
Abstract
DNA methylation is an epigenetic modification that plays an important role in regulating gene expression and therefore a broad range of biological processes and diseases. DNA methylation is tissue-specific, dynamic, sequence-context-dependent and trans-generationally heritable, and these complex patterns of methylation highlight the significance of profiling DNA methylation to answer biological questions. In this review, we surveyed major methylation assays, along with comparisons and biological examples, to provide an overview of DNA methylation profiling techniques. The advances in microarray and sequencing technologies make genome-wide profiling possible at a single-nucleotide or even a single-cell resolution. These profiling approaches vary in many aspects, such as DNA input, resolution, genomic region coverage, and bioinformatics analysis, and selecting a feasible method requires knowledge of these methods. We first introduce the biological background of DNA methylation and its pattern in plants, animals and fungi. We present an overview of major experimental approaches to profiling genome-wide DNA methylation and hydroxymethylation and then extend to the single-cell methylome. To evaluate these methods, we outline their strengths and weaknesses and perform comparisons across the different platforms. Due to the increasing need to compute high-throughput epigenomic data, we interrogate the computational pipeline for bisulfite sequencing data and also discuss the concept of identifying differentially methylated regions (DMRs). This review summarizes the experimental and computational concepts for profiling genome-wide DNA methylation, followed by biological examples. Overall, this review provides researchers useful guidance for the selection of a profiling method suited to specific research questions.
Similar content being viewed by others


Background
DNA methylation, one of the most studied epigenetic modifications, involves the addition of a methyl group to the fifth carbon of cytosine (C), forming 5-methylcytosine (5mC), catalyzed by DNA methyltransferases (Dnmts) [1]. DNA methylation predominantly occurs in CpG dinucleotides (CpGs) but is also found less frequently in non-CpG contexts (e.g., CHG and CHH, where H = A, T or C). These contexts affect gene function and structure differently [2]. The de novo DNA methyltransferases Dnmt3a and Dnmt3b are responsible for catalyzing the methylation of Cs, and the maintenance methyltransferase Dnmt1 enables the propagation of DNA methylation patterns during cell division [3–5]. DNA methylation has been associated with numerous cellular processes, such as transcriptional repression, X chromosome inactivation, embryonic development, genomic imprinting, the alteration of chromatin structure and transposon inactivation [6, 7]. The methyl marks are heritable, that certain methylation patterns have transgenerational effects [8]. The patterns of these marks are also dynamically remodeled during distinct reprogramming phases throughout the life cycle of an organism [9]. DNA methylation does not occur exclusively on C residues; methylation can also present as N6-methyladenine (6mA) in Chlamydomonas reinhardtii (algae) [10], Caenorhabditis elegans (nematode) [11], Drosophila melanogaster (insect) [12] and vertebrates such as Xenopus laevis, mouse and human [13]. In addition to 5mC, 5-hydroxymethylcytosine (5hmC) is another epigenetic mark in the mammalian genome associated with DNA demethylation. 5hmC is produced via the oxidation of 5mC catalyzed by the ten-eleven translocation (TET) family of proteins, and subsequent oxidation results in the formation of 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) [14].
DNA methylation patterns vary across organisms. The mammalian genome is usually highly methylated; in human embryonic stem cells (hESCs), DNA methylation occurs in up to 80 % of CpGs, with the remaining unmethylated CpG residues enriched in CpG islands (CGI) located at gene promoters [15]. Extremely low methylation levels have been reported in invertebrates such as Drosophila [16] and Bombyx [17]. In plants, the methylation level varies in the CpG and non-CpG sites; the levels were found to be 24 % CpG, 7 % CHG and 2 % CHH in Arabidopsis [18] and 86 % CpG, 74 % CHG and 5 % CHH in unfertilized ears of maize [19]. Non-CpG methylation plays key roles in plants, in which this modification can silence exogenous DNA via an RNA-dependent DNA methylation pathway (RdDM) [20]. In fungi, the methylation in black truffle is found exclusively in transposons and is absent from genes [21].
Promoter methylation can potentially down-regulate gene expression by altering the chromatin structure and blocking transcription initiation [7]. For example, in mammals, most CGIs in promoters are unmethylated to facilitate binding between proteins and promoter DNA. Positive correlations between active transcription and gene body methylation have been observed in the active X chromosome [22, 23]. Gene body methylation may also function to silence repetitive DNA elements found within the gene body [24]. In addition, gene body methylation has been found to exhibit dramatic changes at intron–exon boundaries, suggesting an association with splicing [25]. Maunakea et al. found that DNA methylation modulates alternative splicing by recruiting methyl-CpG-binding protein MeCP2 to promote exon recognition [26]. In maize, CpG methylation in transcribed regions is positively correlated with transcription, whereas CHG methylation is negatively correlated [27]. Methylation changes at the intron–exon boundaries have also been observed, suggesting that maize DNA methylation is likely associated with alternative splicing [28].
These important findings regarding DNA methylation would not have been possible without the advancement of various profiling approaches, both experimental and computational. The accelerated development of array and sequencing technologies has significantly improved DNA methylation profiling, providing an unprecedentedly comprehensive view of the DNA methylation landscape. This review provides an overview of the major profiling approaches, with a focus on the recent and promising genome-wide methodologies (see Fig. 1 for a schematic of the major profiling methods).

Commonly used methods for genome-wide DNA methylation analysis. a The procedures may involve fragmentation of genomic DNA by restriction enzyme digestion or sonication. The genomic DNA can be subjected to MBD enrichment, antibody enrichment, bisulfite conversion or TET oxidation before analyzing by microarray or next-generation sequencing platform. b Single-cell DNA methylation analysis that involves the isolation of single cells allows the assessment of methylation heterogeneity in cell populations while other genome-wide DNA methylation profiling methods using pooled heterogeneous cell populations are not capable to dissect the methylation heterogeneity. Blue concrete dots represent 5mC, and hollowed ones represent C. Each track represents 1 read
Experimental techniques for DNA methylation profiling
Early studies of DNA methylation focused on determining the methylation status of the genes of interest and quantifying the total amount of 5mC [29]. Due to the use of microarray hybridization technology, the study of DNA methylation was able to scale up to the genome-wide level. Next-generation sequencing platforms now allow the construction of genomic maps of DNA methylation at single-base resolution [30]. In the following review, we categorize these experimental approaches into enzyme digestion, affinity enrichment and bisulfite conversion and introduce the major methods with their advantages and disadvantages (see Table 1). We also include corresponding biological examples for each method in Table 1 to help readers select suitable profiling methods. Figure 2 shows the workflow of the experimental pipelines with the DNA input requirements. Finally, we introduce the most recent development in the epigenomic profiling of a single-cell methylome, 5hmC and the use of third-generation sequencing in detecting DNA methylation in real time.

Schematic overview of genome-wide DNA methylation profiling methods. a 5mC assays. b 5hmC assays. The actual sample requirement may vary according to the type of sample, genome size and number of PCR cycles
Restriction enzyme-based methods
Restriction enzyme-based methods take advantage of the differential digestion properties of isoschizomers and neoschizomers. A pair of isoschizomers recognizes the same sequence and has the same point of cleavage but exhibit different sensitivities to the DNA methylation state. Methylation-sensitive restriction enzymes (MREs), such as BstUI, HpaII, NotI and SmaI, cleave only their unmethylated target sequences (see [31] for lists of MREs) and leave the methylated DNA intact. MRE digestion has been coupled with sequencing technologies to predict genome-wide DNA methylation levels [32]. In the workflow of MRE digestion followed by sequencing (MRE-seq), the MRE cleaves the unmethylated CpG sites of genomic DNA, and the resulting DNA fragments are size-selected and sequenced. The sequencing results reveal the locations of the unmethylated CpG sites within the recognition sites of the enzyme utilized [33]. MRE-seq allows the estimation of relative DNA methylation levels but has relatively low coverage of the genome because the CpG-containing recognition sites are limited.
Comprehensive high-throughput arrays for relative methylation (CHARM)
The comprehensive high-throughput arrays for relative methylation (CHARM) method first uses McrBC, an enzyme that digests methylated DNA, to fractionate DNA and subsequently utilizes array hybridization [34]. McrBC recognizes RmC(N)55–103RmC and cleaves half of the methylated DNA and all the methylated CGIs [35], and thus, relatively unmethylated DNA will be size-selected and hybridized to the array. Using CHARM, Irizarry et al. discovered that most DNA methylation differences between colon cancer and adjacent normal tissues occurred in sequences up to 2 kb away from CGIs, termed CpG island shores (CGI shores) [36]. Unexpectedly, differentially methylated regions (DMRs) in CGI shores have a strong inverse relationship with differential gene expression. CHARM, as a restriction enzyme-based method, is able to detect DMRs at CGI shores, which are otherwise not detectable with CpG-directed enrichment methods such as methylated DNA immunoprecipitation (MeDIP).
Affinity enrichment-based methods
Affinity enrichment-based methods use either methyl-CpG-binding domain (MBD) proteins or antibodies specific for 5mC (as in MeDIP) to enrich methylated DNA regions. The results from an MBD protein-based approach, which relies on the capacity of MBD proteins to bind specifically to methylated DNA sequences, could be profiled using microarray (MBD-chip) or sequencing (MBDCap-seq/MethylCap-seq [37], methylated DNA capture by affinity purification) technologies. Serre et al. used MBDCap-seq to study 3 isogenic colon cancer cell lines, and the results confirmed known methylated loci and regions and identified differential methylation in ZEB1, VASH2 and PODXL2 between the HCT116 and DICER1-truncated DICERex5 cell lines [38].
Methylated DNA immunoprecipitation (MeDIP)
MeDIP utilizes an anti-methylcytosine antibody to immunoprecipitate DNA with methylated CpG sites [39]. The DNA fractions enriched by MeDIP can be evaluated using tiling arrays (MeDIP-chip) or high-throughput sequencing (MeDIP-seq) [40]. MeDIP-seq typically yields a resolution of 100–300 bp and could not discriminate methylation context. This can be an issue when research topics are context-specific. Because the methylation statuses of neighboring CpG sites are correlated, MeDIP-seq can be a cost-effective approach when single-base resolution is not desired [41] (Table 1). Taiwo et al. reported that a minimum of 1× coverage can cover up to 70 % of all CpGs in human, suggesting that the majority of the methylated CpGs can be interrogated by MeDIP given that 60–80 % of the CpGs are methylated in a genome [42]. MeDIP-seq generates the relative enrichment of methylated DNA across the genome, instead of predicting the absolute DNA methylation level. MeDIP-seq is feasible with even a low amount of starting DNA material (as low as 1 ng); therefore, this method can be utilized to profile DNA methylation in minute DNA samples, rare cell types and microdissected tissues [39, 42].
The affinity enrichment-based methods tend to exhibit biases associated with CpG density and copy number variation. For example, in MeDIP CpG-rich fragments are more likely to be enriched than CpG-poor ones, even when they are both fully methylated [43]. Hence, a computational correction such as BATMAN tool, which attempts to normalize CpG content across a wide range of CpG densities, is needed [44]. Moreover, CpG density might directly cause PCR bias due to the strong hydrogen bond between the CG pair [43]. Bock et al. benchmarked MeDIP-seq and MBDCap-seq [45] and found that MethylCap-seq covers more genomic regions than MeDIP-seq, and MBDCap-seq could detect nearly twice as many DMRs as MeDIP-seq at comparable sequencing depth. In CpG-poor regions, both MeDIP-seq and MBDCap-seq show low statistical power to detect DMRs.
Bisulfite conversion-based methods
Treating genomic DNA with sodium bisulfite deaminates unmethylated C to uracil (U), while methylated C residues remain unaffected [46]. The U eventually converts to thymine (T) in a subsequent polymerase chain reaction (PCR). Bisulfite conversion-based methods provide single-base resolution and are commonly used to investigate specific DNA sequences when coupled with Sanger sequencing. This type of method is also used to study genome-wide methylation via a methylation array, whole-genome bisulfite sequencing (WGBS) and reduced-representation bisulfite sequencing (RRBS) (see Table 1; Figs. 1, 2).
Methylation array
Illumina’s Infinium HumanMethylation450 BeadChip (HM450K) protocol involves the bisulfite conversion of genomic DNA and amplification, followed by the hybridization of the converted DNA to arrays containing predesigned probes to distinguish between methylated and unmethylated Cs (Fig. 2). Each HM450 K BeadChip can interrogate more than 450,000 methylation sites that cover 96 % of the CGIs, 92 % of the CGI shores and 86 % of the CGI shelves (2–4 kb from a CGI) [47]. To date, HM450 K arrays dominate studies investigating the cancer methylome [48] and other epigenome-wide studies. For example, Bakulski et al. isolated 7 cord blood cell types, which were compared according to their specific methylation signatures; these authors found that nucleated red blood cells had the most pronounced differences [49]. The most recent implementation of the Infinium® technology, Infinium MethylationEPIC BeadChip, covers more than 850 K CpG methylation sites, including >90 % of the 450 K sites plus additional CpG sites in the enhancer regions identified by the ENCODE and FANTOM5 projects [50].
Whole-genome bisulfite sequencing (WGBS)
WGBS (BS-seq; MethylC-seq) theoretically covers all the C information [51]. In this method, genomic DNA is purified and sheared into fragments. The fragmented DNAs are end-repaired; adenine bases are added to the 3′ end (A-tailing) of the DNA fragments, and methylated adapters are ligated to the DNA fragments [52]. The DNA fragments are size-selected before sodium bisulfite treatment and PCR amplification, and the resulting library is sequenced. It should be noted that a high number of PCR cycles and inappropriate selection of a uracil-insensitive DNA polymerase may result in an over-representation in the methylated DNA data [53]. Starting with sufficient genomic DNA may avoid a loss of information from regions of interest and over-amplification. The major advantage of WGBS is its ability to assess the methylation state of nearly every CpG site, including low-CpG-density regions, such as intergenic ‘gene deserts’, partially methylated domains and distal regulatory elements. It can also determine absolute DNA methylation level and reveal methylation sequence context. The first WGBS in 2008 reported the composition of CpG, CHG and CHH methylation in the Arabidopsis genome, the bulk methylation level within each context, and the global methylation pattern in wild-type and methylation-related mutants, as well as specific sites associated with gene expression [18, 54]. In 2013, two maize studies reported that the maize genome is highly methylated, and a specific ‘CHH island’ was found upstream of transcription start sites (TSSs) [19, 28]. In addition to global pattern identification, users could determine regions or even loci with differential methylation between groups using bioinformatics tools. For example, Hsieh et al. compared Arabidopsis endosperm and embryo methylomes and found that virtually the entire endosperm genome is demethylated, coupled with extensive local non-CpG hypermethylation of small interfering RNA-targeted sequences [55]. Lu et al. performed WGBS of maize embryo and endosperm, and the results revealed hypomethylation in the endosperm compared to the embryo [27].
WGBS has become the standard profiling method in major epigenome consortiums, such as NIH Roadmap [56], ENCODE [57], Blueprint [58] and IHEC [59]. For studies interested in regions outside of CGIs, targeted approaches such as reduced-representation bisulfite sequencing (RRBS), MeDIP and MethylCap are not applicable, and the best choice is likely to be WGBS.
Reduced-representation bisulfite sequencing (RRBS)
To investigate the mammalian methylome at a lower cost, Meissner et al. developed RRBS, which integrates Msp1 restriction enzyme digestion, bisulfite conversion and next-generation sequencing for the analysis of methylation patterns of specific fragments [60]. A size selection of MspI-digested fragments between 40 and 220 bps was found to cover 85 % of CGIs, mostly in promoters, which compose only 1–3 % of the mammalian genome, thereby significantly decreasing the amount of sequencing [51, 61]. RRBS-based protocols are more cost-effective than WGBS because these methods focus on the enrichment of CpG-rich regions in close proximity to the restriction enzyme’s recognition sequence; however, these protocols may exhibit a lack of coverage at intergenic and distal regulatory elements that are relatively less studied.
RRBS has been widely used in profiling large-scale samples. Orozco et al. performed RRBS in 90 inbred mouse strains, conducted an integrative analysis that included genome-wide expression levels, proteomics, metabolomics, and 68 clinical traits, and performed epigenome-wide association studies (EWAS) [62]. They found associations with numerous clinical traits, including bone density, insulin resistance, expression, and protein and metabolite levels. RRBS has also been used in non-mammalians, such as zebrafish [63], wasp [64], oak populations [65] and Brassica rapa [66].
Commercial DNA methylation assay kits
Another concern for BS-seq is that a large amount of high-quality genomic DNA, e.g., usually 5 μg, is required for WGBS, and RRBS requires 0.01–0.3 μg [51] (see Fig. 2). To study samples with a preciously small amount of DNA, e.g., primordial germ cells (PGCs) and cancer cells, commercial kits for ultralow input were developed. The Ovation® Ultralow Methyl-Seq Library System requires only 10 ng of DNA to construct the WGBS library [67], and the Ovation® RRBS Methyl-Seq Library System requires 100 ng of DNA for RRBS.
For targeted bisulfite sequencing, the SeqCap Epi System from Roche enables the enrichment of a small fraction of the genome containing regions of interest after bisulfite conversion [68]. In addition, the SeqCap Epi CpGiant Enrichment Kit allows the interrogation of more than 5.5 million CpGs in the human genome with a starting DNA input of 1 µg. Roche also provides customization of probe pools according to the type of organism and regions of interest. The SureSelectXT Methyl-Seq Target Enrichment Kit from Agilent Technologies involves the hybridization and enrichment of sequencing libraries with oligonucleotide baits before bisulfite conversion [69]. This platform supports the enrichment of an 84-Mb target covering 3.7 million CpG sites with a DNA input as low as 1 µg.
Single-cell methylome
Most genome-wide DNA methylation profiling techniques have common limitations: the need for bulk cell populations as starting materials and the inability to assess methylation heterogeneity among individual cells [70, 71]. To address these issues, single-cell bisulfite-based techniques have been developed. First, single-cell reduced-representation bisulfite sequencing (scRRBS) integrates the steps of MspI digestion to bisulfite conversion into one tube of cell lysate to minimize DNA loss and to provide methylation information on approximately 1 million CpG sites within an individual mouse or human cell [70]. Another single-cell DNA methylation analysis method, namely single-cell bisulfite sequencing (scBS-seq), is a modified post-bisulfite adapter tagging (PBAT) protocol [72, 73]. PBAT circumvents the issue of a massive bisulfite-induced loss of sequencing templates in WGBS by performing bisulfite treatment ahead of adapter tagging, thereby enabling the use of a lower starting amount of DNA (only 100 ng) and eliminating the need for global amplification [73]. scBS-seq enables the measurement of DNA methylation at up to 48.4 % of the CpG sites and was reported to achieve higher recovery rates than scRRBS [71, 72]. Farlik et al. described single-cell whole-genome bisulfite sequencing (scWGBS) of human and mouse cells and bioinformatics inferences for epigenomic cell-state dynamics in pluripotent and differentiating cells [74]. These single-cell techniques can be applied in studies involving limited cell amounts and heterogeneous cell populations [71, 72] and are particularly useful for specific cell types that play important roles in early development, such as sperm cells, oocytes, PGCs and embryonic stem cells (ESCs).
Genome-wide 5-hydroxylmethylation profiling
The TET family of dioxygenases catalyze the oxidation of 5mC to 5hmC. The detection of 5hmC gained much attention recently after this C modification was identified as an epigenetic mark in mammals (mouse brain and ESCs), and 5hmC has been reported to be an intermediate in DNA demethylation [75, 76]. The detection of 5hmC is technically more challenging than that of 5mC due to the low abundance of 5hmC, and standard bisulfite sequencing does not distinguish between 5mC and 5hmC because both are resistant to bisulfite treatment [77]. Hydroxymethylated DNA immunoprecipitation (hMeDIP), which is modified from MeDIP, characterizes the relative abundance of 5hmC at specific loci or throughout the entire mammalian genome. hMeDIP involves immunoselection and immunoprecipitation using anti-5hmC antibodies and subsequent analysis by qPCR, microarray hybridization or next-generation sequencing [78].
Oxidative bisulfite sequencing (OxBS-seq)
A modified bisulfite sequencing technique, oxidative bisulfite sequencing (OxBS-seq) distinguishes between 5mC and 5hmC via the highly selective chemical oxidation of 5hmC to 5fC [79]. After bisulfite treatment, 5fC is converted to U and is read as T in the sequencing stage. Unlike 5hmC, 5mC does not undergo oxidation upon bisulfite treatment and will be detected as C after sequencing. The 5hmC level can be quantified by comparing the data from BS-seq (which identifies both 5hmC and 5mC) and the data from OxBS-seq (which identifies 5mC). The disadvantages of this technique are the oxidative degradation of DNA and the requirement for multiple bisulfite treatments to completely deaminate 5fC [80].
TET-assisted bisulfite sequencing (TAB-seq)
TET-assisted bisulfite sequencing (TAB-seq) has been used to generate genome-wide 5hmC profiles at a single-base resolution in human and mouse ESCs [81]. In TAB-seq, 5hmC is protected from TET protein-mediated oxidation by the addition of glucose to 5hmC using β-glucosyltransferase (β-GT) to generate β-glucosyl-5-hydroxymethylcytosine (g5hmC). 5mC is oxidized by the Tet1 enzyme to 5caC. 5caC and unmethylated C are susceptible to bisulfite conversion and thus are sequenced as T, whereas 5hmC is sequenced as C. TAB-seq measures 5hmC directly, and information regarding 5mC can be obtained using the same analysis pipeline as BS-seq. Highly active TET proteins are required for the efficient conversion of 5mC to 5caC (more than 96 %), or else the incomplete conversion of 5mC might lead to false identification as 5hmC sites [81]. Both oxidative bisulfite conversion and TET-assisted bisulfite conversion are compatible with microarray and sequencing platforms to generate the 5hmC methylation profile for a whole genome or targeted regions [82]. The relatively low levels of 5hmC and the subtraction step demand an increase in the sequencing coverage and the number of replicates. A study of human PGC epigenome used TAB-seq to reveal the demethylation during epigenetic reprogramming between 57 and 113 days, and the heterogeneity of 5hmc in both individual loci and at individual cells has been identified [83].
Third-generation sequencing
Emerging third-generation sequencing technologies [84], including single-molecule real-time sequencing (SMRT) and Oxford Nanopore technology, have been recently adopted in epigenetics research.
Single-molecule real-time sequencing
Developed by Pacific Biosystems, SMRT allows the direct detection of base modifications by monitoring the activity of DNA polymerase during the incorporation of different fluorescently labeled nucleotides into complementary DNA strands [85, 86]. The direct detection of various base modifications involves the measurement of the kinetics variation in the time between base incorporations. This technology has the following advantages over second-generation sequencing: (1) minimal chemical modification during library preparation; (2) the requirement for DNA amplification is eliminated; (3) reduced requirement for input DNA; (4) the ability to generate longer reads (average read length of 3000 bp); and (5) the ability to detect different types of epigenetic modifications [86, 87]. SMRT has been used in the identification of 6mA in C. elegans, and the recently developed SMRT of chromatin immunoprecipitation enriched DNA (SMRT-ChIP) has resulted in the identification of 6mA and associated demethylase ALKBH1 in mouse ESCs [11, 88].
Nanopore sequencing
In nanopore sequencing, single-stranded DNA is pulled by a phage DNA polymerase through a bacterial pore in single-nucleotide steps, and the ion current through the pore is recorded [89]. C can be distinguished from 5mC and 5hmC based on differences in the current traces. Although the detection of 5mC and 5hmC using nanopore sequencing yielded encouraging results for the DNA methylation profiling of a single locus, the application of this method to genome-wide DNA methylation profiling has yet to be established.
Taken together, these applications of third-generation sequencing open doors to more discoveries of different epigenetic modifications and potentially reveal the novel functions of these epigenetic marks in gene expression. Despite its many promising features, the broad application of third-generation sequencing is still limited by a higher error rate, higher cost and lower throughput than second-generation sequencing technologies [90]. The throughput and accuracy must be substantially improved before applying these approaches to studies involving complex genomes.
Bioinformatics analysis of WGBS and RRBS
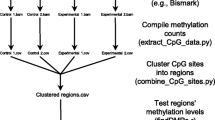
The general workflow for the bioinformatics analysis of DNA methylation data includes data processing, the quantification of DNA methylation levels, general profiling, the identification of DMRs and the visualization of the methylome [91]. Array-based data, such as that from Illumina’s HM450K, are fluorescence intensities that quantify the relative abundance of methylated and unmethylated loci. The data from other non-bisulfite-conversion methods, such as MRE-seq and MeDIP-seq, are usually analyzed by comparing the relative abundance of fragments. Bisulfite-converted data, such as those from WGBS and RRBS, involve methylation calling at individual Cs, and statistical testing is required to assess differential methylation. In this section, we focus on the bioinformatics analyses of bisulfite-converted data, in particular WGBS and RRBS (see Fig. 3 for a general bioinformatics pipeline).

Computational pipeline for genome-wide bisulfite sequencing data analysis. Reads from bisulfite sequencing are first aligned to the reference genome. The alignment data may be visualized in different tracks for comparison. After methylation calling, the bulk methylation level and genome-wide methylation level are calculated and plotted, and DMRs are determined. To perform an integrative analysis, DNA methylation data are coupled with gene expression, e.g., differentially expressed genes (DEGs), to delineate the regulatory role of DNA methylation
Aligning bisulfite-converted reads and data visualization
The bisulfite sequencing data are generally processed with several steps, including adapter trimming [92], a quality assessment of reads [93–95], aligning reads to the reference genome [96–101] and methylation calling [102]. In particular, mapping bisulfite-converted reads is challenging due to reduced sequence complexity, asymmetric C to T alignments, and the fact that the bisulfite-converted Watson and Crick strands are not complementary to each other because bisulfite conversion occurs only at Cs (not G’s) [96, 103]. To address these issues, a number of alignment and post-alignment analysis tools have been developed (see Table 2 for a list of alignment tools and software for post-alignment analysis). Bisulfite sequencing aligners are mostly based on one of two algorithms: wild cards and three-letter algorithms. Wild card aligners substitute Cs with Ys in the reference genome, and reads with both Cs and Ts can then be aligned. This method results in higher genomic coverage together with a bias toward higher methylation levels [91]. However, the three-letter aligners convert all Cs in the reference genome and the read into Ts, and thus, standard aligners with lower mappability can be adopted due to reduced sequence complexity. For example, in the analysis of black truffle methylome, the bisulfite-treated reads were mapped 15–25 % less, comparing to the untreated [21]. In Table 2, we list 7 major bisulfite sequencing aligners along with their features. For example, BS Seeker 2 is a three-letter aligner that supports the local alignment and computational removal of potentially unconverted reads. The alignment profile can be visualized with tools such as the UCSC genome browser [104], WBSA [105], IGV [106] and Methylation plotter [107], which results in greater clarity at a single-base resolution across the genome.
Post-alignment data analysis
Bisulfite aligners will output aligned reads along with the methylation calling information of each C with sequence context information, e.g., the CGmap file in BS Seeker 2 [98]. Users can filter out sites with coverage, calculate the average methylation level and generate informative plots. Table 2 lists 5 post-alignment analysis tools, and each of these tools has specific functions, e.g., BSPAT can detect allele-specific methylation [108], SAAP-RRBS can extract the annotation of each C [94] and MethGo can convert context methylation levels into average and genome-wide plots, as well as extract SNP and CNV profiles [109].
Detection of differentially methylated loci and regions
WGBS and RRBS generate methylation calls at each C as an estimate of the percentage of cells with methylation. Statistical tests are employed to identify differentially methylated loci in comparisons. For studies without replicates, Fisher’s exact test is generally adopted. A comparison with no replicates completely ignores within-group variations, resulting in an overstatement of the differences and a high false-positive rate. Hansen et al. described the need for biological replicates and developed BSmooth to effectively use low-coverage data with biological replicates and to determine DMRs [110].
DMRs are genomic regions that exhibit a different methylation status between two groups of samples. For example, Choufani et al. assessed genome-wide DNA methylation maps in human uniparental samples, a mature cystic ovarian teratoma (MCT) carrying the maternal genome and an androgenetic complete hydatidiform mole (AnCHM) carrying the paternal genome, as references to identify imprinted genes and DMRs. The comparison between the MCT and AnCHM successfully identified AXL as a new imprinted gene [111].
The identification of DMRs relies on both computational power for genome-wide screening and statistical testing. In Table 2, we included tools for implementing statistical methods in DMR screening [110, 112–115]. Generally, the DMR detection algorithm adopts a sliding window across the genome to survey candidate DMRs, and the most common approach is to perform Fisher’s exact test CpG-wise. To detect DMR, as the coverage of each sample may be different, only sites covered by all samples are comparable. To enable the comparison, the comparing statistics such as methylation difference, T-score from T test or P value is needed in the testing. In the BSmooth software, a beta-binomial is assumed to be the suitable model for replicated bisulfite sequencing data. The observation is assumed to be binomially distributed, whereas the methylated proportion at a particular site can vary across samples. The differences at an individual site could be small but may expand and persist across a region, which is a candidate DMR. Therefore, DMRs are determined with greater statistical power and are more informative. When comparing methylomes with weak differences, extending the testing scale from one C to a cluster of neighboring Cs can reduce the number of hypothesis tests to improve the statistical power [91] (e.g., BiSeq takes spatial correlation into account in DMR prediction [115]). Weak DNA methylation differences can be better measured by estimating the standard deviation from biological replicates to obtain more robust P values [91].
Multiple testing in DMR detection
In addition, multiple testing is increased when many sites are simultaneously tested. In Table 2, we include a list of software that enables a correction for multiple testing.
Schmitz et al. performed a large-scale WGBS analysis in which DMRs from many Arabidopsis methylomes were detected [116]. They used the R package methylPipe to scan the genome with 100-bp windows [114], and the methylation level of the sites within a window was compared across all samples using a Kruskal–Wallis test. The P values were then adjusted for multiple testing using the Benjamini–Hochburg method, and only DMRs with an adjusted P value less than 0.01 were selected. In addition to the adjusted P value, a second criterion is used to ensure the differences, and the DMR has to exhibit an eightfold methylation difference between the two groups.
Gkountela et al. devised an in-house method to identify DMRs between each developmental stage during human PGC development [83]. In their comparisons among the ICM (inner cell mass), PGCs and AGCs (advanced germ cells), these authors identified candidate DMRs with at least an 80 % methylation difference in 200-bp windows. To evaluate the FDR, they generated simulated methylomes with the same read coverage per site as the real samples and reproduced the methylation level per site. The methylation levels were generated from a binomial model in which the parameters were the same for all the samples, i.e., all the simulated methylomes were equally methylated in both comparison groups. Any DMRs identified from the simulated methylomes were considered false positives. In total, Gkountela et al. found 3445 DMRs between PGCs and AGCs with an FDR < 0.001 %.
Robinson et al. reviewed some of the major DMR tools and discussed how the statistical significance was assessed [117]; users were advised to select the tool that satisfies their experimental design and data format. For example, most of the tools have been developed based on human and mouse studies; therefore, users studying other organisms should take the flexibility of the tools into account. The accommodation of different data types should also be considered, e.g., BiSeq supports RRBS only, whereas methylPipe supports RRBS and WGBS, as well as low-resolution DNA methylation data.
Conclusions
This review provides an overview of the current techniques for the assessment of genome-wide DNA methylation and the identification of DMRs. The commonly used techniques are primarily based on restriction enzyme digestion, affinity enrichment and bisulfite treatment, coupled with either microarray or sequencing technologies. Because each technique has its own advantages and disadvantages, we summarize in Table 1 a comprehensive evaluation of each technique. In Fig. 2, we provide an overview of these experimental pipelines and their required DNA input amounts. The selection of a technique strongly depends on the research questions, cost, amount of input DNA and the expected degree of methylation changes [118]. In Table 1, readers can also learn from the biological examples in which the profiling techniques were used to determine the experiments that best fit their research topic. For example, for mammalian studies with large-scale samples, one should consider a targeted approach, such as MeDIP or RRBS rather than WGBS, which would allow multiple sample comparisons with limited cost and provide sufficient information from CpG-rich regions. If the study aims to investigate the first methylome of an organism, then WGBS with deep sequencing would be a more suitable method to obtain detailed information in coding regions and intergenic regions. The input DNA amount should also be considered when rare cell types or tissues are studied. To reveal the methylation state of undifferentiated stem cells without heterogeneity, single-cell approaches would be the best choice. The sequencing depth is a key parameter in DMR discovery; the greater the depth, the more power to discover DMRs. However, for studies with a large sample size such as disease-centered research studies, the distribution of limited resources should be considered, e.g., sequencing a few samples deeply or more samples less deeply. A balance may be reached by considering the profiling technique coupling with the data analysis that would provide precise and accurate DMR prediction with low coverage requirements.
The discovery of various forms of C modifications, namely 5hmC, 5fC and 5caC, further expand the efforts to map and quantify these low-abundance bases in different cell and tissue types [76]. The emerging SMRT and nanopore sequencing technologies have enabled the direct reading of C modifications without the pre-treatment of DNA and amplification; however, the throughput and accuracy must be substantially improved before these techniques become contenders against second-generation sequencing technologies [14].
In addition to providing underlying biological insights, DNA methylation assays have great potential for application to different fields, particularly medicine and forensic sciences [119, 120]. In medicine, these methodologies aid in the identification of epigenetic-based biomarkers for cancer and other epigenetic-related diseases, which serve as measurable indicators of biological conditions for predicting the presence or severity of a disease state or treatment response and further contribute to the development of clinical treatments and personalized medicine throughout life [120–124]. DNA methylation has been applied to the discrimination of fetal and maternal DNA in circulating cell-free DNA to obtain more pure fetal DNA for downstream analyses, such as chromosomal abnormality [125]. With improvements in low-input bisulfite sequencing and single-cell techniques, methylomes at an early embryonic developmental stage during pregnancy could be obtained to identify an abnormal fetus. Regarding applications in forensic sciences, DNA methylation analysis may be useful in the verification of DNA samples, body fluid identification and the estimation of ages and phenotypic characteristics [119]. The ongoing advancements in technology allow the development of more accurate and affordable methods for methylation analysis, such as with the application of single-cell noninvasive prenatal tests, and further enhance our understanding of the roles of DNA methylation and its underlying mechanism in disease progression and the modulation of DNA methylation in response to different environmental cues in different cell and tissues types.
Abbreviations
- 5caC:
-
5-carboxylcytosine
- 5fC:
-
5-formylcytosine
- 5mC:
-
5-methylcytosine
- 5hmC:
-
5-hydroxymethylcytosine
- 6mA:
-
N6-methyladenine
- CGI:
-
CpG island
- DMR:
-
differentially methylated region
- ESC:
-
embryonic stem cell
- FDR:
-
false discovery rate
- hESC:
-
human embryonic stem cell
- hMeDIP:
-
hydroxymethylated DNA immunoprecipitation
- MethylCap-seq:
-
methylated DNA capture by affinity purification sequencing
- MRE:
-
methylation-sensitive restriction enzyme
- MBD:
-
methyl-CpG-binding domain
- MeDIP:
-
methylated DNA immunoprecipitation
- OxBS-seq:
-
oxidative bisulfite sequencing
- PGC:
-
primordial germ cell
- RRBS:
-
reduced-representation bisulfite sequencing
- SMRT:
-
single-molecule real-time sequencing
- TAB-seq:
-
TET-assisted bisulfite sequencing
- TE:
-
transposable element
- TET:
-
ten-eleven translocation family
- TSS:
-
transcription start site
- WGBS:
-
whole-genome bisulfite sequencing
References
Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20.
Kim KD, El Baidouri M, Jackson SA. Accessing epigenetic variation in the plant methylome. Brief Funct Genomics. 2014;13:318–27.
Berkyurek AC, Suetake I, Arita K, Takeshita K, Nakagawa A, Shirakawa M, et al. The DNA methyltransferase Dnmt1 directly interacts with the SET and RING finger-associated (SRA) domain of the multifunctional protein Uhrf1 to facilitate accession of the catalytic center to hemi-methylated DNA. J Biol Chem. 2014;289:379–86.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57.
Kinde B, Gabel HW, Gilbert CS, Griffith EC, Greenberg ME. Reading the unique DNA methylation landscape of the brain: non-CpG methylation, hydroxymethylation, and MeCP2. Proc Natl Acad Sci USA. 2015;112:6800–6.
Wilson GA, Dhami P, Feber A, Cortázar D, Suzuki Y, Schulz R, et al. Resources for methylome analysis suitable for gene knockout studies of potential epigenome modifiers. Gigascience. 2012;1:3.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92.
Li Q, Xu W, Cui Y, Ma L, Richards J, Li W, et al. A preliminary exploration on DNA methylation of transgene across generations in transgenic rats. Sci Rep. 2015;5:8292.
Hackett JA, Surani MA. DNA methylation dynamics during the mammalian life cycle. Philos Trans R Soc Lond B Biol Sci. 2013;368:20110328.
Fu Y, Luo GZ, Chen K, Deng X, Yu M, Han D, et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell. 2015;161:879–92.
Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizábal-Corrales D, et al. DNA methylation on N6-Adenine in C. elegans. Cell. 2015;161:868–78.
Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, et al. N6-methyladenine DNA modification in Drosophila. Cell. 2015;161:893–906.
Koziol MJ, Bradshaw CR, Allen GE, Costa AS, Frezza C, Gurdon JB. Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat Struct Mol Biol. 2016;23:24–30.
Plongthongkum N, Diep DH, Zhang K. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat Rev Genet. 2014;15:647–61.
Chen PY, Feng S, Joo JW, Jacobsen SE, Pellegrini M. A comparative analysis of DNA methylation across human embryonic stem cell lines. Genome Biol. 2011;12:R62.
Lyko F, Ramsahoye BH, Jaenisch R. DNA methylation in Drosophila melanogaster. Nature. 2000;408:538–40.
Xiang H, Zhu J, Chen Q, Dai F, Li X, Li M, et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat Biotechnol. 2010;28:516–20.
Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–9.
Gent JI, Ellis NA, Guo L, Harkess AE, Yao Y, Zhang X, et al. CHH islands: de novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013;23:628–37.
Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–20.
Montanini B, Chen PY, Morselli M, Jaroszewicz A, Lopez D, Martin F, et al. Non-exhaustive DNA methylation-mediated transposon silencing in the black truffle genome, a complex fungal genome with massive repeat element content. Genome Biol. 2014;15:411.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22.
Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA. 2010;107:8689–94.
Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–40.
Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–31.
Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23:1256–69.
Lu X, Wang W, Ren W, Chai Z, Guo W, Chen R, et al. Genome-wide epigenetic regulation of gene transcription in maize seeds. PLoS ONE. 2015;10:e0139582.
Regulski M, Lu Z, Kendall J, Donoghue MT, Reinders J, Llaca V, et al. The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res. 2013;23:1651–62.
Lisanti S, Omar WA, Tomaszewski B, De Prins S, Jacobs G, Koppen G, et al. Comparison of methods for quantification of global DNA methylation in human cells and tissues. PLoS ONE. 2013;8:e79044.
Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203.
New England BioLabs. https://www.neb.com/tools-and-resources/selection-charts/dam-dcm-and-cpg-methylation.
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–7.
Li D, Zhang B, Xing X, Wang T. Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods. 2015;72:29–40.
Irizarry RA, Ladd-Acosta C, Carvalho B, Wu H, Brandenburg SA, Jeddeloh JA, et al. Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res. 2008;18:780–90.
Sutherland E, Coe L, Raleigh EA. McrBC: a multisubunit GTP-dependent restriction endonuclease. J Mol Biol. 1992;225:327–48.
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86.
Brinkman AB, Simmer F, Ma K, Kaan A, Zhu J, Stunnenberg HG. Whole-genome DNA methylation profiling using MethylCap-seq. Methods. 2010;52:232–6.
Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38:391–9.
Zhao MT, Whyte JJ, Hopkins GM, Kirk MD, Prather RS. Methylated DNA immunoprecipitation and high-throughput sequencing (MeDIP-seq) using low amounts of genomic DNA. Cell Reprogram. 2014;16:175–84.
Weng YI, Huang TH, Yan PS. Methylated DNA immunoprecipitation and microarray-based analysis: detection of DNA methylation in breast cancer cell lines. Methods Mol Biol. 2009;590:165–76.
Clark C, Palta P, Joyce CJ, Scott C, Grundberg E, Deloukas P, et al. A comparison of the whole genome approach of MeDIP-seq to the targeted approach of the Infinium HumanMethylation450 BeadChip((R)) for methylome profiling. PLoS ONE. 2012;7:e50233.
Taiwo O, Wilson GA, Morris T, Seisenberger S, Reik W, Pearce D, et al. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat Protoc. 2012;7:617–36.
Robinson MD, Stirzaker C, Statham AL, Coolen MW, Song JZ, Nair SS, et al. Evaluation of affinity-based genome-wide DNA methylation data: effects of CpG density, amplification bias, and copy number variation. Genome Res. 2010;20:1719–29.
Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008;26:779–85.
Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 2010;28:1106–14.
Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827–31.
Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–95.
Stirzaker C, Taberlay PC, Statham AL, Clark SJ. Mining cancer methylomes: prospects and challenges. Trends Genet. 2014;30:75–84.
Bakulski KM, Feinberg JI, Andrews SV, Yang J, Brown S, McKenney SL, et al. DNA methylation of cord blood cell types: Applications for mixed cell birth studies. Epigenetics. 2016;11:354–62. doi:10.1080/15592294.2016.1161875
Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016;8:389–99.
Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6:468–81.
Urich MA, Nery JR, Lister R, Schmitz RJ, Ecker JR. MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nat Protoc. 2015;10:475–83.
Ji L, Sasaki T, Sun X, Ma P, Lewis ZA, Schmitz RJ. Methylated DNA is over-represented in whole-genome bisulfite sequencing data. Front Genet. 2014;5:341.
Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–36.
Hsieh TF, Ibarra CA, Silva P, Zemach A, Eshed-Williams L, Fischer RL, et al. Genome-wide demethylation of Arabidopsis endosperm. Science. 2009;324:1451–4.
Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol. 2010;28:1045–8.
Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A, et al. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol. 2012;30:224–6.
International Human Epigenome Consortium. http://ihec-epigenomes.org/. 2016.
Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–70.
Smith ZD, Gu H, Bock C, Gnirke A, Meissner A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009;48:226–32.
Orozco LD, Morselli M, Rubbi L, Guo W, Go J, Shi H, et al. Epigenome-wide association of liver methylation patterns and complex metabolic traits in mice. Cell Metab. 2015;21:905–17.
Chatterjee A, Ozaki Y, Stockwell PA, Horsfield JA, Morison IM, Nakagawa S. Mapping the zebrafish brain methylome using reduced representation bisulfite sequencing. Epigenetics. 2013;8:979–89.
Pegoraro M, Bafna A, Davies NJ, Shuker DM, Tauber E. DNA methylation changes induced by long and short photoperiods in Nasonia. Genome Res. 2016;26:203–10.
Platt A, Gugger PF, Pellegrini M, Sork VL. Genome-wide signature of local adaptation linked to variable CpG methylation in oak populations. Mol Ecol. 2015;24:3823–30.
Chen X, Ge X, Wang J, Tan C, King GJ, Liu K. Genome-wide DNA methylation profiling by modified reduced representation bisulfite sequencing in Brassica rapa suggests that epigenetic modifications play a key role in polyploid genome evolution. Front Plant Sci. 2015;6:836.
Ovation® Ultralow Methyl-Seq Library Systems. http://www.nugen.com/products/ngs/ovation-ultralow-methyl-seq-library-systems. 2016.
SeqCap Epi CpGiant Enrichment Kit. http://sequencing.roche.com/products/nimblegen-seqcap-target-enrichment/seqcap-epi-system/seqcap-epi-cpgiant-enrichment-kit.html.
SureSelect Methyl-Seq. http://www.genomics.agilent.com/article.jsp?pageId=3038.
Guo H, Zhu P, Guo F, Li X, Wu X, Fan X, et al. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat Protoc. 2015;10:645–59.
Schwartzman O, Tanay A. Single-cell epigenomics: techniques and emerging applications. Nat Rev Genet. 2015;16:716–26.
Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11:817–20.
Miura F, Enomoto Y, Dairiki R, Ito T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012;40:e136.
Farlik M, Sheffield NC, Nuzzo A, Datlinger P, Schönegger A, Klughammer J, et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015;10:1386–97.
Bachman M, Uribe-Lewis S, Yang X, Williams M, Murrell A, Balasubramanian S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat Chem. 2014;6:1049–55.
Wu H, Zhang Y. Charting oxidized methylcytosines at base resolution. Nat Struct Mol Biol. 2015;22:656–61.
Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE. 2010;5:e8888.
Nestor CE, Meehan RR. Hydroxymethylated DNA immunoprecipitation (hmeDIP). Methods Mol Biol. 2014;1094:259–67.
Booth MJ, Ost TW, Beraldi D, Bell NM, Branco MR, Reik W, et al. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat Protoc. 2013;8:1841–51.
Song CX, Yi C, He C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat Biotechnol. 2012;30:1107–16.
Yu M, Hon GC, Szulwach KE, Song CX, Jin P, Ren B, et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc. 2012;7:2159–70.
Nazor KL, Boland MJ, Bibikova M, Klotzle B, Yu M, Glenn-Pratola VL, et al. Application of a low cost array-based technique—TAB-Array—for quantifying and mapping both 5mC and 5hmC at single base resolution in human pluripotent stem cells. Genomics. 2014;104:358–67.
Gkountela S, Zhang KX, Shafiq TA, Liao WW, Hargan-Calvopina J, Chen PY, et al. DNA Demethylation Dynamics in the Human Prenatal Germline. Cell. 2015;161:1425–36.
Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing. Hum Mol Genet. 2010;19:R227–40.
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–8.
Chen PY, Pellegrini M. Methylomes. In: Meyers RA (ed) Reviews in cell biology and molecular medicine. Hoboken, NJ: Wiley; 2012.
Roberts RJ, Carneiro MO, Schatz MC. The advantages of SMRT sequencing. Genome Biol. 2013;14:405.
Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, et al. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature. 2016;532:329–33.
Laszlo AH, Derrington IM, Brinkerhoff H, Langford KW, Nova IC, Samson JM, et al. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proc Natl Acad Sci USA. 2013;110:18904–9.
Rhoads A, Au KF. PacBio sequencing and its applications. Genomics Proteomics Bioinf. 2015;13:278–89.
Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 2012;13:705–19.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17;10–12. doi:10.14806/ej.17.1.200.
Benoukraf T, Wongphayak S, Hadi LH, Wu M, Soong R. GBSA: a comprehensive software for analysing whole genome bisulfite sequencing data. Nucleic Acids Res. 2013;41:e55.
Sun Z, Baheti S, Middha S, Kanwar R, Zhang Y, Li X, et al. SAAP-RRBS: streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing. Bioinformatics. 2012;28:2180–1.
Gao S, Zou D, Mao L, Zhou Q, Jia W, Huang Y, et al. SMAP: a streamlined methylation analysis pipeline for bisulfite sequencing. Gigascience. 2015;4:29.
Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–2.
Harris EY, Ponts N, Le Roch KG, Lonardi S. BRAT-BW: efficient and accurate mapping of bisulfite-treated reads. Bioinformatics. 2012;28:1795–6.
Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, et al. BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genom. 2013;14:774.
Pedersen B, Hsieh TF, Ibarra C, Fischer RL. MethylCoder: software pipeline for bisulfite-treated sequences. Bioinformatics. 2011;27:2435–6.
Wu TD, Reeder J, Lawrence M, Becker G, Brauer MJ. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. Methods Mol Biol. 2016;1418:283–334.
Kielbasa SM, Wan R, Sato K, Horton P, Frith MC. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011;21:487–93.
Sun Z, Cunningham J, Slager S, Kocher JP. Base resolution methylome profiling: considerations in platform selection, data preprocessing and analysis. Epigenomics. 2015;7:813–28.
Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinf. 2009;10:232.
Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, et al. The UCSC genome browser database: update 2011. Nucleic Acids Res. 2011;39:D876–82.
Liang F, Tang B, Wang Y, Wang J, Yu C, Chen X, et al. WBSA: web service for bisulfite sequencing data analysis. PLoS ONE. 2014;9:e86707.
Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
Mallona I, Díez-Villanueva A, Peinado MA. Methylation plotter: a web tool for dynamic visualization of DNA methylation data. Source Code Biol Med. 2014;9:11.
Hu K, Ting AH, Li J. BSPAT: a fast online tool for DNA methylation co-occurrence pattern analysis based on high-throughput bisulfite sequencing data. BMC Bioinformatics. 2015;16:220.
Liao WW, Yen MR, Ju E, Hsu FM, Lam L, Chen PY. MethGo: a comprehensive tool for analyzing whole-genome bisulfite sequencing data. BMC Genom. 2015;16(Suppl 12):S11.
Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83.
Choufani S, Shapiro JS, Susiarjo M, Butcher DT, Grafodatskaya D, Lou Y, et al. A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res. 2011;21:465–76.
Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87.
Park Y, Figueroa ME, Rozek LS, Sartor MA. MethylSig: a whole genome DNA methylation analysis pipeline. Bioinformatics. 2014;30:2414–22.
Kishore K, de Pretis S, Lister R, Morelli MJ, Bianchi V, Amati B, et al. methylPipe and compEpiTools: a suite of R packages for the integrative analysis of epigenomics data. BMC Bioinf. 2015;16:313.
Hebestreit K, Dugas M, Klein HU. Detection of significantly differentially methylated regions in targeted bisulfite sequencing data. Bioinformatics. 2013;29:1647–53.
Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, et al. Patterns of population epigenomic diversity. Nature. 2013;495:193–8.
Robinson MD, Kahraman A, Law CW, Lindsay H, Nowicka M, Weber LM, et al. Statistical methods for detecting differentially methylated loci and regions. Front Genet. 2014;5:324.
Kurdyukov S, Bullock M. DNA methylation analysis: choosing the right method. Biology. 2016;5:3. doi:10.3390/biology5010003.
Kader F, Ghai M. DNA methylation and application in forensic sciences. Forensic Sci Int. 2015;249:255–65.
Mikeska T, Craig JM. DNA methylation biomarkers: cancer and beyond. Genes. 2014;5:821–64.
Legendre C, Gooden GC, Johnson K, Martinez RA, Liang WS, Salhia B. Whole-genome bisulfite sequencing of cell-free DNA identifies signature associated with metastatic breast cancer. Clin Epigenetics. 2015;7:100.
Farkas SA, Milutin-Gašperov N, Grce M, Nilsson TK. Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics. 2013;8:1213–25.
Uribe-Lewis S, Stark R, Carroll T, Dunning MJ, Bachman M, Ito Y, et al. 5-hydroxymethylcytosine marks promoters in colon that resist DNA hypermethylation in cancer. Genome Biol. 2015;16:69.
Agardh E, Lundstig A, Perfilyev A, Volkov P, Freiburghaus T, Lindholm E, et al. Genome-wide analysis of DNA methylation in subjects with type 1 diabetes identifies epigenetic modifications associated with proliferative diabetic retinopathy. BMC Med. 2015;13:182.
Nygren AO, Dean J, Jensen TJ, Kruse S, Kwong W, van den Boom D, et al. Quantification of fetal DNA by use of methylation-based DNA discrimination. Clin Chem. 2010;56:1627–35.
Authors’ contributions
WSY and FMH drafted the manuscript; PYC coordinated the study and edited the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by a grant obtained from Academia Sinica and grants obtained from MOST-103-2313-B-001-003-MY3, MOST-103-2633-B-001-002 and NHRI-EX104-10324SC. FMH is supported by the Todai scholarship. We thank Dr. Matteo Pellegrini and Dr. Tzung-Fu Hsieh for reviewing the manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Wai-Shin Yong and Fei-Man Hsu contributed equally to this work
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yong, WS., Hsu, FM. & Chen, PY. Profiling genome-wide DNA methylation. Epigenetics & Chromatin 9, 26 (2016). https://doi.org/10.1186/s13072-016-0075-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13072-016-0075-3