
Abstract
Background
The Plasmodium zygote-to-ookinete developmental transition is an essential step for establishing an infection in the mosquito vector, and antigens expressed during this stage are potential targets for transmission-blocking vaccines (TBVs). The secreted ookinete protein 26 (PSOP26) is a newly identified ookinete surface protein. The anti-PSOP26 serum has moderate transmission-blocking activity, indicating the benefit of further investigating this protein as a target for TBVs.
Methods
The function of psop26 was analyzed by targeted gene disruption. A chimeric PSOP25-PSOP26 protein was expressed in the Escherichia coli system. The PSOP25-PSOP26 fusion protein, along with mixed (PSOP25 + PSOP26) or single proteins (PSOP26 or PSOP25), were used for the immunization of mice. The antibody titers and immunogenicity of individual sera were analyzed by enzyme-linked immunoassay (ELISA), indirect immunofluorescence assay (IFA), and Western blot. The transmission-blocking activity of sera from different immunization schemes was assessed using in vitro and in vivo assays.
Results
PSOP26 is a surface protein expressed in Plasmodium gametes and ookinetes. The protein is dispensable for asexual blood-stage development, gametogenesis, and zygote formation, but is essential for the zygote-to-ookinete developmental transition. Specifically, both the prevalence of infections and oocyst densities were decreased in mosquitoes fed on psop26-null mutants. Mixtures of individual PSOP25 and PSOP26 fragments (PSOP25 + PSOP26), as well as chimeras (PSOP25-PSOP26), elicited high antibody levels in mice, with no immunological interference. Antisera against the mixed and fusion proteins elicited higher transmission-reducing activity (TRA) than antisera against the single PSOP26 antigen, but comparable to antisera against PSOP25 antigen alone.
Conclusions
PSOP26 plays a critical role in the zygote-to-ookinete developmental transition. PSOP25 is a promising TBV candidate that could be used alone to target the ookinete stage.
Similar content being viewed by others

Background
Malaria deaths in Africa have decreased by 44% since 2000, but progress has slowed in recent years (WHO, 2020). This may be due to factors such as shortfalls in funding, resistance to widely used insecticides and anti-malaria drugs, and region-specific inadequacies in health systems [1]. Therefore, new technologies to eliminate malaria from all areas are urgently needed, which can be developed in part by a better understanding of disease transmission.
The parasite sexual stages mediate transmission to mosquitoes, and thereby the spread of malaria. Gametes fertilize in the mosquito midgut to form zygotes, which develop into motile invasive ookinetes that traverse the midgut epithelium to differentiate into oocysts containing hundreds of sporozoites. Following release from oocysts, sporozoites pass into the salivary glands, in wait for transmission to a new vertebrate host during the next blood meal by the infected mosquito. The zygote-to-ookinete developmental transition and the following oocyst development are essential for the malaria parasite to complete its transmission life cycle. Antigens expressed during these key phases have been analyzed extensively; such as the major ookinete surface proteins P25 and P28 [2], secreted ookinete adhesive protein (SOAP) [3], the Plasmodium perforin-like protein (PPLP) family [4], and the putative secreted ookinete proteins (PSOPs) [5, 6]. PSOPs have been implicated to play roles in the zygote-to-ookinete and ookinete-to-oocyst developmental transitions; for example, psop2- and psop25-null mutants are defective for in vivo ookinete development, PIMMS2/57/22 promotes ookinete invasion [7, 8], pimms1/57/22-null mutants are defective for the ookinete-to-oocyst transition, and pimms22-null sporozoites are unable to initiate transmission [8]. Together, these results highlight the role of PSOPs in malaria parasite transmission.
Vaccines designed to induce antibodies that interrupt malaria transmission by targeting sexual stages are referred to as transmission-blocking vaccines (TBVs). Instead of providing direct protection against infection or disease in the human host, these vaccines are intended to prevent parasite transmission from an infected human to a feeding mosquito. At present, a broad range of antibodies against sexual-stage proteins have been screened for transmission-blocking activity (TBA). Among them, P48/45, P230, and P25/28 are leading candidates for vaccines preventing malaria parasite transmission to mosquitoes [9,10,11]. Pfs230 and Pfs48/45 are pre-fertilization TBV candidates, while the P25/28 pair are post-fertilization TBV candidates which are expressed on the surface of ookinetes. The P25/28 antigens exhibit promising TBA in animal studies, but when formulated with alum as an adjuvant, both Plasmodium falciparum and Plasmodium vivax P25 showed poor immunogenicity and no transmission-blocking effect in a human trial [12, 13]. With the use of Montanide ISA 51 as an adjuvant, Pfs25 and Pvs25 were able to induce transmission-blocking immunity in humans, but the higher formulated dose used for immunization was associated with the risk of systemic adverse events, such as erythema nodosum and leukopenia [14]. A clinical trial for Pfs25-IMX313 proved its safety and tolerance for malaria-naïve adults, but this vaccine exhibits weak TBA [15]. Another clinical trial carried out in the United States and Mali using Pfs25-ExoProtein A (EPA) formulated with Alhydrogel™ showed that four doses are required to generate antibodies that possess effective TBA, and the protective antibody titers decreased rapidly after the final immunization [16, 17]. Further, Pfs25-EPA combined with Pfs230D1-EPA did not increase activity over Pfs230D1-EPA alone [18]. These studies highlight that alternative antigens or combinations should be assessed to improve TBA.
Several studies have shown that TBA can be improved by taking advantage of the synergy between two antigens, such as Pfs25 and Pfs28 [19]. However, mixed or fusion antigens carry the risk of immunological interference between antigens, which could lead to a reduced immune response to one or both of the components. Such a case was observed for MSP1 and AMA1, in which a lower antibody titer of AMA1 was observed as compared to AMA1 administered alone [20]. In another example, although dual-antigen vaccines of Pfs25 fused or mixed with Pfs28 or Pfs230C did not cause immunological interference, they could not improve the transmission-reducing activity (TRA) compared to mono-antigen vaccines [21]. Collectively, these findings suggest that the TBA may be dependent on specific antigens used in the combinations.
Our previous study revealed two ookinete surface proteins, PSOP25 and PSOP26, as new targets for transmission-blocking interventions. Mice immunized with either recombinant PSOP25 or PSOP26 resulted in sera which in mosquito feeding assays provoked a moderate reduction of oocyst density (60.0–70.7%) and prevalence of infected mosquitoes (20.1–37.4%) [6, 22]. Since neither PSOP25 nor PSOP26 antigen alone could produce 100% TBA efficacy, functional mapping and testing of the combination of these two surface antigens to improve TBA is warranted. Thus, in this study, we characterized the function of PSOP26 protein during sexual-stage development and evaluated the fusion and mixed forms of PSOP26 and PSOP25 antigen compared to mono-antigens in terms of immunogenicity and TRA.
Methods
Mice, parasites, and mosquitoes
The Plasmodium berghei ANKA 2.34 line was used for genome editing. All parasite lines were maintained in 6-week-old female BALB/c mice (Beijing Animal Institute, Beijing, China). Plasmodium berghei schizonts, gametocytes, zygotes, and ookinetes were cultured and purified as described previously [22]. Anopheles stephensi mosquitoes of the Hor strain were fed on a 10% (w/v) glucose solution, maintained at 25 °C with 50–80% humidity, and a 12/12 h light/dark cycle. All animal procedures were performed in accordance with the welfare and ethical review standards of China Medical University.
Quantitative real-time polymerase chain reaction (qRT-PCR)
To determine the transcription profile of the psop26 gene (PlasmoDB ID: PBANKA_1457700), total RNA was isolated from purified P. berghei schizonts, gametocytes, and ookinetes parasites using an RNeasy Plus Universal Kit (Qiagen, Dusseldorf, Germany) followed by DNase I (Invitrogen, Waltham, MA, USA) treatment to remove genomic (gDNA). Complementary DNA (cDNA) was synthesized using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher, Waltham, MA, USA). qRT-PCR analysis was conducted using PrimeScript™ RT Master Mix (Perfect Real Time, Thermo Fisher) and gene-specific primers according to the kit protocol. Gene expression of psop26 was normalized against β-tubulin (PBANKA_120690) in P. berghei using the 2−ΔΔCt method (see Additional file 1: Table S1 for primer sequences). Analysis was conducted using a 7500 Fast PCR System (Thermo Fisher).
Generation of transgenic parasites
The strategy for generating transgenic parasite lines involved electroporation of gene target vectors into purified schizonts, followed by infection of mice by intravenous injection as described previously [23]. To tag the endogenous psop26 with a 3 × HA tag, a 769-base-pair (bp) fragment [nucleotide (nt) positions 1554–2322 bp] of the psop26 gene was amplified from P. berghei genomic DNA gDNA and ligated into the ApaI and SacII sites of the pL0034 plasmid as the 5′ homologous region (5R). Then, 526 bp of the psop26 3′ untranslated region (UTR) (nt + 1– + 526 bp) was amplified and inserted between the KpnI and EcoRI sites as the 3′ homologous region (3R), to yield the final plasmid pL0034-PSOP26-3 × HA. To disrupt the psop26 gene, a ∆psop26 transgenic parasite was generated using the PbGEM-325763 plasmid (kindly provided by PlasmoGEM; http://plasmogen.sanger.ac.uk/). The linearized plasmid (10 µg) was transfected into purified P. berghei schizonts using a Nucleofector II device (Lonza, Basel, Switzerland) and the Basic Parasite Nucleofector® kit (Lonza). The transfected parasites were injected into BALB/c mice via the tail vein and selected using pyrimethamine (70 µg/ml, Sigma-Aldrich, St. Louis, MO, USA) in the drinking water. Parasite gDNAs were isolated from blood-stage parasites using a DNeasy Blood kit (Qiagen), and the correct integration of the 5′ and 3′ homologous regions was detected using genotype PCR analysis. All primers used in this study are listed in Additional file 1, Table S1. Parasite clones with targeted modifications were obtained after limiting dilution. At least two clones for each gene-modified parasite were used for phenotype analysis.
Western blot analysis
For stage-specific protein expression assays, purified schizonts, gametocytes, zygotes, and ookinetes were treated with 0.15% saponin (Sigma) in phosphate-buffered saline (PBS) for 10 min on ice, followed by three washes with PBS containing 1× protease inhibitor cocktail (PBS-PI, Thermo Fisher) to remove hemoglobin. To detect the expression of recombinant proteins in transgenic parasites, mixed stages of wild-type (WT) and transgenic parasites were directly lysed using 0.15% saponin. Protein samples were boiled and separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions and transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA). The PVDF membranes were blocked with 5% skim milk and then probed using mouse anti-HA monoclonal antibody (mAb, 1:1000; Invitrogen, Carlsbad, CA) and anti-rHSP70 sera (1:2000; PlasmoDB ID: PBANKA_0711900, produced in our laboratory). After washing three times, the membranes were probed with horseradish peroxidase (HRP)-conjugated goat anti-mouse immunoglobulin G (IgG) antibodies (1:5000, Thermo Fisher). The blots were then detected using an ECL Western Blotting Kit (Pierce, Rockford, IL, USA).
Indirect immunofluorescence assay (IFA)
The subcellular localization of PSOP26 and PSOP25 was analyzed by IFA as described previously [24]. The HA-tagged transgenic or WT P. berghei parasites were fixed with 4% paraformaldehyde and 0.0075% glutaraldehyde (Sigma) in PBS for 30 min at room temperature (RT). Cells were either permeabilized with 0.1% v/v Triton X-100 or directly processed without permeabilization. Then the parasites were neutralized with 0.1 mg/ml of sodium borohydride and blocked in 3% w/v BSA (diluted in PBS). Parasites were then stained with anti-HA mAb (1:500, Invitrogen) or antisera against PSOP26 or PSOP25. Parasites were co-labeled with rabbit antisera against PbMSP1 (1:500), Pbg377 (1:500), α-tubulin II (1:500), or PSOP25 (1:500) as stage-specific markers for schizonts, female gametocytes/gametes, male gametocytes/gametes, and zygotes/ookinetes, respectively. The non-permeabilized parasites were treated with 0.1% v/v Triton X-100 before labeling with these stage-specific markers. Alexa Fluor 488-conjugated goat anti-mouse IgG antibodies (1:500; Invitrogen) and Alexa Fluor 555-conjugated goat anti-rabbit IgG antibodies (1:500; Abcam, Cambridge, UK) were used as secondary antibodies. Parasite nuclei were stained with Hoechst 33258 (Invitrogen) at a final concentration of 5 μg/ml. Negative controls were HA-tagged ookinetes incubated with the secondary antibodies alone, or WT ookinetes incubated with the anti-HA mAb. The antibodies against PbMSP1, Pbg377, α-tubulin II, and PSOP25 were made in our laboratory [25]. Parasites were visualized using a Nikon C2 fluorescence confocal laser scanning microscope (Nikon, Tokyo, Japan).
Phenotypic analysis of the ∆psop26 transgenic parasite
To determine the function of PSOP26 in the P. berghei life cycle, 5 × 106 P. berghei wild-type (WT) or ∆psop26 infected red blood cells (iRBCs) were intravenously administered to mice pretreated with 6 mg/ml phenylhydrazine (PHZ). Parasitemia and mortality of the infected mice were monitored daily. To assess the function of parasite sexual-stage development, gametocytemias (mature gametocytes per 104 RBCs) and gametocyte sex ratios (female to male gametocyte ratio) were counted at 3 days post-infection (dpi). Exflagellation centers and male–female gamete interactions were assessed as described previously [26]. Briefly, 10 μl of infected blood containing equal numbers of mature gametocytes was added to 40 μl ookinete medium (RPMI 1640, 50 mg/l penicillin, 50 mg/l streptomycin, 20% [v/v] FCS, 6 U/ml heparin, pH 8.0) for 15 min at 25 °C; then 1 μl of the culture mixture was smeared onto a glass slide (Matsunami Glass Ind., Ltd., Japan) and exflagellation centers (an exflagellating male gametocyte with more than four adhered RBCs) were counted using a light microscope. The male–female gamete interactions were calculated as male gametes attached to female gametes for more than 3 s in 10 fields within 20 min [27]. To count the number of macrogametes, 10 μl of infected blood was mixed with 90 μl ookinete medium for 15 min at 25 °C; then 0.5 μl of the mixture was placed on a slide, fixed, and labeled with anti-Pbs21 mAb (clone 13.1, 1:500, a gift from Dr. Hiroyuki Matsuoka from Jichi Medical University, Japan), and Pbs21-positive macrogametes were counted using a fluorescence microscope. The culture was subsequently incubated at 19 °C for 2 h and 24 h, labeled with anti-Pbs21 mAb (1:500), and the numbers of zygotes and ookinetes, respectively, in 0.5 μl of the cultures were counted using a fluorescence microscope. For oocyst quantification, at 3 dpi, WT or ∆psop26 parasite-infected mice were fed to starved female anopheline mosquitoes for 1 h. Ten days after feeding, ~ 30 fed mosquitoes from each group were dissected for counting the oocyst numbers per infected mosquitoes and to determine the prevalence and intensity of infection.
Recombinant protein expression and immunization
To generate a chimeric PSOP25 and PSOP26 protein (PSOP25-PSOP26), gene fragments of PSOP25 [45–245 amino acids (aa)] and PSOP26 (50–254 aa) were fused with a flexible linker (GGGGS)3 between the two sequences by overlapping PCR using primers described in Additional file 1: Table S1. The PCR products were cloned into the vector pET-32a (+) (Novagen, Darmstadt, Germany). Recombinant proteins were expressed in E. coli Rosetta-gami B (DE3) cells under induction with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG; Sigma) at 19 °C for 16 h. The His-tagged proteins were affinity purified using Ni–NTA His-Bind Superflow resin (Novagen), followed by dialysis in 0.1 M PBS at 4 °C overnight. Purified recombinant proteins were cleaved by enterokinases at 25 °C for 16 h, and further purified with Ni–NTA His-Bind Superflow resin. The final recombinant proteins were analyzed by 10% SDS-PAGE to determine purity.
For the generation of serum against recombinant PSOP25, PSOP26, PSOP25 + PSOP26, and PSOP25-PSOP26, five female BALB/c mice for each group were immunized subcutaneously with the emulsified product of recombinant protein (50 μg per mouse for PSOP25 and PSOP26, and PSOP25-PSOP26 groups; 50 µg PSOP25 + 50 µg PSOP26 per mice for the PSOP25 + PSOP26 group) and complete Freund’s adjuvant (Sigma). The immunizations were enhanced at weeks 2 and 4 with recombinant proteins emulsified in incomplete Freund’s adjuvant (Sigma). At 14 days after the final immunization, blood was collected by cardiac puncture and agglutinated at room temperature to obtain antisera, and stored at −80 °C for the subsequent trials.
Enzyme-linked immunosorbent assay (ELISA)
Serum antibody titers were measured by ELISA as described previously [28]. The 96-well plates were coated with 5 μg/ml recombinant PSOP25 or PSOP26 in 0.05 M sodium carbonate buffer (pH 9.6) at 4 °C overnight. The plates were then washed three times with 200 μl PBS containing 0.02% Tween-20 (PBS-T) and blocked with 1% bovine serum albumin (Sigma) for 1 h at 37 °C. After washing an additional three times with PBS-T, 100 μl serial dilutions of antisera (from 1:200 to 1:25,600) were added to the 96-well plates and incubated at 37 °C for 2 h. After three washes, HRP-conjugated goat anti-mouse IgG antibodies (1:5000 dilutions) were added to the plates and incubated for 1 h at 37 °C. After six washes, 100 μl of tetramethylbenzidine (TMB, Sigma) was added and incubated in the dark for 5 min. The reaction was stopped with 50 μl of 1 mM H2SO4, and the absorbance at 490 nm was measured using a Bio-Rad ELISA microplate reader.
Transmission blocking analysis
Polyclonal antibodies generated against the above recombinant proteins were utilized in transmission-blocking assays. For the in vitro assay, PHZ pretreated mice were infected intraperitoneally with 5 × 106 P. berghei iRBC. At 3 dpi, parasitemias were determined, and 10 μl of blood was taken from appropriate hosts and added to 40 μl ookinete culture medium containing anti-PSOP26, PSOP25, PSOP25 + PSOP26, and PSOP25-PSOP26 sera or negative control mouse serum at final dilutions of 1:5 and 1:10. The exflagellation centers per field were quantified as described above. For ookinete formation analysis, ookinete cultures were incubated at 19 °C for 24 h and the number of matured ookinetes was determined by anti-Pbs mAb staining as described above. The ookinete conversion rates were calculated as the percentage of Pbs21-positive ookinete to Pbs21-positive macrogametes and ookinetes as described previously [29, 30].
For direct mosquito feeding assays (DFA), PHZ pretreated mice were infected with 5 × 106 P. berghei iRBC as described above. At 3 dpi, 4-day-old An. stephensi mosquitoes pre-starved for 12 h were allowed to feed on infected mice for 1 h (~ 50 mosquitoes/mouse). The unfed mosquitoes were removed and the remaining mosquitoes were maintained at 19–22 °C and 50–80% relative humidity. At 12 days post-feeding, ~ 30 mosquitoes in each group were dissected and stained with 0.5% Mercurochrome to count the number of oocysts per mosquito and assess the oocyst infection intensity and oocyst infection prevalence.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software 8.0. Parasitemias, gametocytemias, and gametocyte sex ratios were compared by Student’s t-test. The zygote and ookinete numbers between groups were compared using the Kruskal–Wallis test followed by Dunn’s multiple comparisons test. The Kaplan–Meier method was used to analyze the survival of mice. The prevalence of infection (proportion of infected mosquitoes) was analyzed by the Fisher’s exact test (with Bonferroni correction), while the oocyst density (oocyst number per midgut) was analyzed by the Mann–Whitney U-test. A P-value less than 0.05 was considered statistically significant.
Results
PSOP26::HA localizes to the surface of gamete and ookinete
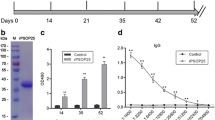
Psop26 transcripts were detected in schizonts, non-activated gametocytes, and ookinetes using real-time RT-PCR analysis (Fig. 1a). To assess protein expression, we targeted psop26 for in situ 3 × HA-tagging by double-crossover homologous recombination of a plasmid encoding a human dihydrofolate reductase:yeast cytosine deaminase and uridyl-phosphoribosyltransferase (hDHFR:yFCU) selectable marker (Additional file 2: Fig. S1a). PCR genotypic analysis was used to confirm successful modification of the endogenous genomic locus (Additional file 2: Fig. S1b). Despite the presence of transcripts in schizonts and non-activated gametocytes, the expression of the PSOP26::HA fusion protein was only detectable in zygotes and ookinetes; indicating that psop26 messenger RNA (mRNA) translation starts after gametocyte activation (Fig. 1b and Additional file 3: Fig. S2). Examination of PSOP26::HA fusion protein by immunofluorescence assays showed parasite plasma membrane localization of PSOP26::HA protein in male gametes. The PSOP26::HA fusion protein was detectable both with and without Triton X-100 permeabilization; and was co-expressed with α-tubulin II (male gamete marker), Pbg377 (female gamete marker), and ookinete surface protein PSOP25 (Fig. 1c). Together, these data strongly support that PSOP26 is a membrane-associated protein in gametes and ookinetes.

PSOP26 transcript levels, protein expression, and localization on the surface of gametes and ookinetes in P. berghei. a PSOP26 RT-PCR transcription profiles. Real-time RT-PCR analysis of the expression profile of psop26 transcripts in schizonts (Sch), non-activated gametocytes (Gam), and ookinetes (Ook). The β-tubulin gene, which is constitutively expressed at all three stages, was used as a loading control. b Expression of PSOP26::HA during parasite development. Western blot analysis of proteins extracted from purified P. berghei schizonts (Sch), gametocytes (Gam), zygotes (Zyg), and ookinetes (Ook) of PSOP26-HA parasites. Reduced lysates were separated by SDS-PAGE, transferred to a PVDF membrane, and labeled with anti-HA antibody and anti-Hsp70 sera (loading control). Non-infected erythrocytes (EC) were used as a negative control. c Immunofluorescence assays of Triton X-100 permeabilized (+Triton X-100, left) or non-permeabilized (−Triton X-100, right) P. berghei ring, trophozoite, schizont, gametocytes, gametes, zygotes, and ookinetes. Stained with α-HA monoclonal antibody (green), α-msp1 (merozoite surface protein, red) and/or α-tubulin II (male gametocyte/gamete protein, red), and/or α-Pbg377 (female gametocyte/gamete protein, red), and/or α-PSOP25 (ookinete surface protein). DNA was stained with DAPI. Only the secondary antibodies or WT ookinetes incubated with the α-HA mAb were used as a negative control (AF488, Alexa Fluor 488, AF555, and Alexa Fluor 555). Merge denotes Alexa Flour 488 + Alexa Flour 555 + DAPI. Scale bars correspond to 5 μm
PSOP26 is required for the zygote-to-ookinete developmental transition
To identify the biological function of the PSOP26 protein, we generated two independent gene knockout mutants, designated Δpsop26 clones C1 and C2. Integration of the disruption cassette (hDHFR:yFCU) was confirmed by diagnostic PCR (Additional file 2: Fig. S1c, d). Western blot analysis of total protein extracts prepared from purified P. berghei (WT) and Δpsop26 ookinetes using anti-PSOP26 sera revealed a band of ~ 90.8 kDa in WT strain parasites, corresponding to the predicted molecular weight of endogenous PSOP26 protein (Additional file 2: Fig. S1e). This band was absent from Δpsop26 ookinetes, indicating successful deletion of the psop26 gene (Additional file 2: Fig. S1e). The absence of PSOP26 protein did not affect asexual blood-stage development, gametocytemia, or male-to-female gamete ratios; and the capability of Δpsop26 gametocytes to produce exflagellation centers and macrogametes was comparable to that of the WT line (Fig. 2a–f). The number of zygotes formed remained unchanged, but the number of mature ookinete was reduced by 69% after 24 h in vitro culture, compared with WT parasites, suggesting that psop26 deletion affected the zygote-to-ookinete developmental transition (Fig. 2g, h). Following a mosquito blood meal, mosquitoes fed on mice infected with the Δpsop26 parasites exhibited 66% prevalence of infection, whereas the control group was 97%, a decrease of 31% (Table 1). Moreover, the mean number of oocysts per midgut was 23 in mosquitoes fed on Δpsop26-infected mice, compared with 115 in control mosquitoes, which indicates an 80% reduction (P < 0.01; Fig. 2i).

Phenotypic analyses of ∆psop26 mutants in asexual and sexual stages. Mice were injected with WT or ∆psop26 parasites clone 1 (C1) and clone 2 (C2). a Survival rates of mice. b Parasitemia was monitored by Giemsa smears after day 3 infection. c Gametocytemia (mature gametocytes per 104 RBCs) on day 3 post-infection. d Gametocyte sex ratios (female: male) on day 3 post-infection with different parasite lines. Error bars indicate mean ± SEM (standard error of the mean) (n = 3). Data in the phenotypic analysis of pbs54 and psop26 are representative of three independent experiments. e Exflagellation centers in 10 fields at × 100 magnification with equal number of mature male gametocytes. f Female gamete numbers in 0.5 µl of ookinete culture at 15 min post-activation. g Zygote numbers formed at 2 h during in vitro ookinete culture. h Ookinete numbers cultured in vitro for 24 h. i Oocyst numbers per midgut in mosquitoes 12 days after feeding on WT, ∆pbs54, or ∆psop26 parasite-infected mice. **P < 0.01 (Mann–Whitney U-test). Error bars indicate mean ± SEM (n = 3). Data in the phenotypic analysis of psop26 are representative of three independent experiments
The chimeric PSOP25-PSOP26 protein is immunogenic
We previously reported that anti-PSOP26 sera, which recognizes a 242-aa fragment of PSOP26 (50–245 aa), blocked or reduced parasite transmission in DFA [22]. Thus, to investigate the potential benefit of combining sera specific to PSOP25 and PSOP26, to represent a combination vaccine, a chimeric construct within a pET-32a plasmid was designed containing the Asp50-Gln245 fragment of PSOP26 fused to the Met45-Glu245 fragment of PSOP25 using a flexible linker sequence (GGGGS)3. The chimeric PSOP25-PSOP26 protein (PSOP25-PSOP26) was induced in E. coli by 1 mM IPTG at 19 °C overnight, and purified using Ni–NTA chromatography (Fig. 3a). The expression of the recombinant protein was detected using an anti-His tag mAb, showing that the purified recombinant PSOP25-PSOP26 was approximately 70 kDa, consistent with the predicted combined molecular weight (Fig. 3b). To allow for better control in TBA analysis, recombinant PSOP25, PSOP26, and Trx-His proteins were expressed using the pET-32a plasmid (data not shown). The purified recombinant PSOP26 alone (PSOP26), mixed/fused with PSOP25 (PSOP25 + PSOP26 and PSOP25-PSOP26), or PSOP25 alone (PSOP25) was used to immunize mice to raise polyclonal antibodies. As expected, immunization with individual antigens yielded only antibodies specific for the respective antigens used for immunization. The antibody titers against PSOP25 induced by recombinant PSOP25 alone or mixed/fused with PSOP26 were similar as determined in ELISA analysis using immune serum collected 2 weeks after the final immunization (Fig. 3c). Similarly, the antibody titers were comparable against PSOP26 induced by recombinant PSOP26 alone, mixed, or fused with PSOP25 (Fig. 3d); indicating that no immune interference occurred between the two antigens.

Purification of recombinant cPSOP25/26 protein and antibody response in mice immunized with rPSOP25, rPSOP26, rPSOP25 + PSOP26, and PSOP25-PSOP26. a Diagram illustrating the expressed regions of the PSOP26, PSOP25, and chimeric PSOP25-PSOP26. The signal peptide (red box), low complexity (green box), and transmembrane region (blue box) are highlighted. The pink line denotes the linker. b Purified recombinant PSOP25-PSOP26 (indicated by an arrow) was separated on a 10% SDS-PAGE gel and stained with Coomassie blue (left) and probed with anti-His mAb on a Western blot (right). BALB/c mice (n = 10) were immunized three times with Trx-His tag (immunization control) and recombinant proteins (rPSOP25, rPSOP26, rPSOP25 + PSOP26, and rPSOP25-PSOP26). Total IgG titers after final immunization were measured by ELISA analysis coated with recombinant PSOP25 (c) and recombinant PSOP26 (d) polypeptides after removal of the Trx tag. Error bars indicate standard deviation. **P < 0.01 indicates a significant difference between the immunization and control groups (Student’s t-test)
Reactivity of the antisera against mixed or fused PSOP25/PSOP26 with the native parasite proteins
Western blot analysis was performed using lysates from ookinetes to verify that the antisera produced against the antigen mixture or chimera reacted with parasite native proteins. Antisera against both mixed (PSOP25 + PSOP26) and fused (PSOP25-PSOP26) recombinant proteins recognized bands of 40 and 91 kDa in ookinete lysates, corresponding to PSOP25 and PSOP26 proteins, respectively (Fig. 4a). Next, we tested the ability of anti-PSOP25-PSOP26 sera and anti-PSOP25 + PSOP26 sera to recognize PSOP25 and/or PSOP26 expressed in different sexual stages of the parasite. The anti-PSOP25 sera and anti-PSOP26 sera were used as controls to ensure PSOP25 or PSOP26 expression. Both antisera against the mixed and fused antigen reacted with male gametes, female gametes/zygotes, and ookinetes (Fig. 4b). Comparable to the IFA results of the PSOP26-HA fusion protein, anti-PSOP25-PSOP26 and anti-PSOP25 + PSOP26 sera bound exflagellating male gametes, female gamete/zygotes, and ookinetes (Fig. 4b). Strong fluorescence signals were detected at the plasma membranes of female gamete/zygotes and ookinetes (Fig. 4b). Negative controls performed with antibodies against Trx-His or with the secondary antibodies alone did not react with these sexual stages (Fig. 4b). Collectively, antisera against the mixed or fused antigens from the pre- and post-fertilization stages were able to recognize the respective proteins expressed during the gamete to ookinete development.

Reactivity of vaccine-induced antisera to native parasite antigen. a Western blot analysis under reducing conditions of gametocyte (GC) and ookinete (Ook) parasite lysates using anti-PSOP26 + PSOP26 sera (left) and anti-PSOP25-PSOP26 sera. PSOP25 and PSOP26 protein bands are indicated with arrows. Equal loading was estimated using anti-rHsp70 sera. b Indirect immunofluorescence analysis using the bivalent immune sera. Parasites of different developmental stages were fixed, permeabilized with 0.1% Triton X-100, and stained with antisera against PSOP25 + PSOP26 (left) and PSOP25-PSOP26 (right) (1:200) as the primary antibodies (green). The parasites were also co-labeled with antibodies against the markers for different stages (red), including α-tubulin (α) for male gametocytes/gametes, P47 for female gametocytes, Pbs21 for zygotes and ookinetes, and SET for the nucleus of gametocytes. Alexa Fluor 488-conjugated goat anti-mouse IgG antibodies and Alexa Fluor 555-conjugated goat anti-rabbit IgG antibodies were used as the secondary antibodies. The sera from mice receiving Trx-His recombinant proteins were included as negative controls. Antibody binding was detected by Alexa Fluor 555-conjugated goat anti-rabbit IgG (red) (lane 1), and Alexa Fluor 488-conjugated goat anti-mouse IgG (green) (lane 2). DNA was stained with DAPI (blue) (lane 3). DIC images (lane 4), and merged view (lane 5) are shown. Scale bar, 5 µm
PSOP25 and PSOP26 fusion proteins elicit strong transmission-blocking antibody responses against P. berghei
The biological activity of specific antisera was assessed in a TBA assay as described in the methodology section. Although expressed on the surface of male gamete flagella, neither anti-PSOP25-PSOP26 sera nor anti-PSOP25 + PSOP26 sera supplements could inhibit male gametocyte exflagellation; suggesting that PSOP25 and PSOP26 do not play essential roles in male gametogenesis (Fig. 5a). Ookinete cultures supplemented with pooled immune sera raised against PSOP25, PSOP26, PSOP25 + PSOP26, or PSOP25-PSOP26 reduced ookinete numbers by 55%, 49%, 59%, and 59% at 1:5 dilutions, compared to the Trx-His group, respectively (Fig. 5b). The ookinete numbers of the cultures supplied with antisera against PSOP25, PSOP26, PSOP25 + PSOP26, or PSOP25-PSOP25 at 1:10 dilution were 71, 86, 66, and 66, corresponding to a reduction of ookinete formation by 53% (54%), 42% (43%), 55% (56%), and 56% (56%), respectively, compared to the Trx-His (control) group (Fig. 5b). Only serum raised against PSOP25 + PSOP26 and PSOP25-PSOP26 provided significantly improved TBA versus the Trx-His group at 1:5 dilution. (Kruskal–Wallis test followed by Dunn’s multiple comparisons test, P < 0.05, Fig. 5b). At a 1:10 dilution, only the antisera against PSOP25 + PSOP26 significantly reduced ookinete numbers by 56% compared to the control group (Kruskal–Wallis test followed by Dunn’s multiple comparisons test, P < 0.05, Fig. 5b). No significant difference between anti-PSOP25-PSOP26 sera and anti-PSOP25 + PSOP26 sera was observed (Fig. 5B). Together, these results showed that the sera against mixed and fused antigens of PSOP25 and PSOP26 produced stronger inhibition effects on in vitro ookinete formation than antisera against the single PSOP25 or PSOP26 antigen.

Transmission-blocking activity of the antisera assessed in vitro and in vivo. a Exflagellation centers per 10 fields. Plasmodium berghei-infected blood collected at 3 dpi was incubated with the respective control and immune sera at dilutions of 1:5 and 1:10. b Ookinete formation. The ookinete numbers after 24 h were stained with Pbs21 mAb and counted using a fluorescence microscope. The data were representative of three independent experiments. c Midgut oocyst numbers in mosquitoes fed on relative recombinant protein-immunized mice. The results were collected from three mice in each immunization group and two separate experiments (n = 180). Data points represent midgut oocyst numbers in individual mosquitoes. Horizontal bars indicate the mean number of oocysts per midgut. d Mosquito infection prevalence was calculated at 12 days after the blood meal. Data points represent the prevalence of infection in mosquitoes from three mice per group and in two separate experiments. Error bars indicate mean ± SD. *P < 0.05 and ***P < 0.001 represent the significant difference between the respective immunization group and the Trx-His control group. #P < 0.05 # represents a significant difference between two immunization groups
To further examine the transmission-blocking effect of antisera recognizing PSOP25, PSOP26, mixed, or fused antigens in vivo, mice immunized with the respective recombinant antigens were infected with P. berghei and used for DFA. Ten days after feeding, mosquitoes were dissected and midgut oocysts were counted. The midgut oocyst densities for all immunization groups were significantly reduced by 52–66%, compared to the Trx-His immunized control (P < 0.001, Kruskal–Wallis test followed by Dunn’s multiple comparisons test; Fig. 5c and Table 2). There were significant reductions in oocyst density in the mixed-antigen (66%) and fused-antigen (66%) immunization groups compared with the PSOP26 (52%) immunization group (Kruskal–Wallis test followed by Dunn’s multiple comparisons test, P < 0.05; Fig. 5c and Table 2). The mean prevalence of mosquito infection in all immunization groups was significantly reduced by 6.7–14.5%, compared to the Trx-His immunized group (P < 0.05 for PSOP26; P < 0.01 for PSOP25, PSOP25-PSOP26, and PSOP25 + PSOP26 group; Fisher’s exact test, Fig. 5c, Table 2). However, the infection prevalence in mixed-antigen (82%) and fused-antigen (81%) immunization groups were comparable to PSOP25 (83%) or the PSOP26 (89%) immunization groups (Fig. 5d). Collectively, the mosquito feeding assays demonstrated that the ookinete surface antigens PSOP25 and PSOP26, when mixed or fused for immunization, produced significantly higher TRA than when PSOP26 was used individually; whereas PSOP25 alone could induce a promising TRA, comparable to the mixed or fusion antigens.
Discussion
Here we have identified the P. berghei PSOP26 protein as an important factor in the zygote-to-ookinete developmental transition. We demonstrated that psop26 transcription begins in schizonts and the expressed PSOP26 protein localizes on the surface of gametes and ookinetes, comparable to our previous report [6]. While the pre-fertilization life cycle stages are not affected in psop26-null mutants (Δpsop26), a strong reduction (> 69%) in mature ookinete numbers was observed, consistent with our report that antisera against PSOP26 could inhibit ookinete maturation [22]. However, Ukegbu et al. reported that PSOP26 is not essential for ookinete differentiation [8]. Thus, to validate the specificity of our results, we counted the zygote numbers in Δpsop26 at 2 h after incubating at 19 °C, and found comparable zygote numbers in Δpsop26 and WT; confirming that the reduced matured ookinete number observed in Δpsop26 mutants is indeed caused by an impairment of the zygote-to-ookinete developmental transition. We further observed evidence of Δpsop26 mutants having a reduction in oocyst formation, indicating that the ookinete-to-oocyst transition is also impaired. Numerous ookinete proteins have been shown to be involved in the ookinete-to-oocyst transition; including proteins which function in ookinete motility (CTRP [31], PPKL [32], CDPK3 [33], DHHC3 [34], and the IMC1 family members [35]), the Plasmodium CHT1 proteins involved in ookinete penetration of the midgut epithelium [36,37,38,39], and ookinete invasion-related proteins (SOAP [3], PIMMS2 [7], PSOP1/2/7/9 [5, 40], P25/P28 [2], MAOP proteins [41], and PPLP5 [42]). One report indicated that psop26 disruption does not adversely affect ookinete motility, but mosquito midgut invasion ability was impaired [8]; suggesting that PSOP26 is an ookinete invasion-related protein. However, the function of PSOP26 during ookinete invasion remains unclear. The mosquito midgut is proposed to be an immune-competent organ, which could generate several antibacterial peptide defensins against P. berghei infection [43]. Thus, additional experiments will be necessary to rule out the impact of these mosquito immune factors on the function of PSOP26 protein during ookinete invasion. Taken together, these data indicate that PSOP26 is a promising target for the intervention of malaria parasite transmission.
Antigens expressed during the post-fertilization stages commonly show less amino acid polymorphism than blood-stage antigens, as they are not subjected to selective pressure by an adaptive immune response that might drive diversity. Parasites in the mosquito midgut are exposed to immune factors taken in with the vertebrate host blood, and thus are vulnerable points to disrupt the transmission cycle of the malaria parasite [44, 45]. Our previous study demonstrated that antisera against PSOP26 exhibited a moderate TBA [22]. It has been proposed that TBVs must induce sustained high antibody titers of high-affinity binding antibodies to possess high TBA [46]. Thus, to attempt to improve the TBA in the current study, we generated a chimera PSOP25-PSOP26 protein. One concern when generating multivalent vaccines is that the immunodominant component could compromise the immune response against an accompanying antigen [47,48,49]. To investigate this, we tested the specific antibody responses to both PSOP25 and PSOP26 recombinant proteins by ELISA analysis. We found that the antibody responses were not affected when the two antigens were presented either as a PSOP25-PSOP26 chimera protein or an equal amount mixture (PSOP25 + PSOP26). The antibody titer raised against PSOP25-PSOP26 and PSOP25 + PSOP26 antigens was not significantly different from mice that received a prime-boost with PSOP25 or PSOP26 alone. Together, these results suggest that the combination of two malaria parasite ookinete antigens did not result in substantial immune interference.
Direct feeding assays allow the assessment of TBA to determine the functionality of antibodies elicited by vaccination, and are considered the best proxy for evaluating TBV candidates [50]. Our rabbit antisera raised against both PSOP25 + PSOP26 and PSOP25-PSOP26 had significantly higher TBA, as shown by the lower percentages of infected mosquitoes and the number of oocysts per mosquito midgut, compared with PSOP26 single immunized sera and pre-immune sera. However, the antisera against fusion and mixed antigens did not exhibit significantly improved TBA compared to antisera against PSOP25 alone. One explanation is that PSOP25 is dominantly expressed on the surface of the ookinete, compared to PSOP26, as shown by our Western blot observation that antisera detected a thicker band for PSOP25 compared to PSOP26. Alternatively, PSOP25 may play a more essential role during the ookinete-to-oocyst transition, which is a hypothesis for further analysis. These results highlight the potential of selecting PSOP25 as a TBV candidate that targets the ookinete-to-oocyst developmental transition.
Conclusions
Our study expands the role of the ookinete surface protein PSOP26 in malaria parasite transmission, and specifically demonstrates that PSOP26 has a clear and vital role in the P. berghei zygote-to-ookinete developmental transition. In addition, the usage of antisera against two ookinete surface antigens in the form of both fusion (PSOP25-PSOP26) and mixed (PSOP25 + PSOP26) proteins improved TBA compared to antisera against PSOP26 alone, but was comparable to the TBA antisera against PSOP25 alone. Our results confirm the previous finding that PSOP25 is a promising ookinete stage TBV candidate, and additionally found that PSOP25 alone is as good as the combination vaccine in inhibiting the ookinete-to-oocyst developmental transition. Furthermore, our data support the continued development of antisera (or antibody) against multi-stage antigens as malaria vaccine candidates to address the need for effective vaccines for the intervention of malaria parasite transmission.
Availability of data and materials
The data supporting the conclusions of this article are included within the article.
Abbreviations
- SOAP:
-
Secreted ookinete adhesive protein
- PSOPs:
-
Putative secreted ookinete proteins
- PPLP:
-
Plasmodium perforin-like protein
- TBVs:
-
Transmission-blocking vaccines
- TRA:
-
Transmission-reducing activity
- An. stephensi :
-
Anopheles stephensi
- nt:
-
Nucleotide
- gDNA:
-
Genomic DNA
- UTR:
-
Untranslated region
- WT:
-
Wild-type
- SDS-PAGE:
-
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- ELISA:
-
Enzyme-linked immunosorbent assay
- PHZ:
-
Phenylhydrazine
- DFA:
-
Direct mosquito feeding assay
- hDHFR:yFCU :
-
Human dihydrofolate reductase:yeast cytosine deaminase and uridyl-phosphoribosyltransferase
References
Challenger JD, Olivera Mesa D, Da DF, Yerbanga RS, Lefevre T, Cohuet A, et al. Predicting the public health impact of a malaria transmission-blocking vaccine. Nat Commun. 2021;12:1494. https://doi.org/10.1038/s41467-021-21775-3.
Tomas AM, Margos G, Dimopoulos G, van Lin LH, de Koning-Ward TF, Sinha R, et al. P25 and P28 proteins of the malaria ookinete surface have multiple and partially redundant functions. EMBO J. 2001;20:3975–83. https://doi.org/10.1093/emboj/20.15.3975.
Dessens JT, Siden-Kiamos I, Mendoza J, Mahairaki V, Khater E, Vlachou D, et al. SOAP, a novel malaria ookinete protein involved in mosquito midgut invasion and oocyst development. Mol Microbiol. 2003;49:319–29. https://doi.org/10.1046/j.1365-2958.2003.03566.x.
Sassmannshausen J, Pradel G, Bennink S. Perforin-like proteins of apicomplexan parasites. Front Cell Infect Microbiol. 2020;10:578883. https://doi.org/10.3389/fcimb.2020.578883.
Ecker A, Bushell ES, Tewari R, Sinden RE. Reverse genetics screen identifies six proteins important for malaria development in the mosquito. Mol Microbiol. 2008;70:209–20. https://doi.org/10.1111/j.1365-2958.2008.06407.x.
Zheng W, Liu F, He Y, Liu Q, Humphreys GB, Tsuboi T, et al. Functional characterization of Plasmodium berghei PSOP25 during ookinete development and as a malaria transmission-blocking vaccine candidate. Parasit Vectors. 2017;10:8. https://doi.org/10.1186/s13071-016-1932-4.
Ukegbu CV, Akinosoglou KA, Christophides GK, Vlachou D. Plasmodium berghei PIMMS2 promotes ookinete invasion of the Anopheles gambiae mosquito midgut. Infect Immun. 2017;85:e00139-e217. https://doi.org/10.1128/IAI.00139-17.
Ukegbu CV, Christophides GK, Vlachou D. Identification of three novel Plasmodium factors involved in ookinete to oocyst developmental transition. Front Cell Infect Microbiol. 2021;11:634273. https://doi.org/10.3389/fcimb.2021.634273.
Singh K, Burkhardt M, Nakuchima S, Herrera R, Muratova O, Gittis AG, et al. Structure and function of a malaria transmission blocking vaccine targeting Pfs230 and Pfs230-Pfs48/45 proteins. Commun Biol. 2020;3:395. https://doi.org/10.1038/s42003-020-01123-9.
Lee SM, Wu Y, Hickey JM, Miura K, Whitaker N, Joshi SB, et al. The Pfs230 N-terminal fragment, Pfs230D1+: expression and characterization of a potential malaria transmission-blocking vaccine candidate. Malar J. 2019;18:356. https://doi.org/10.1186/s12936-019-2989-2.
Marin-Mogollon C, van de Vegte-Bolmer M, van Gemert GJ, van Pul FJA, Ramesar J, Othman AS, et al. The Plasmodium falciparum male gametocyte protein P230p, a paralog of P230, is vital for ookinete formation and mosquito transmission. Sci Rep. 2018;8:14902. https://doi.org/10.1038/s41598-018-33236-x.
Miura K, Keister DB, Muratova OV, Sattabongkot J, Long CA, Saul A. Transmission-blocking activity induced by malaria vaccine candidates Pfs25/Pvs25 is a direct and predictable function of antibody titer. Malar J. 2007;6:107. https://doi.org/10.1186/1475-2875-6-107.
Kaslow DC. Transmission-blocking vaccines. Chem Immunol. 2002;80:287–307. https://doi.org/10.1159/000058850.
Wu Y, Ellis RD, Shaffer D, Fontes E, Malkin EM, Mahanty S, et al. Phase 1 trial of malaria transmission blocking vaccine candidates Pfs25 and Pvs25 formulated with Montanide ISA 51. PLoS ONE. 2008;3:e2636. https://doi.org/10.1371/journal.pone.0002636.
de Graaf H, Payne RO, Taylor I, Miura K, Long CA, Elias SC, et al. Safety and immunogenicity of ChAd63/MVA Pfs25-IMX313 in a phase I first-in-human trial. Front Immunol. 2021;12:694759. https://doi.org/10.3389/fimmu.2021.694759.
Talaat KR, Ellis RD, Hurd J, Hentrich A, Gabriel E, Hynes NA, et al. Safety and immunogenicity of Pfs25-EPA/Alhydrogel®), a transmission blocking vaccine against Plasmodium falciparum: an open label study in malaria naive adults. PLoS ONE. 2016;11:e0163144. https://doi.org/10.1371/journal.pone.0163144.
Sagara I, Healy SA, Assadou MH, Gabriel EE, Kone M, Sissoko K, et al. Safety and immunogenicity of Pfs25H-EPA/Alhydrogel, a transmission-blocking vaccine against Plasmodium falciparum: a randomised, double-blind, comparator-controlled, dose-escalation study in healthy Malian adults. Lancet Infect Dis. 2018;18:969–82. https://doi.org/10.1016/S1473-3099(18)30344-X.
Healy SA, Anderson C, Swihart BJ, Mwakingwe A, Gabriel EE, Decederfelt H, et al. Pfs230 yields higher malaria transmission-blocking vaccine activity than Pfs25 in humans but not mice. J Clin Invest. 2021;131:e146221. https://doi.org/10.1172/JCI146221.
Duffy PE, Kaslow DC. A novel malaria protein, Pfs28, and Pfs25 are genetically linked and synergistic as falciparum malaria transmission-blocking vaccines. Infect Immun. 1997;65:1109–13. https://doi.org/10.1128/IAI.65.3.1109-1113.1997.
Sheehy SH, Duncan CJ, Elias SC, Choudhary P, Biswas S, Halstead FD, et al. ChAd63-MVA-vectored blood-stage malaria vaccines targeting MSP1 and AMA1: assessment of efficacy against mosquito bite challenge in humans. Mol Ther. 2012;20:2355–68. https://doi.org/10.1038/mt.2012.223.
Menon V, Kapulu MC, Taylor I, Jewell K, Li Y, Hill F, et al. Assessment of antibodies induced by multivalent transmission-blocking malaria vaccines. Front Immunol. 2017;8:1998. https://doi.org/10.3389/fimmu.2017.01998.
Zheng W, Kou X, Du Y, Liu F, Yu C, Tsuboi T, et al. Identification of three ookinete-specific genes and evaluation of their transmission-blocking potentials in Plasmodium berghei. Vaccine. 2016;34:2570–8. https://doi.org/10.1016/j.vaccine.2016.04.011.
Janse CJ, Ramesar J, Waters AP. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc. 2006;1:346–56. https://doi.org/10.1038/nprot.2006.53.
Tonkin CJ, van Dooren GG, Spurck TP, Struck NS, Good RT, Handman E, et al. Localization of organellar proteins in Plasmodium falciparum using a novel set of transfection vectors and a new immunofluorescence fixation method. Mol Biochem Parasitol. 2004;137:13–21. https://doi.org/10.1016/j.molbiopara.2004.05.009.
Yang F, Liu F, Yu X, Zheng W, Wu Y, Qiu Y, et al. Evaluation of two sexual-stage antigens as bivalent transmission-blocking vaccines in rodent malaria. Parasit Vectors. 2021;14:241. https://doi.org/10.1186/s13071-021-04743-0.
Tewari R, Straschil U, Bateman A, Bohme U, Cherevach I, Gong P, et al. The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe. 2010;8:377–87. https://doi.org/10.1016/j.chom.2010.09.006.
van Dijk MR, van Schaijk BC, Khan SM, van Dooren MW, Ramesar J, Kaczanowski S, et al. Three members of the 6-cys protein family of Plasmodium play a role in gamete fertility. PLoS Pathog. 2010;6:e1000853. https://doi.org/10.1371/journal.ppat.1000853.
Miura K, Orcutt AC, Muratova OV, Miller LH, Saul A, Long CA. Development and characterization of a standardized ELISA including a reference serum on each plate to detect antibodies induced by experimental malaria vaccines. Vaccine. 2008;26:193–200. https://doi.org/10.1016/j.vaccine.2007.10.064.
Yoshida S, Matsuoka H, Luo E, Iwai K, Arai M, Sinden RE, et al. A single-chain antibody fragment specific for the Plasmodium berghei ookinete protein Pbs21 confers transmission blockade in the mosquito midgut. Mol Biochem Parasitol. 1999;104:195–204. https://doi.org/10.1016/s0166-6851(99)00158-9.
Sinden RE, Winger L, Carter EH, Hartley RH, Tirawanchai N, Davies CS, et al. Ookinete antigens of Plasmodium berghei: a light and electron-microscope immunogold study of expression of the 21 kDa determinant recognized by a transmission-blocking antibody. Proc R Soc Lond B Biol Sci. 1987;230:443–58. https://doi.org/10.1098/rspb.1987.0028.
Dessens JT, Beetsma AL, Dimopoulos G, Wengelnik K, Crisanti A, Kafatos FC, et al. CTRP is essential for mosquito infection by malaria ookinetes. EMBO J. 1999;18:6221–7. https://doi.org/10.1093/emboj/18.22.6221.
Guttery DS, Poulin B, Ferguson DJ, Szoor B, Wickstead B, Carroll PL, et al. A unique protein phosphatase with kelch-like domains (PPKL) in Plasmodium modulates ookinete differentiation, motility and invasion. PLoS Pathog. 2012;8:e1002948. https://doi.org/10.1371/journal.ppat.1002948.
Siden-Kiamos I, Ecker A, Nyback S, Louis C, Sinden RE, Billker O. Plasmodium berghei calcium-dependent protein kinase 3 is required for ookinete gliding motility and mosquito midgut invasion. Mol Microbiol. 2006;60:1355–63. https://doi.org/10.1111/j.1365-2958.2006.05189.x.
Hopp CS, Balaban AE, Bushell ES, Billker O, Rayner JC, Sinnis P. Palmitoyl transferases have critical roles in the development of mosquito and liver stages of Plasmodium. Cell Microbiol. 2016;18:1625–41. https://doi.org/10.1111/cmi.12601.
Tremp AZ, Khater EI, Dessens JT. IMC1b is a putative membrane skeleton protein involved in cell shape, mechanical strength, motility, and infectivity of malaria ookinetes. J Biol Chem. 2008;283:27604–11. https://doi.org/10.1074/jbc.M801302200.
Dessens JT, Mendoza J, Claudianos C, Vinetz JM, Khater E, Hassard S, et al. Knockout of the rodent malaria parasite chitinase pbCHT1 reduces infectivity to mosquitoes. Infect Immun. 2001;69:4041–7. https://doi.org/10.1128/IAI.69.6.4041-4047.2001.
Vinetz JM, Dave SK, Specht CA, Brameld KA, Xu B, Hayward R, et al. The chitinase PfCHT1 from the human malaria parasite Plasmodium falciparum lacks proenzyme and chitin-binding domains and displays unique substrate preferences. Proc Natl Acad Sci USA. 1999;96:14061–6. https://doi.org/10.1073/pnas.96.24.14061.
Vinetz JM, Valenzuela JG, Specht CA, Aravind L, Langer RC, Ribeiro JM, et al. Chitinases of the avian malaria parasite Plasmodium gallinaceum, a class of enzymes necessary for parasite invasion of the mosquito midgut. J Biol Chem. 2000;275:10331–41. https://doi.org/10.1074/jbc.275.14.10331.
Langer RC, Vinetz JM. Plasmodium ookinete-secreted chitinase and parasite penetration of the mosquito peritrophic matrix. Trends Parasitol. 2001;17:269–72. https://doi.org/10.1016/s1471-4922(01)01918-3.
Tachibana M, Iriko H, Baba M, Torii M, Ishino T. PSOP1, putative secreted ookinete protein 1, is localized to the micronemes of Plasmodium yoelii and P. berghei ookinetes. Parasitol Int. 2021;84:102407. https://doi.org/10.1016/j.parint.2021.102407.
Kadota K, Ishino T, Matsuyama T, Chinzei Y, Yuda M. Essential role of membrane-attack protein in malarial transmission to mosquito host. Proc Natl Acad Sci USA. 2004;101:16310–5. https://doi.org/10.1073/pnas.0406187101.
Ecker A, Pinto SB, Baker KW, Kafatos FC, Sinden RE. Plasmodium berghei: plasmodium perforin-like protein 5 is required for mosquito midgut invasion in Anopheles stephensi. Exp Parasitol. 2007;116:504–8. https://doi.org/10.1016/j.exppara.2007.01.015.
Barillas-Mury C, Wizel B, Han YS. Mosquito immune responses and malaria transmission: lessons from insect model systems and implications for vertebrate innate immunity and vaccine development. Insect Biochem Mol Biol. 2000;30:429–42. https://doi.org/10.1016/s0965-1748(00)00018-7.
Smith RC, Vega-Rodriguez J, Jacobs-Lorena M. The Plasmodium bottleneck: malaria parasite losses in the mosquito vector. Mem Inst Oswaldo Cruz. 2014;109:644–61.
Sauerwein RW, Bousema T. Transmission blocking malaria vaccines: assays and candidates in clinical development. Vaccine. 2015;33:7476–82. https://doi.org/10.1016/j.vaccine.2015.08.073.
Yenkoidiok-Douti L, Canepa GE, Barletta ABF, Barillas-Mury C. In vivo characterization of Plasmodium berghei P47 (Pbs47) as a malaria transmission-blocking vaccine target. Front Microbiol. 2020;11:1496. https://doi.org/10.3389/fmicb.2020.01496.
Theisen M, Cox G, Hogh B, Jepsen S, Vuust J. Immunogenicity of the Plasmodium falciparum glutamate-rich protein expressed by vaccinia virus. Infect Immun. 1994;62:3270–5. https://doi.org/10.1128/iai.62.8.3270-3275.1994.
Johansson BE, Kilbourne ED. Dissociation of influenza virus hemagglutinin and neuraminidase eliminates their intravirionic antigenic competition. J Virol. 1993;67:5721–3. https://doi.org/10.1128/JVI.67.10.5721-5723.1993.
Huang WC, Deng B, Mabrouk MT, Seffouh A, Ortega J, Long C, et al. Particle-based, Pfs230 and Pfs25 immunization is effective, but not improved by duplexing at fixed total antigen dose. Malar J. 2020;19:309. https://doi.org/10.1186/s12936-020-03368-5.
Bompard A, Da DF, Yerbanga RS, Biswas S, Kapulu M, Bousema T, et al. Evaluation of two lead malaria transmission blocking vaccine candidate antibodies in natural parasite-vector combinations. Sci Rep. 2017;7:6766. https://doi.org/10.1038/s41598-017-06130-1.
Acknowledgements
We are grateful to Ms. Jun Liu for technical support and to Dr. Thomas Templeton for proofreading the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31900674 and 81971961) and by the National Institutes of Health grants (R01AI150533 and U19AI089672).
Author information
Authors and Affiliations
Contributions
FL, XZ, and YC designed the studies and drafted the manuscript. PW, XJ, JB, and FY took part in the genomic DNA extraction and PCR amplification. YW, YZ, JB, and FY participated in phenotype analysis. PW, FY, XY, WZ, and FL participated in TBV analysis and writing of the manuscript. LC and YC revised the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal procedures were reviewed and approved by the animal ethics committee of China Medical University and carried out according to the guidelines of Laboratory Animal Welfare and Ethics under the License Number CMU2021346.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests. The funders had no role in the design of the study, in the collection, analysis, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primer information and sequences.
Additional file 2: Figure S1.
Generation of HA-tagged PSOP26 transgenic parasites in the P. berghei ANKA line. a Schematic representation of the posp26 locus (psop26::ha) HA tagging by double-crossover homologous recombination. The primers used for diagnostic PCR are indicated by black arrows. b Diagnostic PCR analysis of PSOP26::HA transgenic parasites. PCR analysis was performed using genomic DNA extracts from wild-type P. berghei (WT) and PSOP26::HA transgenic parasites. The native locus was detected using primers p1 + p2 (lane 1: WT, 1471 bp; PSOP26::HA, null). The 5′ and 3′ integration of a modified locus (psop26::ha) was detected using primers p1 + p3 (lane 2: WT, null; PSOP26::HA, 1272 bp) and p4 + p5 (lane 3: WT, null; PSOP26::HA, 1815 bp), respectively. c Schematic representation of psop26 locus disruption by double-crossover homologous recombination. Primers used to detect either the WT locus or the replaced locus are marked. d PCR analysis of the genomic DNA from the WT and ∆psop26 parasite. Predicted DNA fragment sizes: lane 1, QCR1 + QCR2 (525 bp from WT only); lane 2, QCR2 + GW2 (753 bp from ∆psop26 only); lane 3, GW1 + GT (3500 bp from ∆psop26 only). e Western blot analysis shows the deletion of PSOP26. Lysates extracted from WT and Δpsop26-C1 parasites were incubated with α-PSOP26 or α-Hsp70. Non-infected erythrocytes (EC) were used as a negative control. PSOP26 protein bands are indicated with arrows.
Additional file 3: Figure S2.
Giemsa staining of purified parasites. Image showing schizonts, gametocytes, zygotes, and ookinetes of the PSOP26::HA parasites.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Pp., Jiang, X., Bai, J. et al. Characterization of PSOP26 as an ookinete surface antigen with improved transmission-blocking activity when fused with PSOP25. Parasites Vectors 15, 175 (2022). https://doi.org/10.1186/s13071-022-05294-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05294-8