
Abstract
Background
Ticks, which are ectoparasites of animals, may carry multiple pathogens. The cattle tick Rhipicephalus microplus is an important bovine parasite in China. However, the midgut microbiome of R. microplus from China has not been characterized via metagenomic methods.
Methods
Rhipicephalus microplus were collected from cattle in the city of Changsha in Hunan province, China. The DNA of the midgut contents was extracted from fully engorged adult female R. microplus. A DNA library was constructed and sequenced using an Illumina HiSeq sequencing platform. SOAPdenovo software was used to assemble and analyze the clean data. The latent class analysis algorithm applied to system classification by MEGAN software was used to annotate the information on the species’ sequences. DIAMOND software was used to compare unigenes with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, and functional annotation was carried out based on the results of the comparison.
Results
The dominant phyla in the five samples were Firmicutes, Proteobacteria, and Actinobacteria. Streptococcus, Mycobacterium, Anaplasma, Enterococcus, Shigella, Lactobacillus, Brachyspira, Pseudomonas, Enterobacter, Bacillus, and Lactococcus were the dominant genera in the five samples. The endosymbiotic bacterium Wolbachia was also detected in all of the samples. Mycobacterium malmesburyense, Streptococcus pneumoniae, Anaplasma phagocytophilum, Enterococcus faecium, Shigella sonnei, Enterococcus faecalis, Lactobacillus casei, Brachyspira hampsonii, Pseudomonas syringae, Enterobacter cloacae, and Lactococcus garvieae were the dominant species in the five samples. In addition to these bacterial species, we also detected some eukaryotes, such as Rhizophagus irregularis, Enterospora canceri, Smittium culicis, Zancudomyces culisetae, Trachipleistophora hominis, and viruses such as orf virus, human endogenous retrovirus type W, enzootic nasal tumor virus of goats, bovine retrovirus CH15, and galidia endogenous retrovirus in all of the samples at the species level. The results of the annotated KEGG pathway predictions for the gene functions of the midgut microflora of R. microplus indicated genes involved in lipid and amino acid metabolism, infectious diseases (e.g., Streptococcus pneumonia infection, human granulocytic anaplasmosis, Shigella sonnei infection, Salmonella enterica infection, and pathogenic Escherichia coli infection), and cancer.
Conclusions
Our study revealed that the midgut microbiome of R. microplus is not only composed of a large number of bacteria, but that a portion also comprises eukaryotes and viruses. The data presented here enhance our understanding of this tick’s midgut microbiome and provide fundamental information for the control of ticks and tick-borne diseases.
Graphical Abstract

Similar content being viewed by others

Background
The tick species Rhipicephalus microplus belongs to the family Ixodidae. Rhipicephalus microplus is widely distributed throughout the world, and epidemics of the diseases for which it is a vector, such as anaplasmosis and babesiosis, are often reported in Brazil, India, tropical and subtropical Asia, the Caribbean, Central and South America, China, and Mexico [1,2,3,4,5]. Rhipicephalus microplus is a single-host tick that infects cattle and buffaloes. Although R. microplus only infests cattle once at each stage of development (larva, nymph, and adult), it feeds on blood for several days at a time. This tick harms its hosts by biting and sucking blood, which lead to pruritus, emaciation, loss of fur quality, anemia, reduced milk production, and other clinical characteristics in infested animals [6].
Rhipicephalus microplus is not only a blood-sucking parasite but also the vector for a variety of pathogens [7, 8]. It is considered to be the most important vector of bovine tick-borne diseases in global agroecosystems [9, 10]. Among the pathogens that R. microplus can carry, we can cite Anaplasma phagocytophilum, Anaplasma marginale, Babesia bigemina, Babesia bovis, Ehrlichia chaffeensis, Ehrlichia canis, Ehrlichia sp. Tibet, Rickettsia spp., Borrelia spp., and severe fever with thrombocytopenia syndrome virus [11,12,13,14]. Furthermore, some researchers have detected a novel human pathogen, Anaplasma capra, in R. microplus [15]. Among the pathogens carried by this tick, B. bigemina, B. bovis, and A. marginale threaten the health of cattle and cause economic losses to the cattle industry [16, 17]; A. phagocytophilum can cause human granulocytic anaplasmosis, which can be a serious threat to human health [18,19,20].
Previously, the identification of microorganisms generally depended on culturing. With the rapid development of sequencing technology, bacterial communities associated with entire ticks, the midgut, and the ovary of R. microplus have been studied using non-culture methods [9]. The dominant bacteria encountered in these studies of R. microplus were Wolbachia, Coxiella, and Borrelia burgdorferi [9]. Polymerase chain reaction (PCR) combined with denaturing gradient gel electrophoresis, established by Fisher and Lerman in 1983 [21], was first applied to the analysis of microbial populations in 1993 [22], and has been extensively used for the direct identification of the microflora of ticks [23,24,25]. The bacterial community of the midgut of R. microplus collected from cattle in Jiangxi and Hunan provinces of China was analyzed by PCR combined with denaturing gradient gel electrophoresis, and the dominant bacteria were found to be Rickettsia peacockii and Coxiella [25]. In recent years, new ideas and approaches for the study of the gut microbiome of ticks have been developed due to the rise of metagenomics [26, 27].
Metagenomics can be used to identify new and emerging human pathogens circulating in tick vectors [20, 28, 29]. Adegoke et al. [30] analyzed the microbial composition of two tick species (Hyalomma anatolicum and R. microplus) in Pakistan using metagenomic sequencing. In the present study, metagenomics was used to analyze and determine the species of the midgut microbiome of five fully engorged adult female R. microplus collected from cattle in the city of Changsha in Hunan province, China. Furthermore, the Kyoto Encyclopedia of Genes and Genomes (KEGG) was used to predict the gene functions of the midgut microflora of R. microplus.
Methods
Sample collection and DNA extraction
Twenty fully engorged adult female R. microplus were collected from the body surfaces of cattle located in the city of Changsha in Hunan province (28º12′N, 112º59′E), China. All of the R. microplus samples were immediately transferred to Hunan Agricultural University. Five fully engorged adult female R. microplus were utilized for the analysis. Before dissection, the five ticks were surface disinfected with 70% (volume/volume) ethanol for 60 s followed by immersion in three reagents, 100% ethanol, 10% sodium hypochlorite solution, and distilled water, to remove the disinfectant. All of the dissecting apparatuses, plasticware, glassware, buffers (including phosphate-buffered saline), and solutions were sterilized by autoclaving and UV treatment. All of the procedures were conducted in a biosafety cabinet after UV sterilization to protect the samples from environmental contamination.
The five ticks were stabilized with fine-tipped forceps by holding the rear portion. The rear of each tick was cut using sterile ophthalmic scissors, and the midgut contents from each tick (100 μl) were pooled into a single tube. Then 1000 μl of hexadecyltrimethylammonium bromide lysate and 20 μl lysozyme were added to each tube, and the five tubes labeled as follows: R.M.1, R.M.2, R.M.3, R.M.4, and R.M.5. The five tubes were placed in a water bath at 65 °C for 2 h, during which time the tubes were inverted several times to ensure that the samples were fully lysed. After centrifugation (5022 g for 10 min), 950 μl supernatant of each sample was absorbed, and the same volume of phenol (pH 8.0):chloroform:isoamyl alcohol (25:24:1) mixture was added and mixed. After centrifugation (8609 g for 10 min), the supernatants were absorbed, and the same volume of chloroform:isoamyl alcohol (24:1) was added and mixed. After centrifugation (8609 g for 10 min), the supernatant of each sample was transferred to a 1.5-ml centrifuge tube. Then, a 3/4 volume of isopropanol was added to the supernatant of each sample; the tube was shaken and then the sample precipitated at − 20 °C. The sample tubes were centrifuged at 8609 g for 10 min, the liquid removed, and the sample washed twice with 1 ml of 75% (volume/volume) ethanol, after which the ethanol was removed. After that, the precipitate of each sample was allowed to dry naturally at room temperature, and then 50 μl double distilled H2O was added to dissolve the DNA. The sample was incubated at 60 °C for 10 min, and 1 μl RNase A was added to digest RNA. The sample was then incubated at 37 °C for 15 min.
Library construction and sequencing
A total of 1 μg DNA per sample was used as input material for the DNA sample preparation. Sequencing libraries were generated using a NEBNext Ultra DNA Library Prep Kit (New England Biolabs, Ipswich, MA) following the manufacturer’s protocol. Briefly, the DNA samples were fragmented by sonication to a size of 350 base pairs (bp), after which the DNA fragments were end-polished, A-tailed, and ligated with the full-length adaptor for Illumina sequencing with further PCR amplification. Finally, the PCR products were purified by the AMPure XP system (Beckman Coulter, Brea, CA). The libraries were analyzed for size distribution using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA) and quantified using real-time PCR (Thermo Fisher Scientific, Waltham, MA). The clustering of the index-coded samples was performed on a cBot Cluster Generation System (Illumina, San Diego, CA) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina HiSeq platform (Illumina), and paired-end reads were generated.
Pretreatment of sequencing results
Preprocessing of the raw data obtained from the Illumina HiSeq platform (Illumina) using Readfq (v8; https://github.com/cjfields/readfq) was conducted to acquire the clean data for subsequent analysis. The specific processing steps were as follows: removal of the reads that contained low-quality bases (default quality threshold value ≤ 38) above a certain portion (default length of 40 bp); removal of the reads in which the N base had reached a certain percentage (default length of 10 bp); removal of the reads that shared an overlap above a certain portion with the adapter (default length of 15 bp); the reads that were of host origin were also filtered by Bowtie 2.2.4 software (http://bowtiebio.sourceforge.net/bowtie2/index.shtml).
Metagenome assembly
The clean data were assembled and analyzed using SOAPdenovo software (v2.04; http://soap.genomics.org.cn/soapdenovo.html) [31]. For a single sample, k-mer = 55 was selected for assembly to obtain scaffolds of the sample, with the following parameters: -d 1, -M 3, -R, -u, -F, -K 55 [32,33,34,35]. Then, the assembled scaffolds were broken from the N connections to obtain scaftigs without N [32, 36, 37]. All the clean data for the samples were compared to the respective scaftigs using Bowtie 2.2.4 software to acquire the paired-end reads not used. The parameters were as follows: –end-to-end, –sensitive, -I 200, -X 400 [32]. All reads not used in the forward step for all the samples were combined. Then, SOAPdenovo software (v2.04; http://soap.genomics.org.cn/soapdenovo.html) was used for mixed assembly with the same parameters as for the single assembly. The mixed assembly scaffolds were broken from the N connection to obtain the scaftigs. Any fragments shorter than 500 bp in the scaftigs were filtered. The filtered scaftigs from the single or mixed assembly were statistically analyzed.
Gene prediction and abundance analysis
MetaGeneMark software (v2.10; http://topaz.gatech.edu/GeneMark/) was used to predict the open reading frame of the scaftigs (≥ 500 bp) from the single and mixed assembly. Sequence information from the predicted results with lengths less than 100 nucleotides [32, 37,38,39,40] was filtered. CD-HIT software (v4.5.8; http://www.bioinformatics.org/cd-hit) [41, 42] was used to remove redundancy and obtain the unique initial gene catalogue (the parameter options were –c 0.95, –G 0, –aS 0.9, –g 1, –d 0 [39, 43]). The longest sequences were selected as the representative sequences, and those sequences with 95% identity and 90% coverage were clustered. The clean data of each sample were mapped to an initial gene catalogue using Bowtie 2.2.4 (the parameter settings were –end-to-end, –sensitive, –I 200, –X 400 [32, 40]) to obtain the number of reads to which genes mapped in each sample. The genes for which the number of reads was ≤ 2 [40, 44] were filtered in each sample to obtain the gene catalogue (unigenes). The abundance of information for each gene in each sample was calculated based on the number of mapped reads and the length of the gene [38, 39, 45,46,47]. The basic informative statistics, core-pan gene analysis, correlation analysis of samples, and Venn diagram analysis of the numbers of genes were carried out based on the abundance of each gene in each sample of unigenes.
Species annotation
DIAMOND software (v0.9.9; https://github.com/bbuchfink/diamond/) [48] was used to compare the unigenes with the sequences of bacteria, fungi, archaea, and viruses that were extracted from the nr database (v2018-01–02; https://www.ncbi.nlm.nih.gov/) of the National Center for Biotechnology Information (NCBI) [the parameter settings were Basic Local Alignment Search Tool Program (BLASTP) E value ≤ 1e−5]. The previous alignment results were employed for the latent class analysis algorithm [49] that was applied to the classification from MEGAN software [50] to verify the species annotation information of the sequences. A table containing the number of genes and the abundance information for each sample in a taxonomic hierarchy (kingdom, phylum, class, order, family, genus, and species) was obtained based on the latent class analysis annotation results and the gene abundance table. The abundance of a species in one sample was equal to the sum of the gene abundances annotated for the species [38, 45, 51]; the number of genes of a species in a sample was equal to the number of genes with nonzero abundance. Krona analysis, the generation of relative abundance, and the construction of an abundance cluster heat map were carried out based on the abundance table of each taxonomic hierarchy.
Common functional database annotations
DIAMOND software (v0.9.9) was used to compare unigenes with the KEGG [52, 53] database (v2018-01-01; http://www.kegg.jp/kegg/) with the parameter setting of BLASTP, E value ≤ 1e−5 [33, 40]. For each sequence’s alignment result, the best BLAST hit (one high-scoring segment pair > 60 bits) was used for subsequent analysis [33, 40, 54]. The relative abundances of different functional hierarchies were calculated (the relative abundance of each functional level was equal to the sum of the relative abundances of genes annotated at the functional level) [32, 39]. The table of the number of genes of each sample in each taxonomic hierarchy was based on the results of the functional annotation and the table of gene abundances. The number of genes of a function in a sample was equal to the number of genes that was annotated for this function given that the abundance was nonzero. Based on the abundance table of each taxonomic hierarchy, the number of annotated genes was determined; the general relative abundance and the abundance cluster heat map were constructed, and the metabolic pathways were analyzed.
Results
Test results of DNA quality
A total of 26,678.61 Mbp of clean data were generated by sequencing with the Illumina HiSeq platform. The effective data rate was 99.7%. Specific data output statistics and quality control information are shown in Table 1.
General statistics
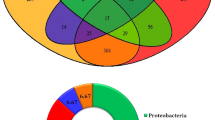
Following quality control, 10,403, 10,253, 10,515, 10,090, and 10,415 genes were obtained from the sequencing data of R.M.1, R.M.2, R.M.3, R.M.4, and R.M.5, respectively. The distribution of the genes of the five samples is shown in Fig. 1. The number of genes common to all five samples was 7,795. The numbers of genes specific to R.M.1, R.M.2, R.M.3, R.M.4, and R.M.5 were 46, 87, 131, 47, and 60, respectively. These results suggest that a large proportion of the microbial population of the five samples was identical, and that relatively small differences existed between individuals.

Venn diagram analysis of the number of genes of the samples of Rhipicephalus microplus. Each circle represents a sample; the numbers in the overlapping areas indicate the number of genes shared between the samples. The numbers in the non-overlapping areas indicate the number of unique genes in the sample
Relative abundance of microorganisms
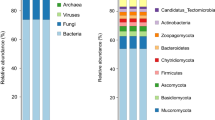
Sixteen phyla were common to the five samples. The microbial population characteristics of the 10 most abundant phyla in the five samples are shown in Fig. 2. Of these, Firmicutes, Proteobacteria, Actinobacteria, and Spirochaetes were the main phyla in all the samples. Firmicutes was predominant (≥ 25% of genes) in all samples.

Microbial population characteristics of the 10 most abundant phyla in the five samples of Rhipicephalus microplus
Ninety-five genera were common to the five samples. The relative abundances of these 95 genera are shown in Additional file 1: Table S1. Of these, Streptococcus, Mycobacterium, Anaplasma, Enterococcus, Shigella, Lactobacillus, Brachyspira, Pseudomonas, Enterobacter, Bacillus, and Lactococcus were the dominant genera in the five samples (Fig. 3). Five genera (Mycobacterium, Brachyspira, Campylobacter, Occidentia, and Neisseria) that were not reported in previous studies of R. microplus were thus, to the best of our knowledge, found for the first time in this species in the present study, despite their abundances being relatively low. Coxiella was not found in the midgut of R. microplus in the present study.

Microbial population characteristics of the 10 most abundant genera in the five samples of Rhipicephalus microplus
At the species level, 144 species were common to the five samples. These included 76 bacterial species, 47 eukaryotes, and 21 viral species. The relative abundances of the most common 35 bacterial species, the 47 eukaryotes, the 21 viral species, and other bacterial species are shown in Tables 2, 3, 4 and Additional file 1: Table S2, respectively. Of the bacterial species, Mycobacterium malmesburyense, Streptococcus pneumoniae, Anaplasma phagocytophilum, Enterococcus faecium, Shigella sonnei, Enterococcus faecalis, Lactobacillus casei, Brachyspira hampsonii, Pseudomonas syringae, Enterobacter cloacae, and Lactococcus garvieae were the dominant ones in the five samples. The dominant eukaryotes were Rhizophagus irregularis, Enterospora canceri, Smittium culicis, Zancudomyces culisetae, and Trachipleistophora hominis. Orf virus, human endogenous retrovirus (HERV) type W (HERV-W), enzootic nasal tumor virus of goats, bovine retrovirus CH15, and galidia endogenous retrovirus were the dominant viruses.
Cluster analysis of the number and relative abundance of annotated genes
The 35 most common genera and their abundance information for each sample were selected from the relative abundance tables at the different taxonomic levels to draw a heat map. The clustering was conducted at the species level to facilitate the result display and information discovery in order to identify the species clustering in the sample. The unigenes of Anaplasma were the most concentrated among all 35 genera in all of the samples, clustering into a single branch (Fig. 4a). The relative abundances of Anaplasma and Enterobacter were lowest in R.M.1, while the relative abundance of Enterobacter was high in R.M.3 (Fig. 4b).

Number of genes and abundance clustering heat map at the genus level. a Heat map of the annotated unigene statistics. Sample names are given on the horizontal axis, and species’ information on the vertical axis; different colors represent the number of unigenes. b Relative abundance clustering heat map at the genus level. Sample information is given on the horizontal axis; the vertical axis gives the species’ information. The cluster tree on the left of the figure is the species cluster tree; the value corresponding to the middle heat map is the Z-value obtained after the relative abundance of each species row is standardized
Gene function prediction of microflora
Unigenes were compared with the KEGG database using DIAMOND software. Based on the alignment results, the relative abundance of different functional levels was counted. At level 1, the number of KEGG pathways annotated to metabolism-related functional genes in the microbiome of R. microplus was 72; the number of functional genes associated with human diseases was 145 (Fig. 5). At level 2, genes of the microbiome of R. microplus were associated with 11 metabolic processes, among which the functional genes involved in lipid metabolism were the most abundant, followed by those involved in amino acid metabolism (Fig. 6). Genes associated with 11 human diseases, including cancer and infectious diseases (such as Streptococcus pneumonia infection, human granulocytic anaplasmosis, Shigella sonnei infection, Salmonella enterica infection, and pathogenic Escherichia coli infection), were significantly more abundant than the other functional genes (Fig. 7).

Annotations of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways related to the number of genes of the midgut microbiota in the five samples of Rhipicephalus microplus at level 1. The black font on the ordinate indicates KEGG level 1, the colored fonts indicate the specific pathway at this level, and the abscissa indicates the number of genes in the pathway

Relative abundance of midgut microbial genes involved in metabolism in the five samples of Rhipicephalus microplus at level 2 in KEGG pathway annotation

Relative abundance of midgut microbial genes involved in human diseases in the five samples of Rhipicephalus microplus at level 2 in KEGG pathway annotation
Discussion
In this study, the microbial population in the midgut of R. microplus was investigated using a metagenomic method. Several of the main phyla, including Firmicutes, Proteobacteria, and Actinobacteria, have been reported in previous studies [55,56,57,58,59]. The results obtained here were somewhat consistent with those of a previous study [27] in which Proteobacteria were present at a high relative abundance. To the best of our knowledge, Mycobacterium, Occidentia, Brachyspira, Campylobacter, and Neisseria are reported here for the first time in R. microplus. Coxiella was detected in the midgut of R. microplus in a previous study [59], but it was not detected in this study. Another relevant finding of our study was the presence of Ehrlichia, which has previously been confirmed to exist in the midgut of R. microplus [27].
In addition, we discovered that the gene functions of the midgut microflora of R. microplus are related to lipid and amino acid metabolism; this may be due to the fact that ticks, which mostly live in agricultural areas and forests, mainly feed on the blood of their hosts [60]. The functions of some of the genes of the midgut microbiota found in the present study are associated with human diseases, and some of the most abundant microbial species are associated with infectious diseases and cancer, which suggests that ticks may be infected with various pathogens. For example, A. phagocytophilum, a tick-borne fever pathogen in ruminants [61], was detected in all of the samples.
Anaplasma, which belongs to the family Anaplasmataceae and is transmitted by arthropod vectors, can cause severe anemia [62]. Anaplasma phagocytophilum is a zoonotic pathogen found in the granulocytes of animals and humans which can infect human peripheral blood neutrophils and lead to tick-borne diseases and symptoms of human granulocytic anaplasmosis, such as fever accompanied by leukopenia, thrombocytopenia, and functional impairment [63]. Domestic animals are important hosts of A. phagocytophilum [64]. In this study, A. phagocytophilum was detected in the R. microplus samples, which were collected from cattle in Hunan province. In addition, Ehrlichia minasensis, which was previously detected in the hemolymph of R. microplus from Brazil [65], was also detected in this study, at low abundance. Ehrlichia minasensis has also recently been identified from cattle on the French island of Corsica [66], and was also found in the serum of Brazilian dogs [67].
Occidentia, a newly identified genus of the family Rickettsiaceae, is a Gram-negative obligate intracellular bacillus [68] that is intimately associated with its arthropod hosts [69]. Occidentia was isolated from the rodent-associated soft tick Ornithodoros sonrai collected in Senegal [68]. A recent study showed that Occidentia also exists in the hard tick Africaniella transversal [70]. In the present study, Occidentia was detected in the midgut of the five R. microplus examined. These findings show that Occidentia infects ticks of the families Argasidae and Ixodidae. Among the detected species, the endosymbionts Rickettsia and Wolbachia were also found, although at low abundances. Through their symbiosis with their host, they affect not only their host’s ecology and evolution but also its reproductive development [71]. Wolbachia, which is present in 66% of insect species, is probably the most abundant endosymbiont on the planet [72]. The presence of the endosymbionts Rickettsia in host insects, and their extensive horizontal transmission, may have contributed to their widespread occurrence in natural populations of insects [71, 73].
Brachyspira cause porcine intestinal spirochetosis, a condition in which diseased animals display chronic diarrhea, rectal bleeding, and lower abdominal cramps [74]. These pathogens do not cause serious disease in swine, but they do have an impact on humans. Some species of Brachyspira, such as Brachyspira pilosicoli and Brachyspira aalborgi, can cause similar symptoms in humans to those seen in swine [74]. Brachyspira have been isolated from the gastrointestinal tracts of mammals and birds, and from habitats contaminated with feces [75]. However, to date, there have been few reports of Brachyspira-infected cattle developing severe disease. In the present study, Brachyspira was detected in R. microplus at a moderately high relative abundance, which is a another reason why preventative measures should be taken to protect cattle from this tick.
The phylum Microsporidia comprises single-celled eukaryotic obligate intracellular parasites that can infect insects (e.g. Nosema apis and Nosema ceranae), fish, mammals, and even humans with immune deficiency diseases [76]. When microsporidia infect humans [77], they can cause diarrhea, myositis, keratitis, bronchitis, and encephalitis [78]. The microsporidian S. culicis is widely distributed and has been reported to infect Culicidae, Chironomidae, and Simuliidae [79]. In the present study, S. culicis was detected in the midgut of R. microplus, suggesting that this tick can carry this microsporidian, although further research is needed to determine whether R. microplus can transmit it.
Orf virus is a highly epitheliotropic parapoxvirus. It may not only cause great production losses in animal husbandry, but also affect human health [80, 81]. The clinical symptoms of orf virus infection in animals are erythema, papules, and blisters on the lips and tongue, followed by severe ulceration and, finally, the formation of scabs [82]. The pathological features of orf virus infection in humans and animals are similar and are confined to the epidermis [83]. The main clinical manifestations in humans are skin lesions on the fingers and hands after contact with infected animals [84, 85]. In addition to direct transmission, orf virus can also be transmitted by flies and ticks. The present study showed that orf virus exists in the midgut of R. microplus. Therefore, the eradication of R. microplus should be pursued in Hunan province, and orf virus monitored to ensure successful livestock farming and healthy animals.
HERV, which originate from exogenous retrovirus infections in germ cells that occurred millions of years ago, have the potential to cause human disease [86]. There are reports of increased expression of HERV-W in cases of schizophrenia and bipolar disorder, which is associated with distinct clinical or biological characteristics and symptoms [87,88,89]. There have been no previous reports of HERV-W in ticks. In this study, HERV were detected in the midgut of R. microplus, suggesting that R. microplus can carry these viruses.
Conclusions
In this study, we analyzed the midgut microbiome of fully engorged adult female R. microplus from cattle in the city of Changsha in Hunan province, China, using a metagenomic sequencing method. We found that 16 phyla, 95 genera, and 144 species were common to the five samples. The midgut microbiome of this species of tick was not only composed of a large number of bacteria, but also eukaryotes and viruses. These results add to our understanding of the midgut microbiome of R. microplus. The annotated KEGG pathway predictions of the functions of the genes of the midgut microflora of R. microplus indicated that they play a role in lipid and amino acid metabolism, infectious diseases, and cancer. These findings provide fundamental information on the physiology of ticks and their transmission of disease.
Availability of data and materials
The raw Illumina sequencing data generated in this study are available from NCBI Sequence Read Archive, BioProject no. PRJNA748905 with BioSamples accession nos. SRR15276227, SRR15276228, SRR15276229, SRR15276230 and SRR15276231.
Abbreviations
- HERV:
-
Human endogenous retrovirus
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PCR:
-
Polymerase chain reaction
References
de Barros MNDL, Riet-Correa F, Azevedo SS, Labruna MB. Off-host development and survival of Rhipicephalus (Boophilus) microplus in the Brazilian semiarid. Vet Parasitol Reg Stud Reports. 2017;9:17–24.
Vesco U, Knap N, Labruna MB, Avsic-Zupanc T, Estrada-Pena A, Guglielmone AA, et al. An integrated database on ticks and tick-borne zoonoses in the tropics and subtropics with special reference to developing and emerging countries. Exp Appl Acarol. 2011;54:65–83.
Estrada-Peña A, García Z, Sánchez HF. The distribution and ecological preferences of Boophilus microplus (Acari: Ixodidae) in Mexico. Exp Appl Acarol. 2006;38:307–16.
Nava S, Mastropaolo M, Guglielmone AA, Mangold AJ. Effect of deforestation and introduction of exotic grasses as livestock forage on the population dynamics of the cattle tick Rhipicephalus (Boophilus) microplus (Acari: Ixodidae) in northern Argentina. Res Vet Sci. 2013;95:1046–54.
Chen Z, Yang X, Bu F, Yang X, Yang X, Liu J. Ticks (acari: ixodoidea: argasidae, ixodidae) of China. Exp Appl Acarol. 2010;51:393–404.
Jongejan F, Uilenberg G. The global importance of ticks. Parasitology. 2004;129(Suppl):S3-14.
Jabbar A, Abbas T, Sandhu ZU, Saddiqi HA, Qamar MF, Gasser RB. Tick-borne diseases of bovines in Pakistan: major scope for future research and improved control. Parasit Vectors. 2015;8:283.
Ceylan O, Byamukama B, Ceylan C, Galon EM, Liu M, Masatani T, et al. Tick-borne hemoparasites of sheep: a molecular research in Turkey. Pathogens. 2021;10:162.
Andreotti R, Pérez de León AA, Dowd SE, Guerrero FD, Bendele KG, Scoles GA. Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiol. 2011;11:6.
Sungirai M, Baron S, Van der Merwe NA, Moyo DZ, De Clercq P, Maritz-Olivier C, et al. Population structure and genetic diversity of Rhipicephalus microplus in Zimbabwe. Acta Trop. 2018;180:42–6.
Misra BR, Kumar N, Kant R, Deval H, Singh R, Pandey AK, et al. Abundance of ticks (Acari: Ixodidae) and presence of Rickettsia and Anaplasma in ticks infesting domestic animals from northern India. J Med Entomol. 2021;58:1370–5.
Pesquera C, Portillo A, Palomar AM, Oteo JA. Investigation of tick-borne bacteria (Rickettsia spp., Anaplasma spp., Ehrlichia spp. and Borrelia spp.) in ticks collected from Andean tapirs, cattle and vegetation from a protected area in Ecuador. Parasit Vectors. 2015;8:46.
Yawa M, Nyangiwe N, Jaja IF, Kadzere CT, Marufu MC. Prevalence of serum antibodies of tick-borne diseases and the presence of Rhipicephalus microplus in communal grazing cattle in the north-eastern region of the Eastern Cape Province of South Africa. Parasitol Res. 2021;120:1183–91.
Fang LQ, Liu K, Li XL, Liang S, Yang Y, Yao HW, et al. Emerging tick-borne infections in mainland China: an increasing public health threat. Lancet Infect Dis. 2015;15:1467–79.
Guo WP, Zhang B, Wang YH, Xu G, Wang X, Ni X, et al. Molecular identification and characterization of Anaplasma capra and Anaplasma platys-like in Rhipicephalus microplus in Ankang. Northwest China BMC Infect Dis. 2019;19:434.
Kaur R, Yadav A, Rafiqi SI, Godara R, Sudan V, Chakraborty D, et al. Epidemiology, haematology and molecular characterization of haemoprotozoon and rickettsial organisms causing infections in cattle of Jammu region. North India BMC Vet Res. 2021;17:219.
Giglioti R, Okino CH, Azevedo BT, Wedy BCR, Gutmanis G, Veríssimo CJ, et al. Semi-quantitative evaluation of Babesia bovis and B. bigemina infection levels estimated by HRM analysis. Ticks Tick Borne Dis. 2021;12: 101753.
Gao D, Cao W, Zhang X. Investigations on human Ehrlichia infectious people in Daxingan Mountains. Chin J Epidemiol. 2001;22:137–41 (in Chinese).
Dong T, Qu Z, Zhang L. Detection of A. phagocytophilum and E. chaffeensis in patient and mouse blood and ticks by a duplex real-time PCR assay. PLoS ONE. 2013;8: e74796.
Tokarz R, Tagliafierro T, Sameroff S, Cucura DM, Oleynik A, Che X, et al. Microbiome analysis of Ixodes scapularis ticks from New York and Connecticut. Ticks Tick Borne Dis. 2019;10:894–900.
Fischer SG, Lerman LS. DNA fragments differing by single base-pair substitutions are separated in denaturing gradient gels: correspondence with melting theory. Proc Natl Acad Sci USA. 1983;80:1579–83.
Muyzer G, de Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59:695–700.
Schabereiter-Gurtner C, Lubitz W, Rölleke S. Application of broad-range 16S rRNA PCR amplification and DGGE fingerprinting for detection of tick-infecting bacteria. J Microbiol Methods. 2003;52:251–60.
Cheng TY, Li ZB, Zou AD, Liu GH. PCR-DGGE test for direct identification of intestinal bacterial flora in blood feeding ticks in China. Trop Biomed. 2016;33:663–7.
Xu XL, Cheng TY, Yang H, Yan F. Identification of intestinal bacterial flora in Rhipicephalus microplus ticks by conventional methods and PCR-DGGE analysis. Exp Appl Acarol. 2015;66:257–68.
Lima RAT, De Oliveira G, Souza AA, Lopes FAC, Santana RH, Istvan P, et al. Functional and structural characterization of a novel GH3 beta-glucosidase from the gut metagenome of the Brazilian Cerrado termite Syntermes wheeleri. Int J Biol Macromol. 2020;165(Pt A):822–34.
Segura JA, Isaza JP, Botero LE, Alzate JF, Gutiérrez LA. Assessment of bacterial diversity of Rhipicephalus microplus ticks from two livestock agroecosystems in Antioquia, Colombia. PloS ONE. 2020;15: e0234005.
Bouquet J, Melgar M, Swei A, Delwart E, Lane RS, Chiu CY. Metagenomic-based surveillance of Pacific coast tick Dermacentor occidentalis identifies two novel bunyaviruses and an emerging human ricksettsial pathogen. Sci Rep. 2017;7:12234.
Carpi G, Cagnacci F, Wittekindt NE, Zhao F, Qi J, Tomsho LP, et al. Metagenomic profile of the bacterial communities associated with Ixodes ricinus ticks. PLoS ONE. 2011;6: e25604.
Adegoke A, Kumar D, Bobo C, Rashid MI, Durrani AZ, Sajid MS, et al. Tick-borne pathogens shape the native microbiome within tick vectors. Microorganisms. 2020;8:1299.
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18.
Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513:59–64.
Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun. 2015;6:6528.
Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2: e01202.
Brum JR, Ignacio-Espinoza JC, Roux S, Doulcier G, Acinas SG, Alberti A, et al. Ocean plankton. Patterns and ecological drivers of ocean viral communities. Science. 2015;348:1261498.
Mende DR, Waller AS, Sunagawa S, Järvelin AI, Chan MM, Arumugam M, et al. Assessment of metagenomic assembly using simulated next generation sequencing data. PloS ONE. 2012;7: e31386.
Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 2014;32:822–8.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65.
Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol. 2014;10:766.
Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–41.
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9.
Fu L, Niu B, Zhu Z, Wu S, Li W. Cd-hit: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2.
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Ocean plankton. Structure and function of the global ocean microbiome. Science. 2015;348:1261359.
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60.
Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245.
Villar E, Farrant GK, Follows M, Garczarek L, Speich S, Audic S, et al. Ocean plankton. Environmental characteristics of Agulhas rings affect interocean plankton transport. Science. 2015;348:1261447.
Aron-Wisnewsky J, Prifti E, Belda E, Ichou F, Kayser BD, Dao MC, et al. Major microbiota dysbiosis in severe obesity: fate after bariatric surgery. Gut. 2019;68:70–82.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Oh J, Byrd AL, Deming C, Conlan S, Kong HH, Segre JA. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514:59–64.
Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–60.
Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99–103.
Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, et al. From genomics to chemical genomics new developments in KEGG. Nucleic Acids Res. 2006;34(Database issue):D354-7.
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42(Database issue):D199-205.
Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:852.
Budachetri K, Browning RE, Adamson SW, Dowd SE, Chao CC, Ching WM, et al. An insight into the microbiome of the Amblyomma maculatum (Acari: Ixodidae). J Med Entomol. 2014;51:119–29.
Karim S, Budachetri K, Mukherjee N, Williams J, Kausar A, Hassan MJ, et al. A study of ticks and tick-borne livestock pathogens in Pakistan. PLoS Negl Trop Dis. 2017;11: e0005681.
Narasimhan S, Rajeevan N, Liu L, Zhao YO, Heisig J, Pan J, et al. Gut microbiota of the tick vector Ixodes scapularis modulate colonization of the Lyme disease spirochete. Cell Host Microbe. 2014;15:58–71.
Travanty NV, Ponnusamy L, Kakumanu ML, Nicholson WL, Apperson CS. Diversity and structure of the bacterial microbiome of the American dog tick, Dermacentor variabilis, is dominated by the endosymbiont Francisella. Symbiosis. 2019;79:239–50.
Xiang L, Poźniak B, Cheng TY. Bacteriological analysis of saliva from partially or fully engorged female adult Rhipicephalus microplus by next-generation sequencing. Antonie Van Leeuwenhoek. 2017;110:105–13.
Toprak U, Musselman LP. From cellular biochemistry to systems physiology: new insights into insect lipid metabolism. Insect Biochem Mol Biol. 2021;133: 103585.
Bakken JS, Krueth J, Tilden RL, Dumler JS, Kristiansen BE. Serological evidence of human granulocytic ehrlichiosis in Norway. Eur J Clin Microbiol Infect Dis. 1996;15:829–32.
Diaz-Cao JM, Adaszek L, Dziegiel B, Paniagua J, Caballero-Gomez J, Winiarczyk S, et al. Prevalence of selected tick-borne pathogens in wild ungulates and ticks in southern Spain. Transbound Emerg Dis. 2021. https://doi.org/10.1111/tbed.14065.
Kovryha N, Tsyhankova A, Zelenuchina O, Mashchak O, Terekhov R, Rogovskyy AS. Prevalence of Borrelia burgdorferi and Anaplasma phagocytophilum in ixodid ticks from southeastern Ukraine. Vector Borne Zoonotic Dis. 2021;21:242–6.
Asman M, Witecka J, Korbecki J, Solarz K. The potential risk of exposure to Borrelia garinii, Anaplasma phagocytophilum and Babesia microti in the Wolinski National Park (north-western Poland). Sci Rep. 2021;11:4860.
Cabezas-Cruz A, Zweygarth E, Vancová M, Broniszewska M, Grubhoffer L, Passos LMF, et al. Ehrlichia minasensis sp. nov., isolated from the tick Rhipicephalus microplus. Int J Syst Evol Microbiol. 2016;66:1426–30.
Cicculli V, Masse S, Capai L, de Lamballerie X, Charrel R, Falchi A. First detection of Ehrlichia minasensis in Hyalomma marginatum ticks collected from cattle in Corsica, France. Vet Med Sci. 2019;5:243–8.
Melo ALT, Luo T, Zhang X, Muraro LS, Pereira NA, Cabezas-Cruz A, et al. Serological evidence of Ehrlichia minasensis infection in Brazilian dogs. Acta Trop. 2021;219: 105931.
Mediannikov O, Nguyen TT, Bell-Sakyi L, Padmanabhan R, Fournier PE, Raoult D. High quality draft genome sequence and description of Occidentia massiliensis gen. nov., sp nov, a new member of the family Rickettsiaceae. Stand Genomic Sci. 2014;9:9.
Yu XJ, Walker DH. Family I. Rickettsiaceae. In: Yu XJ, Walker DH, editors. Bergeys’s manual of systematic bacteriology, (The Proteobacteria), part C (the Alpha-, Beta-, Delta-, and Epsilonproteobacteria), vol. 2. 2nd ed. New York: Springer; 2005. p. 1–324.
Hornok S, Kontschán J, Takács N, Chaber AL, Halajian A, Szekeres S, et al. Rickettsiaceae in two reptile-associated tick species, Amblyomma exornatum and Africaniella transversale: first evidence of Occidentia massiliensis in hard ticks (Acari: Ixodidae). Ticks Tick Borne Dis. 2021;13: 101830.
Caspi-Fluger A, Inbar M, Mozes-Daube N, Katzir N, Portnoy V, Belausov E, et al. Horizontal transmission of the insect symbiont Rickettsia is plant-mediated. Proc Biol Sci. 2012;279:1791–6.
Landmann F. The Wolbachia endosymbionts. Microbiol Spectr. 2019;7:1–15.
Himler AG, Adachi-Hagimori T, Bergen JE, Kozuch A, Kelly SE, Tabashnik BE, et al. Rapid spread of a bacterial symbiont in an invasive whitefly is driven by fitness benefits and female bias. Science. 2011;332:254–6.
Mikosza AS, Hampson DJ. Human intestinal spirochetosis: Brachyspira aalborgi and/or Brachyspira pilosicoli? Anim Health Res Rev. 2001;2:101–10.
Mappley LJ, La Ragione RM, Woodward MJ. Brachyspira and its role in avian intestinal spirochaetosis. Vet Microbiol. 2014;168:245–60.
Kaczmarek A, Boguś MI. Fungi of entomopathogenic potential in Chytridiomycota and Blastocladiomycota, and in fungal allies of the Oomycota and Microsporidia. IMA Fungus. 2021;12:29.
Weber R, Bryan RT. Microsporidial infections in immunodeficient and immunocompetent patients. Clin Infect Dis. 1994;19:517–21.
Ghosh K, Weiss LM. Molecular diagnostic tests for Microsporidia. Interdiscip Perspect Infect Dis. 2009;2009: 926521.
Alencar YB, Rios-Velasquez CM, Lichtwardt RW, Hamada N. Trichomycetes (Zygomycota) in the digestive tract of arthropods in Amazonas, Brazil. Mem Inst Oswaldo Cruz. 2003;98:799–810.
Büttner M, Rziha HJ. Parapoxviruses: from the lesion to the viral genome. J Vet Med B Infect Dis Vet Public Health. 2002;49:7–16.
Maclachlan NJ, Dubovi EJ, Maclachlan JN, Dubovi EJ, et al. Fenner’s veterinary virology. 4th ed. San Diego: Elsevier; 2011. p. 226–31.
Wise LM, Stuart GS, Jones NC, Fleming SB, Mercer AA. Orf virus IL-10 and VEGF-E act synergistically to enhance healing of cutaneous wounds in mice. J Clin Med. 2020;9:1085.
Savory LJ, Stacker SA, Fleming SB, Niven BE, Mercer AA. Viral vascular endothelial growth factor plays a critical role in orf virus infection. J Virol. 2000;74:10699–706.
Zhou Y, Guan J, Gao F, Li Z, Lan Y, Lu H, et al. Orf virus ORF120 protein positively regulates the NF-κB pathway by interacting with G3BP1. J Virol. 2021;95: e0015321.
Nougairede A, Fossati C, Salez N, Cohen-Bacrie S, Ninove L, Michel F, et al. Sheep-to-human transmission of orf virus during Eid al-Adha religious practices, France. Emerg Infect Dis. 2013;19:102–5.
Zhou Y, Liu L, Liu Y, Zhou P, Yan Q, Yu H, et al. Implication of human endogenous retrovirus W family envelope in hepatocellular carcinoma promotes MEK/ERK-mediated metastatic invasiveness and doxorubicin resistance. Cell Death Discov. 2021;7:177.
Tamouza R, Meyer U, Foiselle M, Richard JR, Wu CL, Boukouaci W, et al. Identification of inflammatory subgroups of schizophrenia and bipolar disorder patients with HERV-W ENV antigenemia by unsupervised cluster analysis. Transl Psychiatry. 2021;11:377.
Perron H, Hamdani N, Faucard R, Lajnef M, Jamain S, Daban-Huard C, et al. Molecular characteristics of human endogenous retrovirus type-W in schizophrenia and bipolar disorder. Transl Psychiatry. 2012;2: e201.
Karlsson H, Schröder J, Bachmann S, Bottmer C, Yolken RH. HERV-W-related RNA detected in plasma from individuals with recent-onset schizophrenia or schizoaffective disorder. Mol Psychiatry. 2004;9:12–3.
Acknowledgements
Not applicable.
Funding
This study was supported by a grant from the National Science Foundation of China (no. 31902294), the Planned Programme of Hunan Province Science and Technology Innovation (grant no. 2018RS3085), the Training Programme for Excellent Young Innovators of Changsha (grant no. KH2002001), the Natural Science Foundation of Hunan Province, China (no. 2020JJ5230), and the Research Foundation of the Education Bureau of Hunan Province, China (no. 19A235).
Author information
Authors and Affiliations
Contributions
DYD and GHL conceived and designed the study and critically revised the manuscript. XLZ performed the experiments. YPD and TY analyzed the data. XLZ drafted the manuscript. LYL and TYC helped with the study design, study implementation, and manuscript preparation. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Animal Ethics Committee of Hunan Agricultural University (no. 43321503).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Relative abundance of microflora at the genus level of the five sampled ticks. Table S2. Relative abundance of the bacterial species of the five tick samples with the exception of the 35 most abundant species.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, XL., Deng, YP., Yang, T. et al. Metagenomics of the midgut microbiome of Rhipicephalus microplus from China. Parasites Vectors 15, 48 (2022). https://doi.org/10.1186/s13071-022-05161-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05161-6