
Abstract
Background
Mosquito-borne diseases have a devastating impact on human civilization. A few species of Anopheles mosquitoes are responsible for malaria transmission, and while there has been a reduction in malaria-related deaths worldwide, growing insecticide resistance is a cause for concern. Aedes mosquitoes are known vectors of viral infections, including dengue, yellow fever, chikungunya, and Zika. Aminoacyl-tRNA synthetases (aaRSs) are key players in protein synthesis and are potent anti-infective drug targets. The structure–function activity relationship of aaRSs in mosquitoes (in particular, Anopheles and Aedes spp.) remains unexplored.
Methods
We employed computational techniques to identify aaRSs from five different mosquito species (Anopheles culicifacies, Anopheles stephensi, Anopheles gambiae, Anopheles minimus, and Aedes aegypti). The VectorBase database (https://vectorbase.org/vectorbase/app) and web-based tools were utilized to predict the subcellular localizations (TargetP-2.0, UniProt, DeepLoc-1.0), physicochemical characteristics (ProtParam), and domain arrangements (PfAM, InterPro) of the aaRSs. Structural models for prolyl (PRS)-, and phenylalanyl (FRS)-tRNA synthetases—were generated using the I-TASSER and Phyre protein modeling servers.
Results
Among the vector species, a total of 37 (An. gambiae), 37 (An. culicifacies), 37 (An. stephensi), 37 (An. minimus), and 35 (Ae. aegypti) different aaRSs were characterized within their respective mosquito genomes. Sequence identity amongst the aaRSs from the four Anopheles spp. was > 80% and in Ae. aegypti was > 50%.
Conclusions
Structural analysis of two important aminoacyl-tRNA synthetases [prolyl (PRS) and phenylanalyl (FRS)] of Anopheles spp. suggests structural and sequence similarity with potential antimalarial inhibitor [halofuginone (HF) and bicyclic azetidine (BRD1369)] binding sites. This suggests the potential for repurposing of these inhibitors against the studied Anopheles spp. and Ae. aegypti.

Similar content being viewed by others


Each year approximately 700,000 deaths (~ 17% of total infectious disease deaths worldwide) are attributed to mosquito-borne diseases such as chikungunya, dengue, malaria, West Nile virus, and Zika [1,2,3]. Mosquitoes transmit diseases by harboring the disease-causing parasites or viruses that are transmitted to the host during blood-feeding [3, 4]. Increased insecticide resistance makes efforts to control mosquitoes challenging [5,6,7]. Worldwide there are over 3500 mosquito species [8]. Among the different species of mosquitoes, Aedes aegypti transmits viruses (e.g., dengue, yellow fever, chikungunya, and Zika), whereas Anopheles spp. transmit the malaria-causing parasites of Plasmodium spp. [9]. Regarding malaria, the three primary Anopheles vectors in India are (i) An. culicifacies (rural areas), which is responsible for the majority of malaria cases in India; (ii) An. stephensi (urban areas); and (iii) An. minimus (northeastern region of India) [10, 11]. Anopheles gambiae is highly effective in spreading malaria and is mainly localized to Africa. In addition to Plasmodium falciparum, An. gambiae hosts and transmits the filarial worm Wuchereria bancrofti that causes lymphatic filariasis [12]. With recent advancements in sequencing technologies, the full genomes of several mosquito species have been decoded and new protein targets identified. These findings hold promise for the discovery and development of novel insecticides [13].
Aminoacyl-tRNA synthetases (aaRSs) are also known as tRNA ligases, and they universally drive the protein translation process [14, 15]. The aminoacylation reaction catalyzed by aaRSs provides an opportunity for the development of protein translation inhibitors [16,17,18,19,20]. The main function of aaRSs is to append an amino acid to the respective tRNAs in an adenosine 5′-triphosphate (ATP)-dependent manner. First, ATP activates an amino acid to form an intermediate molecule known as “aminoacyl-adenylate.” In the next step, the intermediate is attached to the cognate tRNA molecule through covalent bond formation and the reaction is completed with the release of adenosine 5′-monophosphate (AMP) [21]. The aaRSs are multi-domain proteins with (i) a conserved catalytic domain (responsible for tRNA and amino acid ligation events), (ii) an anticodon binding domain (ABD) (responsible for binding of the anticodon region of the tRNA), and other additional domains for (iii) RNA binding and (iv) editing activity, or (v) C-terminal zinc-binding-like domain. The editing domain is responsible for removing incorrectly charged tRNA. In nature, there are 20 amino acids, and in general, for each amino acid there is at least one aaRS present. These 20 aaRSs can be broadly classified into either class I or class II. Class I and class II enzyme annotation is based on two factors: (i) possession of conserved structural motifs and (ii) their mode of substrate binding. Typical characteristics of class I enzymes are possession of a Rossmann fold, which is composed of two highly conserved motifs, KMSKS and HIGH. Compared to the class I type, class II aaRSs contain three conserved motifs in their domain and carry a unique fold composed of antiparallel beta strands [22]. The aaRSs show high sequence diversity, with structural differences and domain arrangements across organisms; however, aaRS catalytic domains are more conserved across species. Further, noncanonical functions of aaRSs include RNA splicing, transcription regulation, signal processing, immune responses, and apoptosis [23, 24]. For instance, the P. falciparum tyrosyl-tRNA synthetase (PfYRS) possesses cytokine-like activity [25]. The aaRSs are mainly located in the cytoplasm and the mitochondria for protein synthesis [26]. Recently, aaRSs have also emerged as a potential drug target for several eukaryotic pathogens (Leishmania, Plasmodium, and Toxoplasma) via multi-site targeting [16,17,18, 25, 27,28,29,30,31,32,33,34,35].
In the present study, computational tools were employed to characterize aaRSs present in the genomes of Anopheles (An. gambiae, An. culicifacies, An. stephensi, and An. minimus) and Ae. aegypti. All the different aaRSs present in the five studied mosquito species were annotated. Our analysis identified a total of 37 (An. gambiae), 37 (An. culicifacies), 37 (An. stephensi), 37 (An. minimus), and 35 (Ae. aegypti) different aaRSs in these respective genomes. We also investigated individual aaRS sequences in detail and predicted their isoelectric point (pI) and potential subcellular localizations. Furthermore, we determined the domain arrangements of all different aaRS spread across five different mosquito species and generated structural models of several important druggable targets. Overall, this study lays the groundwork for the development of next-generation insecticides against the Anopheles spp. and Ae. aegypti aaRSs.
Protein sequences of the aaRSs from the five mosquitoes (An. culicifacies, An. stephensi, An. minimus, An. gambiae, Ae. aegypti) were retrieved from the VectorBase database (https://vectorbase.org/vectorbase). The VectorBase (https://vectorbase.org/vectorbase) annotated protein sequences were further validated by comparing the sequences from the NCBI (https://www.ncbi.nlm.nih.gov/) and UniProt (https://www.uniprot.org/) databases. ProtoParam (https://web.expasy.org/protparam/) was used to characterize individual aaRS isoelectric points. VectorBase (https://vectorbase.org/vectorbase) was initially used to extract the protein sequence of aaRSs from An. gambiae, and this was used as a reference for genome annotation of the other mosquito species studied here via Protein BLAST. The aaRS sequences from An. culicifacies, An. stephensi, An. minimus, and Ae. aegypti were further screened against each other in various databases, including NCBI (https://www.ncbi.nlm.nih.gov/) and UniProt (https://www.uniprot.org/), for further validation.
Subdomains of individual aaRSs were determined using the online web servers Pfam (http://pfam.xfam.org/) and InterPro (https://www.ebi.ac.uk/interpro/). The same databases were also used to acquire information about domain function. The aaRS signal sequence/peptide and subcellular localization were predicted using the online web servers TargetP-2.0 (http://www.cbs.dtu.dk/services/TargetP/), SignalP 5.0 (http://www.cbs.dtu.dk/services/SignalP/), and DeepLoc-1.0 (http://www.cbs.dtu.dk/services/DeepLoc/). Multiple sequence alignment (MSA) of the drug binding site was performed utilizing MUSCLE (https://www.ebi.ac.uk/Tools/msa/muscle/). MSA for phylogenetic analysis was carried out using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Homology modeling of lysyl (KRS)-, prolyl (PRS)-, and phenylalanyl-tRNA (FRS) synthetases was carried out using two different web-based servers: Phyre (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index) and I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/server). The quality of the protein models was assessed using SAVES v6.0 (https://saves.mbi.ucla.edu/). Protein structure visualization was done using PyMOL (https://pymol.org/2/) and Chimera (https://www.cgl.ucsf.edu/chimera/download.html).
Our analyses identified 37 aaRSs enzymes in all four Anopheles spp. (An. culicifacies, An. stephensi, An. minimus, An. gambiae) and 35 in Ae. aegypti, including the bifunctional aaRSs—glutamyl-prolyl-tRNA synthetase (EPRS) (Fig. 1, Additional file 1: Table S1) [36]. Aedes aegypti carries only one copy of histidyl-(HRS) and lysyl-tRNA synthetase (KRS), whereas the four Anopheles spp. carry two copies each of KRS and HRS (Additional file 1: Table S1). All five mosquito species (four Anopheles spp. and Ae. aegypti) possess two copies of each aaRS, except for glycyl-(GRS), threonyl-(TRS), and glutaminyl-(QRS) tRNA synthetases, with one gene copy; and FRS, which has three copies, with two coding for the one FRS-alpha-like, one for FRS-alpha, and one for the FRS-beta subunit.

The distribution of aminoacyl-tRNA synthetases (aaRSs) in the studied mosquito species. aaRSs from Anopheles (An. culicifacies, An. stephensi, An. minimus, and An. gambiae) and Ae. aegypti were characterized based on topologies and subcellular location. a Class I and II annotation was performed based on the topologies of the central catalytic domain (CCD) and the mode of substrate binding. The number of aaRSs annotated for each studied species and their localization for class I, class II, and bifunctional aaRSs is shown, with exceptions listed in brackets. For class I aaRSs, An. gambiae, An. stephensii, and An. minimus carry nine cytoplasmic and nine mitochondrial aaRSs, whereas An. culicifacies and Ae. aegypti have 10 cytoplasmic and eight mitochondrial aaRSs. b The tentative distribution of the predicted subcellular localizations (cytoplasmic and mitochondrial) for the studied An. culicifacies, An. stephensi, An. minimus, An. gambiae, and Ae. aegypti is shown
The aaRSs are generally classified into two groups, class I and class II, based on the conserved topology of the synthetase core domain. We observed an equal number of proteins belonging to class I and II families (18 each), except for Ae. aegypti, where 18 class I and 16 class II type aaRSs were found (Fig. 1 and Additional file 1: Table S1). Additionally, a single protein copy of the bifunctional EPRS was present in all the studied mosquito species. The subcellular localization of the annotated aaRSs from the mosquito species was predicted using online servers. Anopheles gambiae and An. stephensi have 21 aaRSs localized to the cytoplasm and 16 to the mitochondria (Fig. 1 and Additional file 1: Table S1), whereas in An. minimus and An. culicifacies, out of a total of 37 aaRS enzymes, 22 are localized to the cytoplasm and 15 to the mitochondria. In Ae. aegypti, out of 35 aaRSs, 20 are predicted to localize to the cytoplasm and 15 to the mitochondria (Fig. 1). Consistent with other species such as Babesia spp., Plasmodium spp., and Homo sapiens, a larger number of aaRSs are found in the cytoplasm compared to the mitochondria [34, 37]. Generally, mitochondrial-targeting peptide is present at the N- or C-terminus or at the internal site of the protein [26]. It is worth noting that protein/tRNA migration to the mitochondria has been reported in the absence of signal peptides [38]. For instance, the charged tRNAs of the aaRSs are missing within the mitochondrial compartment, and they are imported from the cytoplasm in protozoa of the genera in Leishmania, Trypanosoma, Plasmodium, and Toxoplasma [39,40,41,42,43].
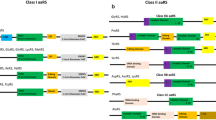
The aaRSs consist of multiple domains, with a core synthetase domain and several other auxiliary domains for RNA binding, editing, and oligomerization, some of which have been targeted for drug discovery [18]. Our analysis of the five mosquito species suggests the presence of a core catalytic domain and several additional domains for all studied aaRSs. For instance, the DALR [aspartate (D), alanine (A), leucine (L), arginine (R)] is an anticodon binding domain, which largely consists of an α-helical structure and was found in arginyl tRNA synthetase (RRS) of all mosquito species (Fig. 2 and Additional file 1: Table S1). The DHHA [aspartate (D), histidine (H), histidine (H), alanine (A)] domain is unique to the cytoplasmic variant of the (alanyl tRNA synthatase) ARS enzyme [44]. Furthermore, Pfam predicted the presence of the glutathione S-transferases (GST)-like domain in the cytoplasmic version of methionyl-tRNA synthetase (MRS). Two GST subdomains at the N- and C-terminus were also present within MRS from all the studied mosquito species (Additional file 1: Table S1). GST or GST-homology domains play a crucial role in aaRS complex formation with multifunctional factors such as p18, p38, and p43 [45, 46]. The second additional domain (SAD), WHEP-TRS (also known as helix-turn helix domain or Wh-T), TGS [TRS (threonyl-tRNA synthetase), GTPase, and SpoT], and the HGTP anticodon domain were also present in several of the studied aaRSs (Fig. 2). For example, the Wh-T domain was mainly detected in bifunctional EPRS of all five mosquito species, and our analysis further revealed the presence of multiple copies of the same (Wh-T) domain within the EPRS structure. Other than EPRS, the Wh-T domain was also found in the cytoplasmic version of MRS in all Anopheles and Aedes mosquitoes (Additional file 1: Table S1). The Wh-T domain was detected in the GRS enzyme of all mosquitoes examined here as well. The same domain was also found in the cytoplasmic variant of HRS in all Anopheles and Aedes species, the only exception being An. culicifacies HRS. These domains are well known for their interaction with the GAIT (interferon-gamma-activated inhibitor of translation) complex [47]. Additionally, these domains are involved in tRNA binding to the aaRS [48]. Intriguingly, one of the three FRS genes (cytoplasmic variants of FRS-alpha) encodes multiple DNA binding domains (DBD) [49] in all four Anopheles spp. and Ae. aegypti analyzed here (Fig. 2). Our analysis detected the presence of a secondary associated domain (tRNA_SAD) in all ARS and TRS examined (Fig. 2 and Additional file 1: Table S1). This domain generally contains a highly conserved HxxxH motif that is frequently present in metal-dependent hydrolases [50]. In addition to the SAD domain, TRS possesses a TGS domain that is located at the N-terminus of the protein [44], and the TGS domain was found to be present in all mosquito species. Additionally, a comparison of class I and class II aaRSs clearly showed the presence of a higher number of subdomains in class II enzymes.

Pfam (http://pfam.xfam.org/) and UniProt (https://www.uniprot.org/) assigned common domain features of all catalogued aaRS enzymes from four different Anopheles species and Aedes spp. Detailed domain arrangement of class I and class II type aaRSs across different mosquito species. Protein sequences of the aaRS gene (both class I and class II) were retrieved from VectorBase (https://vectorbase.org/). For CRS, HRS, and KRS, cytoplasmic and mitochondrial variants are not uniformly distributed across different species analyzed here
Analysis of the physicochemical properties for all aaRSs in this study (isoelectric point [pI]) showed that in general, the core synthetase domain of class II aaRSs is more acidic in nature and carries a large number of negatively charged residues (on the surface) compared to the class I synthetase (Additional file 1: Table S2). However, the length of the core synthetase domain belonging to class I aaRSs was found to be greater than that of the class II family of the aaRS enzymes (Additional file 1: Table S2).
Further analysis of the sequence identity between the core synthetase domain of the annotated aaRSs from An. culicifacies and An. gambiae in comparison with H. sapiens is shown in Additional file 1: Table S3. The core synthetase domain of the aaRSs from An. culicifacies and An. gambiae shared ~ 90–95% sequence identity (data not shown), whereas these two species shared sequence identity of ~ 39–82% with aaRSs from H. sapiens (Additional file 1: Table S3). In the case of cytoplasm variants, the maximum sequence identity (~ 82%) was observed for seryl t-RNA synthatase (SRS) and the minimum sequence identity was found in valyl-tRNA synthetase (VRS) from An. culicifacies (~ 55%). Our analysis showed that mitochondrial aaRSs have poor sequence identity in comparison with their cytoplasmic counterparts when compared to the human homolog (Additional file 1: Table S3). The moderate difference in sequence identity between Anopheles and human aaRSs shows promise for the design of insecticides selectively targeting mosquitoes.
Three of the aaRSs—lysyl (KRS), prolyl (PRS), and phenylanalyl (FRS)—are advanced targets for drug discovery against malaria parasites and have been structurally and functionally validated against either P. falciparum or Plasmodium vivax. The importance of structure-based targeting of orthologous pathogen proteins (STOPP) to accelerate the discovery of novel drugs via the assessment of sequence conservation within the active site residues in aaRSs has been widely explored [28]. The sequence similarity between the aaRSs from the four Anopheles spp. and Ae. aegypti was of a moderate range (i.e., 40–80%). Thus, in order to explore the possibility of exploiting three of the advanced aaRS antimalarial drug targets KRS, PRS, and FRS in Anopheles and Ae. aegypti, we determined the sequence similarity between KRS, PRS, and FRS of different mosquito species compared to P. falciparum and P. vivax. Our analysis showed partial sequence conservation amongst the five mosquito species with both P. falciparum (KRS ~ 32–55%, PRS ~ 19–29% range, FRS-alpha ~ 42–44% range) and P. vivax (KRS ~ 31–54% range, PRS ~ 19–29% range, FRS-alpha ~ 40–42% range), with high active-site conservation that was further explored in our analysis. Hence, as proof of concept, we built three-dimensional models of KRS, PRS, and FRS enzymes from one of the five mosquito species (i.e., An. culicifacies), as previously described in the methods section. Anopheles culicifacies was chosen as it is the most prevalent and the primary malaria-causing vector in India [11]. Analysis of the active site residues of these three (pfKRS, HsKRS, and AcKRS) aaRSs showed poor active site residue conservation in KRS (data not shown). We subsequently analyzed the halofuginone active site in PRS and bicyclic azetidine compound active site in FRS.
Plasmodium falciparum PRS (PfPRS) has been studied as a drug target of halofuginone (HF) [32, 51, 52]. Sequence and structural comparison was performed between the PfPRS (PDB: 4YDQ), HsPRS (PDB: 4K88), and AcPRS (modeled three-dimensional structure) (Fig. 3a). The analyses revealed the HF active site to be partly conserved (Fig. 3b) [32]. Among the active sites, the bulkier residues in PfPRS and HsPRS were replaced with the smaller residues in AcPRS–for example, Tyr with Thr, Thr with Ser, Phe with Tyr (Fig. 3c). In addition to PfPRS, an inhibitor bound structure for the P. vivax FRS (PvFRS) was also analyzed (PfFRS structure is not available). Structural analysis of the PvFRS (PDB: 7BY6), HsFRS (PDB: 3L4G), and the AcFRS was performed to characterize the active site residues of the BRD1389 bound inhibitor of FRS (Fig. 4a) [35]. Our analysis revealed that the active site within the PvFRS, HsFRS, and the modeled AcFRS is highly conserved (Fig. 4b). A comparison with the HsFRScyto revealed that the PvFRScyto occupies a ligand-induced fit model based on open conformation of the loop formed by the ATP binding pocket residues numbered 443–453 to accommodate the methoxymethyl group. On the other hand, closed conformation of residues is observed in the auxiliary pocket [35, 36]. Further, the key BRD1389 interacting residues as shown in Fig. 4c within the active site were highly conserved in comparison to the PvFRS.

Halofuginone binding site in prolyl-tRNA synthetase from An. culicifacies in comparison with P. falciparum and H. sapiens. a Catalytic (C domain), anticodon binding and the C-terminal zinc-binding-like (Z domain) domains are marked on the three-dimensional crystal structure of holo-prolyl-tRNA synthetase from HsPRS (PDB: 4K88) (blue), PfPRS (PDB: 4YDQ3) (yellow), and AcPRS (built structure model, this study) (purple) are shown. Halofuginone (HF) is shown in red and phosphoamniophosphonic acid-adenylate ester (ANP) in green. The chemical structure of HF (halofuginone) is also shown (marked in red) in the same figure. b Structural superposition of the three-dimensional crystal structure with the key HF interacting residues is shown for P. falciparum, H. sapiens, and An. culicifacies. c Multiple sequence alignment of the HF binding site residues (red box) and the important secondary shell residues as determined from the known three-dimensional crystal structures across species is shown. Pf: P. falciparum; Cp: Cryptosporidium parvum; Tg: Toxoplasma gondii; Hs: H. sapiens; Ac: An. culicifacies; As; An. stephensi; Am: An. minimus; Ag: An. gambiae; Ae: Ae. aegypti

The structural analysis of the binding site in the phenylanalyl-tRNA synthetase from An. culicifacies in comparison with P. vivax and H. sapiens. a The three-dimensional crystal structure of the superimposed phenylanalyl-tRNA synthetase from An. culicifacies (built structure model, this study), P. vivax (PDB ID: 7BYG), and H. sapiens (PDB ID: 3L4G) are shown with bound inhibitor BRD1389 (in red). In the same figure chemical structure of the inhibitor BRD1389 is also depicted. b The key residues within 4 Å range of the BRD1389 binding cavity are shown for Pv (yellow), Hs (blue), and Ac (purple). c Multiple sequence alignment of the BRD1389 interacting residues is shown, with the key residues marked in red box. The IC50 value for the Pf-, Pv- and Hs-FRS are shown in µM range [35]. Pv: P. vivax; Hs: H. sapiens; Ac: An. culicifacies; As; An. stephensi; Am: An. minimus; Ag: An. gambiae; Ae: Ae. aegypti
Owing to increased insecticide resistance in several of the mosquito species, there is an immediate need to develop novel strategies to control mosquito populations. The availability of the genomes from several mosquito species has opened new avenues for screening novel insecticides. In the current study, we catalogued aaRSs from four different Anopheles spp. (An. culicifacies, An. stephensi, An. gambiae, and An. minimus) and Ae. aegypti. We show variation in the number of aaRSs present in the four Anopheline species compared to Ae. aegypti, as has been seen for several other species including Plasmodium, H. sapiens, and Babesia [36, 37]. In general, all organisms should have 20 aaRSs for protein synthesis (translation) coding for each of the amino acids, and an additional aaRSs enzyme is attributed to organelle-specific activity [53]. However, there are exceptions to this observation; for example, in bacteria and archaea families there are indirect routes of Gln-tRNAGln and Asn-tRNAAsn synthesis that coexist in parallel with the classical synthesis pathways [54]. In addition, it was observed that organelle-specific tRNA synthetases remain missing (either partially or completely). Ideally, 20 different aaRSs are present within an organism/organelle, as stated earlier. However, occasionally the tRNAs and aaRSs are shared among more than one organelle, for example, FRS [36].
Among all aaRSs analyzed here, FRS is found to be the most interesting, as this is the only protein found in our analysis that consists of two chains, one α and one β, and similar to other species it likely exists as a heterodimer in solution [35]. It is worth noting that there are exceptions, and mitochondrial FRS (yeast and human) can exist as a monomer [55]. The α and β FRS subunits were found in the cytoplasm of all five studied mosquito species. Furthermore, a second copy of α-FRS was also detected in the mitochondria of all examined mosquito genomes. Our analysis of the three aaRSs—KRS, PRS, and FRS—also revealed partial structural conservation of the respective inhibitor binding site topology within the enzyme across species (Figs. 3b, 4b and Additional file 1: Fig. S1). We propose that antiplasmodial inhibitors like HF (PRS) and BRD1389 (FRS) may have the potential to be repurposed against Anopheles spp. and Ae. aegypti.
In conclusion, our analysis provides in-depth data on genome-wide identification and annotation of their potential localization along with domain arrangements of aaRSs from four different Anopheles spp. and Ae. aegypti. The aaRSs are essential for protein synthesis, and their inhibition is detrimental to the organism. AaRSs are a gene family that is considered a high-value drug target against parasites (especially protozoan parasites). Moderate sequence identity (40–80%) within the core synthetase domain of aaRSs from H. sapiens and mosquito species in our analysis suggests that targeting aaRSs in mosquito species can be effectively translated in designing safe (nontoxic) inhibitors against mosquitoes. Furthermore, the partial structural and sequence similarity between Plasmodium and mosquito aaRS binding sites offers a window for drug repurposing. Indeed, targeting any particular aaRS that is conserved between the mosquito (Anopheles) and the parasite (Plasmodium) presents a very unique opportunity. This comprehensive study of aaRSs from Anopheles and Aedes mosquitoes will be beneficial for new insecticide development and thus vector control via targeting aaRSs as potent insecticidal targets.
Availability of data and materials
Individual aminoacyl-tRNA synthetase (aaRS) sequences, molecular weight, detailed domain diagrams including different domain parameters, and other materials are available from the corresponding author upon reasonable request.
Abbreviations
- aaRS:
-
Aminoacyl-tRNA synthetase
- DALR:
-
[Aspartate (D), alanine (A), leucine (L), arginine (R)] anticodon binding domain (ABD) of arginyl tRNA synthetase
- GST:
-
Glutathione S-transferase
- WHEP-TRS:
-
Helix-turn-helix domain
- ProRS-C_1:
-
Prolyl-tRNA synthetase, C-terminal
- FDX_ACB:
-
Ferredoxin-fold anticodon binding domain
- TGS:
-
Threonyl-tRNA synthetase, GTPase, and SpoT
- SAD:
-
Second additional domain
- DHHA:
-
DHH associated domain
- PheRS_DBD1:
-
Phenylalanyl-tRNA synthetase DNA binding domain
- HTS:
-
Homoserine O-transsuccinylase domain
- PhetRS_B1:
-
Phe-tRNA synthetase beta subunit B1 domain
References
Norris EJ, Coats JR. Current and future repellent technologies: the potential of spatial repellents and their place in mosquito-borne disease control. Int J Environ Res Public Health. 2017;14:124.
Dahmana H, Mediannikov O. Mosquito-borne diseases emergence/resurgence and how to effectively control it biologically. Pathogens. 2020;9:310.
Tolle AM. Mosquito-borne diseases. Curr Probl Pediatr Adolesc Health Care. 2009;39:97–140.
Franklinos LHV, Jones KE, Redding DW, Abubakar I. The effect of global change on mosquito-borne disease. Lancet Infect Dis. 2019;19:e302–12.
Wetsman N. Turning up the heat on neglected diseases. Nat Med. 2019;25:1632–3.
Minetti C, Ingham VA, Ranson H. Effects of insecticide resistance and exposure on Plasmodium development in Anopheles mosquitoes. Curr Opin Insect Sci. 2020;39:42–9.
Liu N. Insecticide resistance in mosquitoes: impact, mechanisms, and research directions. Annu Rev Entomol. 2015;60:537–59.
Ecology FJ. A world without mosquitoes. Nature. 2010;466:432–4.
Reinert JF, Harbach RE, Kitching IJ. Phylogeny and classification of Aedini (Diptera: Culicidae), based on morphological characters of all life stages. Zool J Linnean Soc. 2004;142:289–368.
Afrane YA, Githeko AK, Yan G. The ecology of Anopheles mosquitoes under climate change: case studies from the effects of environmental changes in East Africa highlands. Ann N Y Acad Sci. 2012;1249:204–10.
Subbarao SK, Nanda N, Rahi M, Raghavendra K. Biology and bionomics of malaria vectors in India: existing information and what more needs to be known for strategizing elimination of malaria. Malar J. 2019;18:396.
Derua YA, Rumisha SF, Batengana BM, Max DA, Stanley G, Kisinza WN, et al. Lymphatic filariasis transmission on Mafia Islands, Tanzania: evidence from xenomonitoring in mosquito vectors. PLoS Negl Trop Dis. 2017;11:e0005938.
Carolina K, Winzeler EA. The antimalarial resistome—finding new drug targets and their modes of action. Curr Opin Microbiol. 2020;57:49–55.
Gomez MAR, Ibba M. Aminoacyl-tRNA synthetases. RNA. 2020;26:910–36.
Kwon NH, Fox PL, Kim S. Aminoacyl-tRNA synthetases as therapeutic targets. Nat Rev Drug Discov. 2019;18:629–50.
Jain V, Yogavel M, Kikuchi H, Oshima Y, Hariguchi N, Matsumoto M, et al. Targeting prolyl-tRNA synthetase to accelerate drug discovery against malaria, leishmaniasis, toxoplasmosis, cryptosporidiosis, and coccidiosis. Structure. 2017;25:e6.
Das P, Babbar P, Malhotra N, Sharma M, Jachak GR, Gonnade RG, et al. Specific stereoisomeric conformations determine the drug potency of cladosporin scaffold against malarial parasite. J Med Chem. 2018;61:5664–78.
Yogavel M, Chaturvedi R, Babbar P, Malhotra N, Jain V, Sharma A. Drug targeting of one or more aminoacyl-tRNA synthetase in the malarial parasite Plasmodium falciparum. Drug Discov Today. 2018;23:26–33.
Kato N, Comer E, Sakata-Kato T, Sharma A, Sharma M, Maetani M, et al. Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature. 2016;538:344–9.
Lee EY, Kim S, Kim MH. Aminoacyl-tRNA synthetases, therapeutic targets for infectious diseases. Biochem Pharmacol. 2018;154:424–34.
Nie A, Sun B, Fu Z, Yu D. Roles of aminoacyl-tRNA synthetases in immune regulation and immune diseases. Cell Death Dis. 2019;10:901.
Chaliotis A, Vlastaridis P, Mossialos D, Ibba M, Becker HD, Stathopoulos C, et al. The complex evolutionary history of aminoacyl-tRNA synthetases. Nucleic Acids Res. 2017;45:1059–68.
Nyamai DW, Tastan BÖ. Aminoacyl tRNA synthetases as malarial drug targets: a comparative bioinformatics study. Malar J. 2019;18:34.
Paul M, Schimmel P. Essential non-translational functions of tRNA synthetases. Nat Chem Biol. 2013;9:145–53.
Bhatt TK, Khan S, Dwivedi VP, Banday MM, Sharma A, Chandele A, et al. Malaria parasite tyrosyl-tRNA synthetase secretion triggers pro-inflammatory responses. Nat Commun. 2011;2:530.
Garin S, Levi O, Cohen B, Golani-Armon A, Arava YS. Localization and RNA binding of mitochondrial aminoacyl-tRNA synthetases. Genes (Basel). 2020;11:1185.
Baragaña B, Forte B, Choi R, Nakazawa HS, Calabuig JAB, Pisco JP, et al. Lysyl-tRNA synthetase as a drug target in malaria and cryptosporidiosis. Proc Natl Acad Sci U S A. 2019;116:7015–20.
Jain V, Sharma A, Singh G, Yogavel M, Sharma A. Structure-based targeting of orthologous pathogen proteins accelerates antiparasitic drug discovery. ACS Infect Dis. 2017;3:281–92.
Mishra S, Malhotra N, Kumari S, Sato M, Kikuchi H, Yogavel M, et al. Conformational heterogeneity in apo and drug-bound structures of Toxoplasma gondii prolyl-tRNA synthetase. Acta Crystallogr F Struct Biol Commun. 2019;75:714–24.
Sharma A, Sharma M, Yogavel M, Sharma A. Protein translation enzyme lysyl-tRNA synthetase presents a new target for drug development against causative agents of loiasis and schistosomiasis. PLoS Negl Trop Dis. 2016;10:e0005084.
Jain V, Yogavel M, Sharma A. Dimerization of arginyl-tRNA synthetase by free heme drives its inactivation in Plasmodium falciparum. Structure. 2016;24:1476–87.
Jain V, Yogavel M, Oshima Y, Kikuchi H, Touquet B, Hakimi MA, et al. Structure of prolyl-tRNA synthetase-halofuginone complex provides basis for development of drugs against malaria and toxoplasmosis. Structure. 2015;23:819–29.
Khan S, Garg A, Camacho N, Van Rooyen J, Kumar Pole A, Belrhali H, et al. Structural analysis of malaria-parasite lysyl-tRNA synthetase provides a platform for drug development. Acta Crystallogr D Biol Crystallogr. 2013;69:785–95.
Bhatt TK, Kapil C, Khan S, Jairajpuri MA, Sharma V, et al. A genomic glimpse of aminoacyl-tRNA synthetases in malaria parasite Plasmodium falciparum. BMC Genomics. 2009;10:644–57.
Sharma M, Malhotra N, Yogavel M, Harlos K, Melillo B, Comer E, et al. Structural of malaria parasite phenylalanine tRNA-synthetase inhibition by bicyclic azetidines. Nat Commun. 2021;12:343.
Bullwinkle TJ, Ibba M. Emergence and evolution. Top Curr Chem. 2014;344:43–87.
Goel JC, Joshi S, Sharma A. Aminoacyl tRNA synthetases as potential drug targets of the Panthera pathogen Babesia. Parasit Vectors. 2019;12:482.
Rinehart J, Horn EK, Wei D, Soll D, Schneider A. Non-canonical eukaryotic glutaminyl- and glutamyl-tRNA synthetases form mitochondrial aminoacyl-tRNA in Trypanosoma brucei. J Biol Chem. 2004;279:1161–6.
Khan S, Sharma A, Jamwal A, Sharma V, Pole AK, Thakur KK, et al. Uneven spread of cis- and trans-editing aminoacyl-tRNA synthetase domains within translational compartments of P. falciparum. Sci Rep. 2011;1:188.
Jackson KE, Habib S, Frugier M, Hoen R, Khan S, Pham JS, et al. Protein translation in Plasmodium parasites. Trends Parasitol. 2011;27:467–76.
Esseiva AC, Naguleswaran A, Hemphill A, Schneider A. Mitochondrial tRNA import in Toxoplasma gondii. J Biol Chem. 2004;279:42363–8.
Hancock K, Jahduk SL. The mitochondrial tRNAs of Trypanosoma brucei are nuclear encoded. J Biol Chem. 1990;265:19208–15.
Schneider A. Mitochondrial tRNA import and its consequences for mitochondrial translation. Annu Rev Biochem. 2011;80:1033–53.
Wolf YI, Aravind L, Grishin NV, Koonin EV. Evolution of aminoacyl-tRNA synthetases analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999;9:689–710.
Ko YG, Park H, Kim HJ, Lee JW, Kim S, Kim JY, et al. p38 is essential for the assembly and stability of macromolecular tRNA synthetase complex: implications for its physiological significance. Proc Natl Acad Sci. 2002;99:7912–6.
Gupta S, Chhibber-Goel J, Sharma M, Parvez S, Harlos K, Sharma A, et al. Crystal structures of the two domains that constitute the Plasmodium vivax p43 protein. Acta Crystallogr D Struct Biol. 2020;76:135–46.
Arif A, Yao P, Terenzi F, Jia J, Ray PS, Fox PL. The GAIT translational control system. Wiley Interdiscip Rev RNA. 2018;9:e1441.
Ray PS, Sullivan JC, Jia J, Francis J, Finnerty JR, Fox PL. Evolution of function of a fused metazoan tRNA synthetase. Mol Biol Evol. 2011;28:437–47.
Chakraborty S, Banerjee R. Phenylalanyl-tRNA synthetase. Res Rep Biochem. 2016;6:25–38.
Sankaranarayanan R, Dock-Bregeon AC, Romby P, Caillet J, Springer M, et al. The structure of threonyl-tRNA synthetase-tRNA(Thr) complex enlightens its repressor activity and reveals an essential zinc ion in the active site. Cell. 1999;97:371–81.
Jain V, Kikuchi H, Oshima Y, Sharma A, Yogavel M. Structural and functional analysis of the anti-malarial drug target prolyl-tRNA synthetase. J Struct Funct Genomics. 2014;15:181–90.
Herman JD, Pepper LR, Cortese JF, Estiu G, Galinsky K, Zuzarte-Luis V, et al. The cytoplasmic prolyl-tRNA synthetase of the malaria parasite is a dual-stage target of febrifugine and its analogs. Sci Transl Med. 2015;7:288ra77.
Francklyn C, Perona JJ, Puetz J, Hou YM. Aminoacyl-tRNA synthetases: versatile players in the changing theater of translation. RNA. 2002;8:1363–72.
Ibba M, Söll D. The renaissance of aminoacyl-tRNA synthesis. EMBO Rep. 2001;2:382–7.
Sharma A, Sharma A. Plasmodium falciparum mitochondria import tRNAs along with an active phenylalanyl-tRNA synthetase. Biochem J. 2015;465:459–69.
Acknowledgements
SC thankfully acknowledges the Ramalingaswami Fellowship (BT/RLF/Re-entry/09/2019) received from the Department of Biotechnology, India. AS is recipient of JC BOSE National fellowship (JCB-41). We are also thankful to NIMR-ICMR for providing necessary infrastructural support; RIC approval number of this MS is RIC-21/2021.
Funding
SC is supported by Ramalingaswami Fellowship (BT/RLF/Re-entry/09/2019) by DBT. JCG is funded by Department of Biotechnology (DBT) under the Bio-CARe scheme (BT/PR30603/BIC/101/1104/2018). AS is supported by Department of Science and Technology (DST) JC Bose fellowship (JCB-41).
Author information
Authors and Affiliations
Contributions
AS designed the study. SC and JCG collected the genomic data. SC, JCG, and AS wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Annotation of aminoacyl-tRNA synthetases (aaRSs) in five different mosquito genomes. Domain annotations were made using Pfam and InterPro. Physicochemical parameters of domains, for instance isoelectric points (pI), were calculated using ProtoParam. Table S2. Class I and II type aaRS average size (core synthetase domain) and isoelectric point. Table S3. Sequence-level comparison of core synthetase domain between human and An. gambiae and An. culicifacies aaRSs. Figure S1. Phylogenetic trees were constructed for three representative aminoacyl tRNA synthetases (lysyl-, KRS; prolyl-, PRS and phenylalanyl-, FRS). a For lysyl and prolyl aminoacyl tRNA synthetases, a phylogenetic tree was generated using the maximum likelihood method (MLM). b In the case of FRS, both alpha and beta subunits were considered for phylogenetic analysis. Sc: Saccharomyces cerevisiae; Pf: P. falciparum; Hs: H. sapiens; Ec: Escherichia coli; Bv: Babesia bovis; Ae: Ae. aegypti; Ag: An. gambiae; Ac: An. culicifacies; As: An. stephensi; Am: An. minimus; Mtb: Mycobacterium tuberculosis; Pfu: Pyrococcus furiosus.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chakraborti, S., Chhibber-Goel, J. & Sharma, A. Drug targeting of aminoacyl-tRNA synthetases in Anopheles species and Aedes aegypti that cause malaria and dengue. Parasites Vectors 14, 605 (2021). https://doi.org/10.1186/s13071-021-05106-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-05106-5