histone variant H2A.Z-interacting proteins by bPPI-seq. Cell Res. 2017;27(10):1258–74. https://doi.org/10.1038/cr.2017.112.
Kusakabe M, Oku H, Matsuda R, Hori T, Muto A, Igarashi K, et al. Genetic complementation analysis showed distinct contributions of the N-terminal tail of H2A.Z to epigenetic regulations. Genes Cells. 2016;21(2):122–35. https://doi.org/10.1111/gtc.12327.
Eberl HC, Spruijt CG, Kelstrup CD, Vermeulen M, Mann M. A map of general and specialized chromatin readers in mouse tissues generated by label-free interaction proteomics. Mol Cell. 2013;49(2):368–78. https://doi.org/10.1016/j.molcel.2012.10.026.
Garay PM, Wallner MA, Iwase S. Yin-yang actions of histone methylation regulatory complexes in the brain. Epigenomics. 2016;8(12):1689–708. https://doi.org/10.2217/epi-2016-0090.
Raisner RM, Hartley PD, Meneghini MD, Bao MZ, Liu CL, Schreiber SL, et al. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell. 2005;123(2):233–48. https://doi.org/10.1016/j.cell.2005.10.002.
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–37. https://doi.org/10.1016/j.cell.2007.05.009.
Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132(5):887–98. https://doi.org/10.1016/j.cell.2008.02.022.
Chen J, Miller A, Kirchmaier AL, Irudayaraj JM. Single-molecule tools elucidate H2A.Z nucleosome composition. J Cell Sci. 2012;125(Pt 12):2954–64. https://doi.org/10.1242/jcs.101592.
Won KJ, Choi I, LeRoy G, Zee BM, Sidoli S, Gonzales-Cope M, et al. Proteogenomics analysis reveals specific genomic orientations of distal regulatory regions composed by non-canonical histone variants. Epigenetics Chromatin. 2015;8:13. https://doi.org/10.1186/s13072-015-0005-9.
Yao F, Yu P, Li Y, Yuan X, Li Z, Zhang T, et al. Histone variant H2A.Z is required for the maintenance of smooth muscle cell identity as revealed by single-cell transcriptomics. Circulation. 2018;138(20):2274–88. https://doi.org/10.1161/circulationaha.117.033114.
Jeronimo C, Watanabe S, Kaplan CD, Peterson CL, Robert F. The histone chaperones FACT and Spt6 restrict H2A.Z from intragenic locations. Mol Cell. 2015;58(6):1113–23. https://doi.org/10.1016/j.molcel.2015.03.030.
Rach EA, Winter DR, Benjamin AM, Corcoran DL, Ni T, Zhu J, et al. Transcription initiation patterns indicate divergent strategies for gene regulation at the chromatin level. PLoS Genet. 2011;7(1):e1001274. https://doi.org/10.1371/journal.pgen.1001274.
Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446(7135):572–6. https://doi.org/10.1038/nature05632.
Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453(7193):358–62. https://doi.org/10.1038/nature06929.
Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–9. https://doi.org/10.1038/nature09906.
Cauchy P, Koch F, Andrau JC. Two possible modes of pioneering associated with combinations of H2A.Z and p300/CBP at nucleosome-occupied enhancers. Transcription. 2017;8(3):179–84. https://doi.org/10.1080/21541264.2017.1291395.
Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007;21(12):1519–29. https://doi.org/10.1101/gad.1547707.
Soboleva TA, Nekrasov M, Ryan DP, Tremethick DJ. Histone variants at the transcription start-site. Trends Genet. 2014;30(5):199–209. https://doi.org/10.1016/j.tig.2014.03.002.
Henikoff S, Henikoff JG, Sakai A, Loeb GB, Ahmad K. Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res. 2009;19(3):460–9. https://doi.org/10.1101/gr.087619.108.
Xi Y, Yao J, Chen R, Li W, He X. Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res. 2011;21(5):718–24. https://doi.org/10.1101/gr.117101.110.
Suto RK, Clarkson MJ, Tremethick DJ, Luger K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat Struct Biol. 2000;7(12):1121–4. https://doi.org/10.1038/81971.
Bernstein E, Hake SB. The nucleosome: a little variation goes a long way. Biochem Cell Biol. 2006;84(4):505–17. https://doi.org/10.1139/o06-085.
Abbott DW, Ivanova VS, Wang X, Bonner WM, Ausio J. Characterization of the stability and folding of H2A.Z chromatin particles: implications for transcriptional activation. J Biol Chem. 2001;276(45):41945–9. https://doi.org/10.1074/jbc.m108217200.
Horikoshi N, Arimura Y, Taguchi H, Kurumizaka H. Crystal structures of heterotypic nucleosomes containing histones H2A.Z and H2A. Open Biol. 2016. https://doi.org/10.1098/rsob.160127.
Nekrasov M, Amrichova J, Parker BJ, Soboleva TA, Jack C, Williams R, et al. Histone H2AZ inheritance during the cell cycle and its impact on promoter organization and dynamics. Nat Struct Mol Biol. 2012;19(11):1076–83. https://doi.org/10.1038/nsmb.2424.
Nekrasov M, Soboleva TA, Jack C, Tremethick DJ. Histone variant selectivity at the transcription start site: H2A.Z or H2A.Lap1. Nucleus. 2013;4(6):431–8. https://doi.org/10.4161/nucl.26862.
Thakar A, Gupta P, Ishibashi T, Finn R, Silva-Moreno B, Uchiyama S, et al. H2A.Z and H33 histone variants affect nucleosome structure: biochemical and biophysical studies. Biochemistry. 2009;48(46):10852–7. https://doi.org/10.1021/bi901129e.
Weber CM, Henikoff JG, Henikoff S. H2A.Z nucleosomes enriched over active genes are homotypic. Nat Struct Mol Biol. 2010;17(12):1500–7. https://doi.org/10.1038/nsmb.1926.
Park YJ, Dyer PN, Tremethick DJ, Luger K. A new fluorescence resonance energy transfer approach demonstrates that the histone variant H2AZ stabilizes the histone octamer within the nucleosome. J Biol Chem. 2004;279(23):24274–82. https://doi.org/10.1074/jbc.M313152200.
Thambirajah AA, Dryhurst D, Ishibashi T, Li A, Maffey AH, Ausio J. H2A.Z stabilizes chromatin in a way that is dependent on core histone acetylation. J Biol Chem. 2006;281(29):20036–44. https://doi.org/10.1074/jbc.m601975200.
Ishibashi T, Dryhurst D, Rose KL, Shabanowitz J, Hunt DF, Ausio J. Acetylation of vertebrate H2A.Z and its effect on the structure of the nucleosome. Biochemistry. 2009;48(22):5007–17. https://doi.org/10.1021/bi900196c.
Lantermann AB, Straub T, Stralfors A, Yuan GC, Ekwall K, Korber P. Schizosaccharomyces pombe genome-wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae. Nat Struct Mol Biol. 2010;17(2):251–7. https://doi.org/10.1038/nsmb.1741.
Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature. 2008;456(7218):125–9. https://doi.org/10.1038/nature07324.
Conerly ML, Teves SS, Diolaiti D, Ulrich M, Eisenman RN, Henikoff S. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res. 2010;20(10):1383–90. https://doi.org/10.1101/gr.106542.110.
Edwards JR, O’Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010;20(7):972–80. https://doi.org/10.1101/gr.101535.109.
Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328(5980):916–9. https://doi.org/10.1126/science.1186366.
Murphy PJ, Wu SF, James CR, Wike CL, Cairns BR. Placeholder nucleosomes underlie germline-to-embryo DNA methylation reprogramming. Cell. 2018;172(5):993–1006. https://doi.org/10.1016/j.cell.2018.01.022.
Yang X, Noushmehr H, Han H, Andreu-Vieyra C, Liang G, Jones PA. Gene reactivation by 5-aza-2’-deoxycytidine-induced demethylation requires SRCAP-mediated H2A.Z insertion to establish nucleosome depleted regions. PLoS Genet. 2012;8(3):e1002604. https://doi.org/10.1371/journal.pgen.1002604.
Barzily-Rokni M, Friedman N, Ron-Bigger S, Isaac S, Michlin D, Eden A. Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A). Nucleic Acids Res. 2011;39(4):1326–35. https://doi.org/10.1093/nar/gkq994.
Hua S, Kallen CB, Dhar R, Baquero MT, Mason CE, Russell BA, et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol Syst Biol. 2008;4:188. https://doi.org/10.1038/msb.2008.25.
Svotelis A, Gevry N, Grondin G, Gaudreau L. H2A.Z overexpression promotes cellular proliferation of breast cancer cells. Cell Cycle. 2010;9(2):364–70. https://doi.org/10.4161/cc.9.2.10465.
Baptista T, Graca I, Sousa EJ, Oliveira AI, Costa NR, Costa-Pinheiro P, et al. Regulation of histone H2A.Z expression is mediated by sirtuin 1 in prostate cancer. Oncotarget. 2013;4(10):1673–85. https://doi.org/10.18632/oncotarget.1237.
Dunican DS, McWilliam P, Tighe O, Parle-McDermott A, Croke DT. Gene expression differences between the microsatellite instability (MIN) and chromosomal instability (CIN) phenotypes in colorectal cancer revealed by high-density cDNA array hybridization. Oncogene. 2002;21(20):3253–7. https://doi.org/10.1038/sj.onc.1205431.
Yang B, Tong R, Liu H, Wu J, Chen D, Xue Z, et al. H2A.Z regulates tumorigenesis, metastasis and sensitivity to cisplatin in intrahepatic cholangiocarcinoma. Int J Oncol. 2018;52(4):1235–45. https://doi.org/10.3892/ijo.2018.4292.
Yang HD, Kim PJ, Eun JW, Shen Q, Kim HS, Shin WC, et al. Oncogenic potential of histone-variant H2A.Z.1 and its regulatory role in cell cycle and epithelial-mesenchymal transition in liver cancer. Oncotarget. 2016;7(10):11412–23. https://doi.org/10.18632/oncotarget.7194.
Chen IY, Lypowy J, Pain J, Sayed D, Grinberg S, Alcendor RR, et al. Histone H2A.z is essential for cardiac myocyte hypertrophy but opposed by silent information regulator 2alpha. J Biol Chem. 2006;281(28):19369–77. https://doi.org/10.1074/jbc.m601443200.
Zhao H, Jin S, Gewirtz AM. The histone acetyltransferase TIP60 interacts with c-Myb and inactivates its transcriptional activity in human leukemia. J Biol Chem. 2012;287(2):925–34. https://doi.org/10.1074/jbc.M111.279950.
Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature. 2007;448(7157):1063–7. https://doi.org/10.1038/nature06055.
Chevillard-Briet M, Quaranta M, Grezy A, Mattera L, Courilleau C, Philippe M, et al. Interplay between chromatin-modifying enzymes controls colon cancer progression through Wnt signaling. Hum Mol Genet. 2014;23(8):2120–31. https://doi.org/10.1093/hmg/ddt604.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–308. https://doi.org/10.1038/nprot.2013.143.
Serikawa T, Eberle J, Kurreck J. Effects of genomic disruption of a guanine quadruplex in the 5′ UTR of the Bcl-2 mRNA in melanoma cells. FEBS Lett. 2017;591(21):3649–59. https://doi.org/10.1002/1873-3468.12855.
Song Y, Xu Y, Deng J, Chen M, Lu Y, Wang Y, et al. CRISPR/Cas9-mediated mutation of tyrosinase (Tyr) 3′ UTR induce graying in rabbit. Sci Rep. 2017;7(1):1569. https://doi.org/10.1038/s41598-017-01727-y.
Zhao W, Siegel D, Biton A, Tonqueze OL, Zaitlen N, Ahituv N, et al. CRISPR-Cas9-mediated functional dissection of 3′-UTRs. Nucleic Acids Res. 2017;45(18):10800–10. https://doi.org/10.1093/nar/gkx675.
Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature. 2016;534(7607):402–6. https://doi.org/10.1038/nature18294.
Chang JW, Zhang W, Yeh HS, Park M, Yao C, Shi Y, et al. An integrative model for alternative polyadenylation, IntMAP, delineates mTOR-modulated endoplasmic reticulum stress response. Nucleic Acids Res. 2018;46(12):5996–6008. https://doi.org/10.1093/nar/gky340.
Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25(7):778–85. https://doi.org/10.1038/nbt1319.
Collin J, Mellough CB, Dorgau B, Przyborski S, Moreno-Gimeno I, Lako M. Using zinc finger nuclease technology to generate CRX-reporter human embryonic stem cells as a tool to identify and study the emergence of photoreceptors precursors during pluripotent stem cell differentiation. Stem Cells. 2016;34(2):311–21. https://doi.org/10.1002/stem.2240.
Wang Y, Long H, Yu J, Dong L, Wassef M, Zhuo B, et al. Histone variants H2A.Z and H3.3 coordinately regulate PRC2-dependent H3K27me3 deposition and gene expression regulation in mES cells. BMC Biol. 2018;16(1):107. https://doi.org/10.1186/s12915-018-0568-6.
Wu WH, Wu CH, Ladurner A, Mizuguchi G, Wei D, Xiao H, et al. N terminus of Swr1 binds to histone H2AZ and provides a platform for subunit assembly in the chromatin remodeling complex. J Biol Chem. 2009;284(10):6200–7. https://doi.org/10.1074/jbc.M808830200.
Beck HC, Nielsen EC, Matthiesen R, Jensen LH, Sehested M, Finn P, et al. Quantitative proteomic analysis of post-translational modifications of human histones. Mol Cell Proteomics. 2006;5(7):1314–25. https://doi.org/10.1074/mcp.M600007-MCP200.
Bonenfant D, Coulot M, Towbin H, Schindler P, van Oostrum J. Characterization of histone H2A and H2B variants and their post-translational modifications by mass spectrometry. Mol Cell Proteomics. 2006;5(3):541–52. https://doi.org/10.1074/mcp.M500288-MCP200.
Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23(4):607–18. https://doi.org/10.1016/j.molcel.2006.06.026.
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–40. https://doi.org/10.1126/science.1175371.
Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, et al. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol Cell. 2012;46(2):212–25. https://doi.org/10.1016/j.molcel.2012.01.026.
Boskovic A, Bender A, Gall L, Ziegler-Birling C, Beaujean N, Torres-Padilla ME. Analysis of active chromatin modifications in early mammalian embryos reveals uncoupling of H2A.Z acetylation and H3K36 trimethylation from embryonic genome activation. Epigenetics. 2012;7(7):747–57. https://doi.org/10.4161/epi.20584.
Gallant-Behm CL, Espinosa JM. DeltaNp63alpha utilizes multiple mechanisms to repress transcription in squamous cell carcinoma cells. Cell Cycle. 2013;12(3):409–16. https://doi.org/10.4161/cc.23593.
Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10(7):634–7. https://doi.org/10.1038/nmeth.2518.
Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013;4(4):842–51. https://doi.org/10.1016/j.celrep.2013.07.024.
Wu Q, Cheng Z, Zhu J, Xu W, Peng X, Chen C, et al. Suberoylanilide hydroxamic acid treatment reveals crosstalks among proteome, ubiquitylome and acetylome in non-small cell lung cancer A549 cell line. Sci Rep. 2015;5:9520. https://doi.org/10.1038/srep09520.
Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Scholz C, et al. Time-resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell. 2018;174(1):231–44. https://doi.org/10.1016/j.cell.2018.04.033.
Danielsen JM, Sylvestersen KB, Bekker-Jensen S, Szklarczyk D, Poulsen JW, Horn H, et al. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol Cell Proteomics. 2011;10(3):M110003590. https://doi.org/10.1074/mcp.m110.003590.
Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–20. https://doi.org/10.1093/nar/gku1267.
Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci USA. 2008;105(2):692–7. https://doi.org/10.1073/pnas.0707270105.
Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal. 2011;4(179):5. https://doi.org/10.1126/scisignal.2001497.
Schweppe DK, Rigas JR, Gerber SA. Quantitative phosphoproteomic profiling of human non-small cell lung cancer tumors. J Proteomics. 2013;91:286–96. https://doi.org/10.1016/j.jprot.2013.07.023.
Tsai CF, Wang YT, Yen HY, Tsou CC, Ku WC, Lin PY, et al. Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat Commun. 2015;6:6622. https://doi.org/10.1038/ncomms7622.
Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics. 2014;13(7):1690–704. https://doi.org/10.1074/mcp.M113.036392.
Lumpkin RJ, Gu H, Zhu Y, Leonard M, Ahmad AS, Clauser KR, et al. Site-specific identification and quantitation of endogenous SUMO modifications under native conditions. Nat Commun. 2017;8(1):1171. https://doi.org/10.1038/s41467-017-01271-3.
Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44(2):325–40. https://doi.org/10.1016/j.molcel.2011.08.025.
Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, et al. A proteome-wide quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10(10):M111013284. https://doi.org/10.1074/mcp.m111.013284.
Povlsen LK, Beli P, Wagner SA, Poulsen SL, Sylvestersen KB, Poulsen JW, et al. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat Cell Biol. 2012;14(10):1089–98. https://doi.org/10.1038/ncb2579.
Wagner SA, Beli P, Weinert BT, Scholz C, Kelstrup CD, Young C, et al. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol Cell Proteomics. 2012;11(12):1578–85. https://doi.org/10.1074/mcp.M112.017905.
Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, Qiao JW, et al. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000 s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics. 2013;12(3):825–31. https://doi.org/10.1074/mcp.O112.027094.
Boeing S, Williamson L, Encheva V, Gori I, Saunders RE, Instrell R, et al. Multiomic analysis of the UV-induced DNA damage response. Cell Rep. 2016;15(7):1597–610. https://doi.org/10.1016/j.celrep.2016.04.047.
Surface LE, Fields PA, Subramanian V, Behmer R, Udeshi N, Peach SE, et al. H2A.Z.1 monoubiquitylation antagonizes BRD2 to maintain poised chromatin in ESCs. Cell Rep. 2016;14(5):1142–55. https://doi.org/10.1016/j.celrep.2015.12.100.
Li Z, Gadue P, Chen K, Jiao Y, Tuteja G, Schug J, et al. Foxa2 and H2A.Z mediate nucleosome depletion during embryonic stem cell differentiation. Cell. 2012;151(7):1608–16. https://doi.org/10.1016/j.cell.2012.11.018.
Authors’ contributions
All authors wrote the manuscript. BDG and AH prepared the figures and tables. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors have approved the manuscript.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project number 109546710—TRR81 to T.B. and S.B.H, and the Heisenberg program (BO 1639/5-1) by the DFG (German Research Foundation) and the Excellence Cluster for Cardio Pulmonary System (ECCPS) to T.B and the Cardio-Pulmonary Institute (CPI) to S.B.H. Funding for open access charge was provided by the DFG collaborative research TRR81. B.D.G. is supported by a Research Grant of the University Medical Center Giessen and Marburg (UKGM).
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Giaimo, B.D., Ferrante, F., Herchenröther, A. et al. The histone variant H2A.Z in gene regulation. Epigenetics & Chromatin 12, 37 (2019). https://doi.org/10.1186/s13072-019-0274-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13072-019-0274-9