
Abstract
Background
Schistosoma japonicum infection is an important public health problem, imposing heavy social and economic burdens in 78 countries worldwide. However, the mechanism of transition from chronic to advanced S. japonicum infection remains largely unknown. Evidences suggested that gut microbiota plays a role in the pathogenesis of S. japonicum infection. However, the composition of the gut microbiota in patients with chronic and advanced S. japonicum infection is not well defined. In this study, we compared the composition of the intestinal flora in patients with chronic and advanced S. japonicum infection.
Methods
The feces of 24 patients with chronic S. japonicum infection and five patients with advanced S. japonicum infection from the same area were collected according to standard procedures, and 16S rRNA sequencing technology was used to analyze the intestinal microbial composition of the two groups of patients.
Results
We found that alteration occurs in the gut microbiota between the groups of patients with chronic and advanced S. japonicum infections. Analysis of alpha and beta diversity indicated that the diversity and abundance of intestinal flora in patients with advanced S. japonicum infection were lower than those in patients with chronic S. japonicum infection. Furthermore, Prevotella 9, Subdoligranulum, Ruminococcus torques, Megamonas and Fusicatenibacter seemed to have potential to discriminate different stages of S. japonicum infection and to act as biomarkers for diagnosis. Function prediction analysis revealed that microbiota function in the chronic group was focused on translation and cell growth and death, while that in the advanced group was concentrated on elevating metabolism-related functions.
Conclusions
Our study demonstrated that alteration in gut microbiota in different stages of S. japonicum infection plays a potential role in the pathogenesis of transition from chronic to advanced S. japonicum infection. However, further validation in the clinic is needed, and the underlying mechanism requires further study.

Similar content being viewed by others


Background
Schistosoma japonicum infection is one of the most prevalent neglected tropical diseases, affecting more than 240 million people in 78 countries worldwide and claiming 250,000 lives annually [1]. This progressive and debilitating parasitic disease imposes a heavy social and economic burden on people in pandemic areas. There are five main species of Schistosoma that can infect humans, including S. japonicum, S. mansoni, S. haematobium, S. intercalatum and S. mekongi. However, S. japonicum is the only Schistosoma species responsible for human infection in China, particularly in 12 provinces south of the Yangtze River [2]. Although China has achieved great progress in controlling S. japonicum infection, reducing infected cases by > 99.0%, it remains a public health problem in about 140 counties.
Schistosoma japonicum infection occurs when people come in contact with contaminated water. It is classified into three distinct stages according to the disease progression, including acute, chronic and advanced S. japonicum infection [3]. Patients with advanced infection often suffer from many serious complications such as portal hypertension [4], ascites [5], splenomegaly [6], etc., which lead to physical weakness, loss of labor and even death [7]. Although advances have been achieved on the road to eliminating S. japonicum infection, it has several big challenges in the last mile to conquer S.japonicum infection. There is no effective vaccine in the clinic, and development of new drugs is needed as only praziquantel is available [8]. In addition, praziquantel seems ineffective at preventing chronic S. japonicum infection from developing into the advanced stage [9]. Also, the mechanism of transition from chronic to advanced stages is not well defined. Naturally, it is hard to evaluate the risk and perform prognostic analysis on this progress. Thus, further study is needed in this field, particularly on the underlying mechanism in the transition from chronic to advanced S. japonicum infection.
Evidence has shown that the gut microbiota plays an important role in maintaining human health [10] and is also involved in various diseases [11]. High-quality data from the US Human Microbiome Project (HMP) [12], European Metagenomics of the Human Intestinal Tract (MetaHIT) [13] and other studies have demonstrated that normal gut microbiota can influence human health by affecting gene expression. Evidences also showed that gut microbiota has an impact on human metabolism and immunity [14,15,16]. Particularly Bacteroides thetaiotaomicron can promote the efficiency of lipid hydrolysis [15], while members of the genus Bacteroides can reduce blood lipids and modulate immunity by synthesizing conjugated linoleic acid (CLA) [16]. The occurrence and progression of various diseases often lead to changes in the composition of the gut microbiota, and alterations in gut microbiota also impact the pathogenesis of different diseases [11]. Imbalance in the gut microbiota is often related to an increased risk of various diseases; for example, gut microbiota disrupted by antibiotics seems to increase the risk of new-onset Crohn's disease [17]. Also, alterations in gut microbial composition and diversity have often been observed in patients with gastric cancer, liver cancer and many other tumors [18]. As gut microbiota plays such a significant role, many studies have focused on their role in different diseases and potential in disease diagnosis and prognosis assessment [19]. Animal studies showed that gut microbiota can be used to facilitate the diagnosis of liver cirrhosis in rats with good accuracy [20]. A clinical study showed that the content of Faecali bacterium decreased in patients with bipolar disorder and was closely related to the severity of the disease [21]. In addition, the ratio of different species can also be used in the diagnosis of some diseases; for example, the ratio of Firmicutes/Bacteroidetes is often cited as a marker of obesity [22]. Recent studies have showed that acute S. japonicum infection leads to alteration in gut microbiome composition [23]. In contrast, the gut microbiota in patients with S. japonicum infection-induced liver cirrhosis is similar to that in healthy control groups. However, gut microbiota signatures in patients with chronic and advanced S. japonicum infection remain unclear, and the potential role of gut microbiota in the progression of S. japonicum infection is still unknown. Thus, profiling gut microbiota in different stages of S. japonicum infection will not only deepen our understanding of the mechanism of S. japonicum infection in the aspects of gut microbiome, but also shed light on the potential role of gut microbiota as a non-invasive biomarker for diagnosis as well as prognostic analysis of different stages of S. japonicum infection.
In this study, we use 16S ribosomal RNA gene sequencing to profile the gut microbiota in patients with chronic and advanced S. japonicum infection. Revealing the features of intestinal microbial community with S. japonicum in different stages will contribute to further understanding of the pathogenesis of S. japonicum infection developing from chronic to advanced stage and identifying potential non-invasive biomarkers for the diagnosis and prognosis of S. japonicum infection.
Methods
Study design
The study was conducted at the Third People's Hospital of Hanshou County in Dongting Lake area, Hunan Province, China, from February 2021 to February 2022. In this study, all patients (≥ 18 years old) with Schistosoma japonica infection visiting the Third People's Hospital of Hanshou County from February 2021 to February 2022 who agreed to participate in this study were included. According to the inclusion and exclusion criteria, 24 patients with chronic S. japonicum infection and five patients with advanced S. japonicum infection were included in this study. The fecal specimens and clinical information of the patients with S. japonicum infection in the hospital were collected.
Inclusion criteria for study subjects
According to the National Standardized Diagnostic Criteria for S. japonicum infection (WS261-2006) of the Ministry of Health of China, patients with advanced schistosomiasis should meet the following conditions.
-
(i)
History of repeated or prolonged exposure to water in infected areas.
-
(ii)
History of treatment for S. japonicum infection.
-
(iii)
Positive ovum test in the patient's stool or positive serum immunological test.
-
(iv)
Patients with liver fibrosis.
-
(v)
The patient has one or more complications of splenomegaly, hypersplenism, portal hypertension or ascites.
Exclusion criteria for study subjects
-
(i)
The patient was not currently in an acute infection state.
-
(ii)
The patient did not have a tumor.
-
(iii)
The patient was not infected with hepatitis A,B,C,D and E viruses.
-
(iv)
The patient was not infected with other parasites.
The basic information of the patients is shown in Table 1.
Sample collection
Study participants were trained by healthcare workers. Fresh stool samples were packed in sterile stool collection tubes. All specimens were snap-frozen in liquid nitrogen within half an hour of collection, transferred to sterile Eppendorf tubes and stored in a −80 °C freezer until DNA extraction was performed [24].
Sequencing
Extraction of genome DNA
Total genome DNA from samples was extracted using CTAB method [25]. Magnetic Soil and Stool DNA Kit (TIANGEN DP712) was used. DNA concentration and purity were monitored on 1.0% agarose gels. According to the concentration, DNA was diluted to 1 ng/μl using sterile water.
Amplicon generation
Primer 16S V3-V4: 341F (5′-CCTACGGGNGGCWGCAG-3′) and 806R (5′-GGACTACHVGGGTATCTAAT-3′) 16S rRNA genes were amplified using the specific primer with the barcode. All PCR reactions were carried out in 30-μl reactions with 15 μl Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM forward and reverse primers and about 10 ng template DNA. Thermal cycling consisted of initial denaturation at 98 ℃ for 1 min, followed by 30 cycles of denaturation at 98 ℃ for 10 s, annealing at 50 ℃ for 30 s and elongation at 72 ℃ for 60 s and finally 72 ℃ for 5 min [25].
PCR product quantification and qualification
The same volume of 1× loading buffer (contained SYB green) was mixed with PCR products and electrophoresis operated on 2.0% agarose gel for detection. Samples with a bright main strip between 400 and 450 bp were chosen for further experiments.
PCR product mixing and purification
PCR products were mixed in equidense ratios. Then, the mixture of PCR products was purified with AxyPrep DNA Gel Extraction Kit (AXYGEN).
Library preparation and sequencing
Sequencing libraries were generated using NEB Next® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA) following the manufacturer’s recommendations, and index codes were added. Finally, the library was sequenced on an Illumina Miseq/HiSeq 2500 platform, and 250 bp/300 bp paired-end reads were generated [26].
Data analysis
Paired-end read assemblies
Paired-end reads from the original DNA fragments were merged using FLASH (http://ccb.jhu.edu/software/FLASH/).
OTU cluster and species annotation
Sequences analyses were performed with UPARSE software package (http://drive5.com/uparse/) using the UPARSE-OTU and UPARSE-OUT ref algorithms. In-house Perl scripts were used to analyze alpha (within samples) and beta (among samples) diversity. Sequences with ≥ 97.0% similarity were assigned to the same OTUs [27]. We picked a representative sequence for each OTU and used the RDP classifier to annotate taxonomic information for each representative sequence.
Analysis
Chao1, ACE, Simpson and Shannon diversity indices were calculated by QIIME [28], and comparisons between groups were performed by t-test. Beta diversity analysis was visualized using PCA, PCoA and NMDS [20]. To confirm differences in the abundances of individual taxonomy between the two groups, STAMP software was utilized [29]. LEfSe (http://huttenhower.sph.harvard.edu/lefse/) was used for the quantitative analysis of biomarkers within different groups [30]. To identify differences of microbial communities between the two groups, ANOSIM was performed based on the Bray-Curtis dissimilarity distance matrices [31].
Results
Species composition analysis.
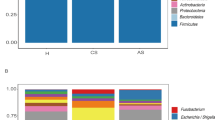
To explore the differences of gut microbiota between patients with chronic and advanced S. japonicum infection, we first analyzed the microbial composition of both groups of patients from three levels: phylum (Fig. 1a), genus (Fig. 1b) and species (Fig. 1c). We identified the top 10 species with differences at each level and their percentages. We found that at the phylum level, Bacteroidetes and Firmicutes were most abundant in both groups of patients. However, compared with patients with chronic S. japonicum infection, patients with advanced infection had a lower percentage of Firmicutes and a higher percentage of Proteobacteria (Fig. 1a). At the genus level, the percentages of Faecalis bacterium and Bacteroides were increased and the content of Prevolla 9 was significantly decreased in patients with advanced S. japonicum infection (Fig. 1b). Escherichia coli and Bacteroides fragilis were significantly increased in patients with advanced S. japonicum infection at species level (Fig. 1c). Thus, the gut microbiota structure altered when chronic S. japonicum infection progressed to advanced S. japonicum infection.

Species distribution of gut microbiota in patients with chronic and advanced schistosomiasis. Phylum level (a). Genus level (b). Species level (c). CS chronic S. japonicum infection, AS advanced S. japonicum infection
Analysis of alpha and beta diversity of gut microbiota
The results of Shannon index analysis showed that all samples were saturated (Additional file 1: Fig.S1), indicating that the amount of sequencing data was reasonable, the number of OTU species in the samples was sufficient, and the average abundance of different species reached the standard. For comparison of the bacterial community within groups, we used four metrics to analyze the data, including the Shannon(t-test,t(27) = 2.05, P = 0.013)(Fig. 2a), Simpson(t-test, t(27) = 2.05, P = 0.038) (Fig. 2b), Chao 1 (t-test, t(27) = 2.77, P = 0.0042) (Fig. 2c) and ACE (t-test, t(27) = 2.77, P = 0.0028) (Fig. 2d). All metrics suggested that there were significant differences in the bacterial community within groups. Patients with advanced S. japonicum infection had lower levels of microbial diversity and microbial abundance compared with patients with chronic S. japonicum infection. We further compared the bacterial community between the two groups using beta diversity analysis. Difference in beta diversity between patients with advanced and chronic S. japonicum infection was significant based on weighted Unifrac (Wilcoxon, Z = −2.44, P = 0.0063) (Fig. 3a). In addition, we performed PCA (Fig. 3b), PCoA (Fig. 3c) and NMDS analysis (Fig. 3d). To determine whether the grouping of this study was meaningful, we used ANOSIM analysis (based on the Bray-Curtis algorithm) to test whether the difference between groups was significantly greater than the difference within groups. ANOSIM analysis (based on the Bray-Curtis algorithm) demonstrated that the difference between the two groups of patients was greater than the difference within groups (R = 0.406; P = 0.011), and the statistical results were credible (Fig. 3e). Therefore, there are differences in the composition of gut microbiota between patients with advanced and chronic S. japonicum infection, and the diversity and abundance of intestinal flora in patients with advanced S. japonicum infection are lower than those in patients with chronic S. japonicum infection.

Alpha diversity analysis of intestinal flora in patients with chronic and advanced S. japonicum infection. The sequencing data were evaluated by four metrics: Shannon index (t-test, t(27) = 2.05, P = 0.013) (a); Simpson index (t-test, t(27) = 2.05, P = 0.038) (b); Chao 1 index (t-test, t(27) = 2.77, P = 0.0042) (c); ACE index (t-test, t(27) = 2.77, P = 0.0028) (d). CS chronic S. japonicum infection, AS advanced S. japonicum infection

Beta diversity of intestinal flora in patients with chronic and advanced S. japonicum infection was analyzed based on weighted UniFrac (Wilcoxon, Z = −2.44, P = 0.0063) (a). PCA (b), PCoA (c) and NMDS analysis (d) are shown. Anosim analysis (R = 0.406; P = 0.011) (e). CS, chronic S. japonicum infection, AS advanced S. japonicum infection, PCA principal component analysis, PcoA principal coordinate analysis, NMDS non-metric multidimensional scaling, ANOSIM analysis of similarities
Discovery of potential diagnostic biomarkers
To explore the species-specific differences in the gut microbiota between the groups of advanced and chronic S. japonicum infection and to find potential diagnostic markers, we next performed LEfSe analysis on the gut microbiota of the two groups of patients. The LEfSe analysis showed that there were differences in the intestinal flora between the two groups of patients at different species classification levels. At the family level, the dominant flora in patients with advanced S. japonicum infection was Bacteroidaceae; however, the Prevotellaceaein only existed in patients with chronic S. japonicum infection. At the genus level, the specific flora of the two groups of patients were Bacteroides and Prevotella 9 (Fig. 4a). In addition, we performed a stamp analysis on the intestinal flora of the two groups of patients at the genus level. The stamp analysis showed that the top five species with the highest percentage of differences between the two groups of patients were Prevotella 9, Subdoligranulum, Ruminococcus torques, Megamonas and Fusicatenibacter (P < 0.05) (Fig. 4b). According to our results, some specific species in the gut of patients with advanced and chronic S. japonicum infection are significantly different, and these species have potential to act as non-invasive biomarkers for differentiation of different stages of S. japonicum infection.

Species-specific differences in the gut microbiota between the groups. The histogram of LDA scores compared the two groups of patients with significant differences in the abundance of intestinal flora (a). The result of stamp analysis was shown (b). CS chronic S. japonicum infection, AS advanced S. japonicum infection, LDA linear discriminant analysis
Function prediction
To explore the functional differences, we used the KEGG database and COG database to analyze the gut microbiota in two groups of patients. KEGG functional prediction analysis is an effective way to study the metabolic function changes of community samples to adapt to environmental changes. First, we applied PCA analysis to reveal the similarities or differences in the microbiota function of different groups at the overall level. The results of KEGG (Fig. 5a) and COG (Fig. 5b) suggested that there were differences in the function of the microbiota between the groups of patients with advanced and chronic S. japonicum infection. We used LEfSe analysis to further explore specific functional differences between the two groups. According to the KEGG database, the function of patients with chronic S. japonicum infection is mainly concentrated in translation and cell growth and death. In the group of patients with advanced S. japonicum infection, functional alterations mainly occurred in the elevation of metabolism-related functions (Fig. 5c). COG database analysis suggested the functional changes in patients with chronic S. japonicum infection were manifested in translation ribosomal structure and biogenesis and cell cycle control, cell division and chromosome partitioning. The functional changes in gut microbiota in the group of patients with advanced S. japonicum infection were mainly manifested as inorganic ion transport, metabolism, RNA processing and modification (Fig. 5d). We further performed a stamp analysis (Fig. 6a and b) on the functional differences between the two groups of patients, and the top five results are shown in Table 2.

Prediction of intestinal flora function in patients with chronic and advanced S. japonicum infection. PCA analysis of two groups of patients based on the KEGG database (a). PCA analysis of two groups of patients based on the COG database (b). Histogram showing the difference in the abundance of gut bacterial function between the two groups of patients based on KEGG database (c). Histogram showing the difference in the abundance of gut bacterial function between the two groups of patients based on COG database (d). CS chronic S. japonicum infection, AS advanced S. japonicum infection, COG cluster of orthologous group, KEGG Kyoto Encyclopedia of Genes and Genomes

Stamp analysis on the functional differences between the two groups of patients. Result based on the KEGG database (a). Result based on the COG database (b). CS chronic S. japonicum infection, AS advanced S. japonicum infection, COG cluster of orthologous group, KEGG Kyoto Encyclopedia of Genes and Genomes
Discussion
Schistosoma. japonicum infection remains an important public health problem in the world, imposing a heavy social and economic burden on 78 countries in endemic areas. Big challenges must be overcomed to conquer this kind of parasitic disease as there is no effective vaccine and only praziquantel is used in the clinic. The mechanism of transition from chronic to advanced S. japonicum infection remains largely unknown and further study is needed. Evidences suggested that gut microbiota plays a role in the pathogenesis of S. japonicum infection and shows potential to act as a non-invasive biomarker in diagnosis and prognostic analysis. However, the composition of the gut microbiota in chronic and advanced S. japonicum infection is not clear. In this study, we for the first time compared the composition of the intestinal flora in patients with chronic and advanced S. japonicum infection. We found that alteration occurs in the gut microbiota between the groups of patients with chronic and advanced S. japonicum infection. Analysis of alpha diversity and beta diversity indicated that the diversity and abundance of intestinal flora in patients with advanced S. japonicum infection were lower than that in patients with chronic S. japonicum infection. Prevotella 9, Subdoligranulum, Ruminococcus torques, Megamonas and Fusicatenibacter could be biomarkers for diagnosis and help to discriminate different stages of S. japonicum infection. Besides, function prediction analysis revealed that microbiota function of the chronic group was focused on translation and cell growth and death, while that in the advanced group was concentrated on the elevation of metabolism-related functions. Our study demonstrated that alteration in gut microbiota in different stages of S. japonicum infection indicated a potential role of gut microbiota in the pathogenesis of transition from chronic to advanced S. japonicum infection. Furthermore, differently expressed bacteria seemed to have potential to facilitate differentiate diagnosis and prognosis analysis. However, further validation in the clinic is needed, and the underlying mechanism requires further study.
Gut microbiota alteration in different stages of S. japonicum infection was observed in our study. This was consistent with previous studies [24]. Numerous studies on liver disease have reported that it is closely related to gut microbes [32]. With the aggravation of liver disease, the alpha diversity of patients' gut microbiota decreased significantly [33]. This phenomenon was also observed in our study; patients with advanced S. japonicum infection had lower levels of gut microbiota alpha diversity than patients with chronic S. japonicum infection. Apart from alpha diversity, we also observed differences between individuals in each group. Although differences within groups will affect statistical analysis, ANOSIM analysis suggested that the difference between the two groups of patients was greater than the difference within the groups and the statistical results were reasonable and credible. In addition, we found that patients with advanced S. japonicum infection had higher beta diversity levels than patients with chronic S. japonicum infection. In another comparison between patients with S. japonicum infection-associated cirrhosis and healthy people, the researchers also found that patients with S. japonicum infection-associated cirrhosis had increased levels of beta diversity [34]. Although the different controls were chosen in our study, both studies demonstrate that S. japonicum infection and disease progression lead to increased beta diversity in the gut microbiota.
Due to the lack of effective diagnostic methods to differentiate patients with chronic S. japonicum infection from those with advanced S. japonicum infection in clinical practice, an important goal of our study was to explore whether there are potential biological targets for disease staging based on gut microbes. Our results showed that the content of Bacteroides in the intestine of patients with advanced S. japonicum infection was much higher than that of patients with chronic S. japonicum infection. Bacteroides is a double-edged sword to the human body [35]. Recent studies showed that elevated levels of Bacteroides can lead to the occurrence of colorectal tumors [36], consistent with the increased incidence of colorectal cancer in patients with advanced S. japonicum infection [37]. Thus, Bacteroides not only has potential to act as a staging marker for S. japonicum infection, but also could be a potential marker for cancer risk assessment. However more studies are needed to validate this function. The most abundant microbiota in people with chronic S. japonicum infection is Prevotella 9, which helps break down proteins and carbohydrates. It can also act as an opportunistic pathogen to induce intestinal [38] and vaginal inflammation [39]. Patients with chronic S. japonicum infection have been in a chronic inflammatory state for a long time. The increase in the content of Prevotella in the intestine is related to the reported changes in intestinal flora in other inflammatory states. Although numerous studies have provided sufficient evidences for its association with inflammation, currently there are insufficient data to confirm the causal relationship between Prevotella and the inflammatory state in patients with chronic S. japonicum infection.
After discovering the differences in intestinal flora between patients with chronic and advanced S. japonicum infection, we further explored the possible differences in physiological functions between the two groups of patients due to differences in gut microbes. The results of the KEGG and COG databases suggested that metabolism-related gene expression changed significantly in patients with advanced S. japonicum infection, while in patients with chronic S. japonicum infection, it was mainly the expression of cell cycle and cell death-related genes, consistent with the development of S. japonicum infection. The liver and intestinal tissues of patients with chronic S. japonicum infection are damaged [3]. However, organ function can still be compensated, so the function of patients during this period is mainly focused on tissue damage, repair and cell death. As the disease progresses, the patient will transit into advanced stage. When many tissues and cells within the liver and intestinal tract develop necrosis or apoptosis, tissue fibrosis occurs [40]. The liver and intestine are the most important digestive and metabolic organs, also as the target organs of S. japonicum infection. When the damage is too serious, it leads to disorders of the metabolism and immune system [41]. This also explains the increased incidence of metabolic and infectious diseases in patients with advanced S. japonicum infection. When the disease continues to progress, due to insufficient energy supply and application, metabolic disorders occur, and then patients become weaker, gradually lose their ability to work [42] and even die.
In recent years, because of the efficient and standardized treatment for patients with S. japonicum infection in China, new cases of S. japonicum infection have been dramatically reduced. The infection in many chronic patients has been stopped from progressing to advanced stage; meanwhile, the number of patients with advanced S. japonicum infection has significantly decreased [43]. Many of the patients with pneumonia, cholecystitis or hepatitis B were excluded, as these diseases affect the composition of the gut microbiota [44,45,46]. Therefore, few patients with advanced S. japonicum infection were included in this study. Although the limited patient number in the advanced group was a weakness of our study, ANOSIM analysis suggested that the within-group difference was acceptable and the statistical results reliable. Of course, an increased number of patients in advanced group would definitely improve the study, and more validation in the future is needed. Although we found some microbiota with potential as biomarkers to distinguish patients with chronic S. japonicum infection from those with advanced S. japonicum infection, more research is needed to clarify the causal relationship between different species and disease progression. The functional changes caused by alterations in the gut microbiota also need further verification in the clinic.
Conclusions
By using 16S rRNA sequencing technology, our study found that there were differences in gut microbiota between patients with chronic and advanced S. japonicum infection. Patients with advanced S. japonicum infection had lower alpha and higher beta diversity when compared with patients with chronic S. japonicum infection. The proportion of Bacteroides in the intestine of patients with advanced schistosomiasis was higher, while the content of Prevotella, especially Prevotella 9, was relatively abundant in patients with chronic S. japonicum infection. Thus, Prevotella 9 has the potential to become a biomarker to distinguish patients with chronic S. japonicum infection from patients with advanced S. japonicum infection. The functions of flora in patients with chronic S. japonicum infection are mainly focused on the cell cycle and cell death, while those in patients with advanced S. japonicum infection are mainly focused on metabolism.
Availability of data and materials
Data supporting the conclusions of this article are included within the article.
Abbreviations
- ANOSIM:
-
Analysis of similarities
- 16S rRNA:
-
16S ribosomal ribonucleic acid
- COG:
-
Cluster of orthologous group
- CTAB:
-
Cetyltrimethylammonium bromide
- DNA:
-
Deoxyribonucleicacid
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LDA:
-
Linear discriminant analysis
- LEfSe:
-
Linear discriminant analysis effect size
- NMDS:
-
Non-metric multidimensional scaling
- OUT:
-
Operational taxonomic unit
- PCR:
-
Polymerase chain reaction
- PCA:
-
Principal component analysis
- PCoA:
-
Principal coordinate analysis
- S. japonicum :
-
Schistosoma japonicum
References
Song LG, Wu XY, Sacko M, Wu ZD. History of schistosomiasis epidemiology, current status, and challenges in China: on the road to schistosomiasis elimination. Parasitol Res. 2016;115:4071–81. https://doi.org/10.1007/s00436-016-5253-5.
Chen C, Guo Q, Fu Z, Liu J, Lin J, Xiao K, et al. Reviews and advances in diagnostic research on Schistosoma japonicum. Acta Trop. 2021;213:105743. https://doi.org/10.1016/j.actatropica.2020.105743.
Gryseels B, Polman K, Clerinx J, Kestens L. Human schistosomiasis. Lancet. 2006;368:1106–18. https://doi.org/10.1016/s0140-6736(06)69440-3.
Klotz F. Portal hypertension and schistosomiasis: “an originally killing entity.” Bull Soc Pathol Exot. 2003;96:191–5.
Xiao-Rong Z, Xiao-Wei S, Li Z, Guo L, Si L. Study on effect of clinical pathway implementation in advanced schistosomiasis with ascites. Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi. 2018;30:278–81. https://doi.org/10.16250/j.32.1374.2017235.
Kong D, Zhou C, Guo H, Wang W, Qiu J, Liu X, et al. Praziquantel targets M1 macrophages and ameliorates splenomegaly in chronic schistosomiasis. Antimicrob Agents Chemother. 2018. https://doi.org/10.1128/aac.00005-17.
Alirol E, Getaz L, Stoll B, Chappuis F, Loutan L. Urbanisation and infectious diseases in a globalised world. Lancet Infect Dis. 2011;11:131–41. https://doi.org/10.1016/s1473-3099(10)70223-1.
Trainor-Moss S, Mutapi F. Schistosomiasis therapeutics: whats in the pipeline? Expert Rev Clin Pharmacol. 2016;9:157–60. https://doi.org/10.1586/17512433.2015.1102051.
Hu F, Xie SY, Yuan M, Li YF, Li ZJ, Gao ZL, et al. The dynamics of hepatic fibrosis related to schistosomiasis and its risk factors in a cohort of China. Pathogens. 2021. https://doi.org/10.3390/pathogens10121532.
Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Nageshwar RD. Role of the normal gut microbiota. World J Gastroenterol. 2015;21:8787–803. https://doi.org/10.3748/wjg.v21.i29.8787.
Sidhu M, van der Poorten D. The gut microbiome. Aust Fam Physician. 2017;46:206–11.
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14. https://doi.org/10.1038/nature11234.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. https://doi.org/10.1038/nature08821.
Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307:1955–9. https://doi.org/10.1126/science.1109051.
Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–4. https://doi.org/10.1126/science.291.5505.881.
Devillard E, McIntosh FM, Paillard D, Thomas NA, Shingfield KJ, Wallace RJ. Differences between human subjects in the composition of the faecal bacterial community and faecal metabolism of linoleic acid. Microbiology (Reading). 2009;155:513–20. https://doi.org/10.1099/mic.0.023416-0.
Margolis DJ, Fanelli M, Hoffstad O, Lewis JD. Potential association between the oral tetracycline class of antimicrobials used to treat acne and inflammatory bowel disease. Am J Gastroenterol. 2010;105:2610–6. https://doi.org/10.1038/ajg.2010.303.
Meng C, Bai C, Brown TD, Hood LE, Tian Q. Human gut microbiota and gastrointestinal cancer. Genomics Proteomics Bioinformatics. 2018;16:33–49. https://doi.org/10.1016/j.gpb.2017.06.002.
Erdrich S, Hawrelak JA, Myers SP, Harnett JE. Determining the association between fibromyalgia, the gut microbiome and its biomarkers: a systematic review. BMC Musculoskelet Disord. 2020;21:181. https://doi.org/10.1186/s12891-020-03201-9.
Li Z, Ni M, Yu H, Wang L, Zhou X, Chen T, et al. Gut microbiota and liver fibrosis: one potential biomarker for predicting liver fibrosis. Biomed Res Int. 2020;2020:3905130. https://doi.org/10.1155/2020/3905130.
Lucidi L, Pettorruso M, Vellante F, Di Carlo F, Ceci F, Santovito MC, et al. Gut microbiota and bipolar disorder: an overview on a novel biomarker for diagnosis and treatment. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22073723.
Magne F, Gotteland M, Gauthier L, Zazueta A, Pesoa S, Navarrete P, et al. The Firmicutes/Bacteroidetes ratio: a relevant marker of gut dysbiosis in obese patients? Nutrients. 2020. https://doi.org/10.3390/nu12051474.
Jiang Y, Yuan Z, Shen Y, Rosa BA, Martin J, Cao S, et al. Alteration of the fecal microbiota in Chinese patients with Schistosoma japonicum infection. Parasite. 2021;28:1. https://doi.org/10.1051/parasite/2020074.
Hu Y, Chen J, Xu Y, Zhou H, Huang P, Ma Y, et al. Alterations of gut microbiome and metabolite profiling in mice infected by Schistosoma japonicum. Front Immunol. 2020;11:569727. https://doi.org/10.3389/fimmu.2020.569727.
Wang D, He Y, Liu K, Deng S, Fan Y, Liu Y. Sodium humate alleviates enterotoxigenic Escherichia coli-induced intestinal dysfunction via alteration of intestinal microbiota and metabolites in mice. Front Microbiol. 2022;13:809086. https://doi.org/10.3389/fmicb.2022.809086.
Hu J, Wang C, Huang X, Yi S, Pan S, Zhang Y, et al. Gut microbiota-mediated secondary bile acids regulate dendritic cells to attenuate autoimmune uveitis through TGR5 signaling. Cell Rep. 2021;36:109726. https://doi.org/10.1016/j.celrep.2021.109726.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8. https://doi.org/10.1038/nmeth.2604.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. https://doi.org/10.1038/nmeth.f.303.
Zhu Q, Jiang S, Du G. Effects of exercise frequency on the gut microbiota in elderly individuals. Microbiologyopen. 2020;9:e1053. https://doi.org/10.1002/mbo3.1053.
Wang LJ, Yang CY, Chou WJ, Lee MJ, Chou MC, Kuo HC, et al. Gut microbiota and dietary patterns in children with attention-deficit/hyperactivity disorder. Eur Child Adolesc Psychiatry. 2020;29:287–97. https://doi.org/10.1007/s00787-019-01352-2.
Chang SC, Lin SF, Chen ST, Chang PY, Yeh YM, Lo FS, et al. Alterations of gut microbiota in patients with graves’ disease. Front Cell Infect Microbiol. 2021;11:663131. https://doi.org/10.3389/fcimb.2021.663131.
Acharya C, Bajaj JS. Gut microbiota and complications of liver disease. Gastroenterol Clin North Am. 2017;46:155–69. https://doi.org/10.1016/j.gtc.2016.09.013.
Oh TG, Kim SM, Caussy C, Fu T, Guo J, Bassirian S, et al. A universal gut-microbiome-derived signature predicts cirrhosis. Cell Metab. 2020;32:878-88.e6. https://doi.org/10.1016/j.cmet.2020.06.005.
Gui QF, Jin HL, Zhu F, Lu HF, Zhang Q, Xu J, et al. Gut microbiota signatures in Schistosoma japonicum infection-induced liver cirrhosis patients: a case-control study. Infect Dis Poverty. 2021;10:43. https://doi.org/10.1186/s40249-021-00821-8.
Zafar H, Saier MH Jr. Gut Bacteroides species in health and disease. Gut Microbes. 2021;13:1–20. https://doi.org/10.1080/19490976.2020.1848158.
Zamani S, Taslimi R, Sarabi A, Jasemi S, Sechi LA, Feizabadi MM. Enterotoxigenic Bacteroides fragilis: a possible etiological candidate for bacterially-induced colorectal precancerous and cancerous lesions. Front Cell Infect Microbiol. 2019;9:449. https://doi.org/10.3389/fcimb.2019.00449.
Almoghrabi A, Mzaik O, Attar B. Schistosoma japonicum associated with colorectal cancer. ACG Case Rep J. 2021;8:e00572. https://doi.org/10.14309/crj.0000000000000572.
Iljazovic A, Roy U, Gálvez EJC, Lesker TR, Zhao B, Gronow A, et al. Perturbation of the gut microbiome by Prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal Immunol. 2021;14:113–24. https://doi.org/10.1038/s41385-020-0296-4.
Randis TM, Ratner AJ. Gardnerella and Prevotella: co-conspirators in the pathogenesis of bacterial vaginosis. J Infect Dis. 2019;220:1085–8. https://doi.org/10.1093/infdis/jiy705.
Jia TW, Utzinger J, Deng Y, Yang K, Li YY, Zhu JH, et al. Quantifying quality of life and disability of patients with advanced Schistosomiasis japonica. PLoS Negl Trop Dis. 2011;5:e966. https://doi.org/10.1371/journal.pntd.0000966.
Gobert GN, Burke ML, McManus DP, Ellis MK, Chuah C, Ramm GA, et al. Transcriptional profiling of chronic clinical hepatic schistosomiasis japonica indicates reduced metabolism and immune responses. Parasitology. 2015;142:1453–68. https://doi.org/10.1017/s0031182015000682.
Wu XH, Wang TP, Lu DB, Hu HT, Gao ZB, Zhu CG, et al. Studies of impact on physical fitness and working capacity of patients with advanced Schistosomiasis japonica in Susong County, Anhui Province. Acta Trop. 2002;82:247–52. https://doi.org/10.1016/s0001-706x(02)00016-5.
Wu W, Feng A, Huang Y. Research and control of advanced Schistosomiasis japonica in China. Parasitol Res. 2015;114:17–27. https://doi.org/10.1007/s00436-014-4225-x.
Wu T, Xu F, Su C, Li H, Lv N, Liu Y, et al. Alterations in the gut microbiome and Cecal Metabolome during Klebsiella pneumoniae-induced pneumosepsis. Front Immunol. 2020;11:1331. https://doi.org/10.3389/fimmu.2020.01331.
Grigor’eva IN, Romanova TI. Gallstone disease and microbiome. Microorganisms. 2020. https://doi.org/10.3390/microorganisms8060835.
Neag MA, Mitre AO, Catinean A, Buzoianu AD. Overview of the microbiota in the gut-liver axis in viral B and C hepatitis. World J Gastroenterol. 2021;27:7446–61. https://doi.org/10.3748/wjg.v27.i43.7446.
Acknowledgements
We thank Professor Guanghui Ren for his insightful and thoughtful suggestions on this manuscript and APTBIO for genome-sequencing services.
Funding
This work was supported by National Natural Science Foundation of China (grant number 81771722 to Y Ming) and the Key Research and Development Plan of Hunan Province (grant number 2021SK2032 to Y Ming).
Author information
Authors and Affiliations
Contributions
The experiment was conceived and designed by CZ, JL and YM. The patient recruitment and screening were completed by YC and JZ. The biological specimen processing was completed by CG and ZZ. The data were processed by ZY, YZ and JJ. The article was written by CZ, JL and YM. YM provided financial support for the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The work has been carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments. All experiments in this study were approved by the Clinical Research Ethics Committee of the Third Xiangya Hospital of Central South University. All participants signed an informed consent form, and patient privacy was fully protected.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Shannon curve showed that all samples were saturated.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, C., Li, J., Guo, C. et al. Comparison of intestinal flora between patients with chronic and advanced Schistosoma japonicum infection. Parasites Vectors 15, 413 (2022). https://doi.org/10.1186/s13071-022-05539-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05539-6