
Abstract
Background
Q fever, a zoonosis caused by Coxiella burnetii, has adverse effects on public health. Ticks are vectors of C. burnetii and they contribute to the transmission of the pathogen. A tool for rapid, sensitive, and accurate detection of C. burnetii from ticks is important for the prevention of Q fever.
Methods
Ultra-rapid real-time PCR (UR-qPCR) as a chip-based real-time PCR system was developed for the detection of C. burnetii from ticks. The UR-qPCR system was established and evaluated for the rapidity, sensitivity, and specificity of C. burnetii detection.
Results
C. burnetii was detected using UR-qPCR from 5644 larval, nymphal, and adult ticks from 408 pools collected from livestock and epidemiologically linked environments in two provinces, Gangwon and Jeju, in Korea. Ticks from three species were identified; Haemaphysalis longicornis accounted for the highest number, present in 333 of 408 pools (81.62%), followed by Haemaphysalis flava in 62 pools (15.19%) and Ixodes nipponensis in 13 pools (3.19%). The rapidity and sensitivity of PCR detection was demonstrated with the sufficient amplification and detection of approximately 56 copies of C. burnetii DNA with only 20 min of PCR amplification. The kappa value for the diagnostic agreement between UR-qPCR and stationary qPCR was in perfect agreement (κ = 1). PCR detection and sequencing indicated that C. burnetii was present in 5 of the 408 pools (1.23%), in which four pools contained H. longicornis and one pool contained H. flava. The infection rates of C. burnetii in the tick pools collected from Gangwon and Jeju Provinces were 1.70% and 0.58%, respectively. Phylogenetic analysis indicated a close relationship between the detected C. burnetii and those originating from goats, humans, and ticks in different countries, such as the USA, France, Germany, and Serbia.
Conclusions
The methods described in this study could be important for the prevention and control of Q fever in the two provinces. The UR-qPCR, with its features of mobility, sensitivity, and rapidity, is helpful for constructing early alert systems in the field for C. burnetii in ticks and could help alleviate the transmission of and economic damage due to Q fever.
Graphical Abstract

Similar content being viewed by others


Background
Coxiella burnetii is an obligate intracellular bacterium that causes flu-like zoonotic disease [1]. C. burnetii infects a variety of vertebrates and is a key threat to veterinary and human health worldwide [2, 3]. The transmission to humans mainly occurs through inhalation of bacteria from contaminated faeces, close contact with livestock, or ingestion of infected animals and animal products [3,4,5,6,7,8].
More than 40 species of hard and soft ticks are identified as vectors of C. burnetii [9, 10]. Body fluids and faeces of ticks contain a large number of infectious C. burnetii [11]; as a result, exposure to tick excreta, direct contact with ticks, or tick bite pose potential risks of C. burnetii transmission [10, 12]. Therefore, a method for sensitive and accurate detection of C. burnetii from ticks is important to prevent infection of humans directly from ticks or indirectly via animals that were hosts of infected ticks [13,14,15].
Polymerase chain reaction (PCR) and related techniques are widely used as sensitive and specific tools for the detection of C. burnetii in ticks, such as conventional PCR [16], restriction fragment length polymorphism (RFLP)-PCR [17, 18], and direct sequencing [19]. The repetitive, transposon-like element, named IS1111, is a specific DNA marker for the sensitive detection of C. burnetii, in transposon (Trans)-PCR [5, 20]. PCR-based detection is fast and does not require handling in biosafety level 3 (BL3) cabinets, unlike the isolation of C. burnetii. Recently, a chip-based PCR system with optimal thermal transfer was developed to reduce the time needed to perform the assay. The system, named ultra-rapid real-time PCR (UR-qPCR), was demonstrated to be useful for detection of honeybee pathogens [21,22,23]. The rapidity and mobility of the UR-qPCR could be useful in developing a molecular tool for point-of-care detection of C. burnetii from its vectors.
The cases of Q fever diagnosed in humans in Korea have rapidly increased in the subsequent years since the first case was recorded in 2006 [24, 25]. However, there is little information on the tick species that carry C. burnetii as well as the regions where C. burnetii is present in ticks [26]. Therefore, the present study aimed to detect C. burnetii in tick samples collected from different regions of Korea. UR-qPCR, a chip-based real-time PCR, was used for the rapid detection of C. burnetii from total nucleic acids extracted from tick samples.
Methods
Tick samples
In total, 5644 larval, nymphal, and adult ticks from 408 pools were collected from livestock (cattle and horse) and wild animals (elk, roe deer, raccoon, badger, wild boar, and wild rabbit) from two provinces, Gangwon and Jeju, in Korea between August and November 2019. The 235 pools collected from Gangwon Province were designated as 19M1–19M235, and the 173 samples collected from Jeju Province as 19T1–19T173.
Identification of tick species
The tick species were identified using a stereo microscope, Discovery.V8 (ZEISS, Oberkochen, Germany). The morphological identification was based on a previously established method [27]. After species identification, samples of the same species collected from the same site were pooled. Each pool contained 1, 10, and 50 ticks of adult, nymph, and larva, respectively. Pools were then stored at −20 °C until the assay for the detection of C. burnetii was performed.
Extraction of total nucleic acid
Total nucleic acids were extracted from tick samples using the Maxwell® RSC Viral Total Nucleic Acid Purification Kit with Maxwell® Instruments (Promega, Madison, WI, USA). One adult tick, 10 nymphs, or 50 larvae were homogenised in a tissue homogeniser using steel beads of diameter 2.381 mm (SNC, Hanam, Korea). The sample was lysed with 330 µl of lysis buffer in a Precellys 24 Tissue Homogeniser (Bertin Instruments, Montigny-le-Bretonneux, France). The homogenate was incubated at 56 °C for 10 min and the following steps were performed using a Maxwell® RSC Instrument, according to the manufacturer’s instructions. Finally, 50 µl of total nucleic acid was obtained from each sample.
Primers and standard DNA of C. burnetii
Coxiella burnetii was detected by targeting a 295-bp long DNA fragment in the transposase of the insertion sequence (IS) element IS1111a, using the primers Cox-F (5′-GTCTTAAGGTGGGCTGCGTG-3′) and Cox-R (5′-CCCCGAATCTCATTGATCAGC-3′), and probe Cox-TM (FAM-AGCGAACCATTGGTATCGGACGTT-TAMRA) [28]. DNA from the C. burnetii strain 493 (Nine Mile Phase I), preserved in our laboratory, was used as the positive control. The PCR product was cloned in the pGEM®-T vector system (Promega, Madison, WI, USA).
PCR performance
UR-qPCR was performed using a GENECHECKER® UF-300 PCR (Genesystem Co., Ltd., Daejeon, Korea) and 2× Rapi:Spec™ Probe Master mix (Cat. No. 9799100500; Genesystem Co., Ltd.). The reaction mix (10 µl) consisted of 0.4 µl (20 pmol) of each primer, 0.4 µl (2 pmol) of probe, 0.8 µl ddH2O, 5 µl PCR premix, and 3 µl of sample total nucleic acid. PCR conditions were set as follows: 95 °C for 30 s, 50 cycles of 95 °C for 5 s, and 60 °C for 10 s. Detection of C. burnetii was carried out in two steps, screening and detection. C. burnetii was screened from a combination of every five pools of adults, nymphs, or larvae of the same tick species from the same collection site, prepared by combining 10 µl of total nucleic acid from each pool. The individual pools from the PCR-positive combination samples were then reanalysed to identify the exact pool that carried the pathogen.
The performance of the UR-qPCR system was compared to that of the CFX96 Touch Real-time PCR Detection System (Bio-Rad, Hercules, CA, USA) by evaluating the amplification for each of the 408 tick pool nucleic acids using the same primers and probe. The 20 µl reaction mixture consisted of 1 µl (10 pmol) of each primer, 1 µl (5 pmol) of probe, 10 µl of iQ™ Supermix as PCR premix (Bio-Rad), 3 µl of DNA template, and 4 µl of ddH2O. PCR conditions were set as follows: 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s, and 60 °C for 30 s. Cohen’s kappa coefficient was used to evaluate the concordance between detection result using UR-qPCR and stationary qPCR. The kappa (κ) value is interpreted as follows: 0–0.20 (slight agreement), 0.21–0.40 (fair agreement), 0.41–0.60 (moderate agreement), 0.61–0.80 (substantial agreement), and 0.81–1 (almost perfect agreement) [29, 30].
Sensitivity and specificity of C. burnetii UR-qPCR
To check the sensitivity of UR-qPCR, serial dilutions of C. burnetii recombinant DNA template, from 2.8 × 108 to 2.8 × 100 copies, were used for PCR in triplicate to identify the minimum copy number at which the amplification was stable.
To evaluate the specificity of UR-qPCR for C. burnetii, DNA from five other tick-borne pathogens (Anaplasma phagocytophilum, Ehrlichia chaffeensis, Ehrlichia canis, Toxoplasma gondii, Borrelia burgdorferi) were tested with C. burnetii-specific primers and probe under the same PCR conditions.
Sequencing and phylogenetic analysis
The accuracy of UR-qPCR for C. burnetii detection was confirmed through sequencing of the PCR products using Cox-F/R primers. The phylogenetic relationship between the detected C. burnetii and other reported strains was identified by analysing a 687 bp DNA fragment of the IS1111 transposon gene, which was amplified using the primer pair Trans 1 (5′-TATGTATCCACCGTAGCCAGTC-3′)/Trans 2 (5′-CCCAACAACACCTCCTTATTC-3′) [31, 32]. The sequences were compared to the available C. burnetii sequences in NCBI using the Basic Local Alignment Search Tool (BLAST). Consensus sequences were aligned using the Clustal X2 program [33], overhanging ends were trimmed using BioEdit 7.2 [34], and a maximum likelihood phylogenetic tree was created using MEGA7 [35], the Kimura two-parameter model, gamma distribution, and bootstrapping 1000 times.
Results
Sensitivity and specificity of C. burnetii-specific UR-qPCR
UR-qPCR could stably detect C. burnetii DNA at 5.6 × 101 copies with a cycle threshold (Ct) of less than 40 (Fig. 1a). The linear regression representing the relationship between initial DNA copy number and Ct from triplicate PCR reactions was determined by y = −3.1797x + 45.079; R2 = 0.9977, where y and x are the Ct value and log10 DNA copy number, respectively (Fig. 1b). The amplification efficiency calculated from the slope of the standard curve (E = 10(−1/slope) −1) was 106.30%.

Sensitivity of UR-qPCR for detection of Coxiella burnetii. a The amplification curves indicate the amplification of C. burnetii recombinant DNA, serially diluted from 5.6 × 108 to 5.6 × 100 copies, denoted by numbers 8 to 0, respectively. “N” is negative control without DNA template. b Standard linear regression graph representing the relationship between initial DNA copy number and Ct value, averaged from PCR reactions performed in triplicate
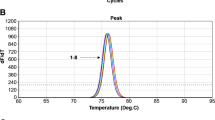
The specificity of the UR-qPCR system for C. burnetii detection was confirmed by the lack of cross detection of any of the five other tick-borne pathogens (Fig. 2). Therefore, the UR-qPCR system could be a potential molecular tool for the rapid, sensitive, and specific detection of C. burnetii from ticks and for the diagnosis of Q fever.

Specificity of UR-qPCR for detection of Coxiella burnetii. The specificity of UR-qPCR for the detection of C. burnetii was confirmed by the lack of amplification from the DNA templates of five other tick-borne pathogens (Anaplasma phagocytophilum, Ehrlichia chaffeensis, Ehrlichia canis, Toxoplasma gondii, Borrelia burgdorferi) and in the negative control, which lacked the DNA template. “P” is the positive control containing 105 copies of C. burnetii recombinant DNA
Infection rate of C. burnetii in tick samples
Ticks from 408 pools were evaluated; the major species was Haemaphysalis longicornis (Asian longhorned tick), present in 333 pools (81.62%), followed by H. flava, in 62 pools (15.19%), and Ixodes nipponensis (Japanese hard tick), in 13 pools (3.19%). I. nipponensis was observed only in the pools from Gangwon Province. Five of the 408 tick pools (1.23%) carried C. burnetii, in which four pools originated from wild animals and epidemiologically linked environments in Gangwon Province (19M22, 19M42, 19M73, and 19M88) and one pool (19T112) was collected from the cattle in Jeju Province (Table 1).
The rate of C. burnetii infection was 1.20% (4/333 pools), 1.61% (1/62 pools), and 0% (0/13 pools) in H. longicornis, H. flava, and I. nipponensis, respectively. C. burnetii infection was detected in the larval and adult stages of H. longicornis at 2.63% (2/76 pools) and 0.87% (2/230 pools), respectively, however, only in the adult stage of H. flava at 2.70% (1/37 pools) (Table 2).
The accuracy of C. burnetii detection in UR-qPCR was consistent with that of conventional real-time PCR (qPCR; CFX96 Touch Real-time PCR; Bio-Rad). The same five tick pools (19M22, 19M42, 19M73, 19M88, and 19T112) were positive (Table 1; Fig. 3). The Cohen’s kappa coefficient calculated from the results of UR-qPCR and qPCR was κ = 1, indicating a near perfect agreement (0.81–1.00).

Confirmation of Coxiella burnetii detection from tick samples. Detection curves indicate that the same five tick samples are positive for C. burnetii when tested using the CFX96 Touch Real-time PCR System (Bio-Rad). a Four samples (19M88, 19M73, 19M42, 19M22) from Gangwon Province. b One sample (19T112) from Jeju Province. “P” is the positive control using C. burnetii recombinant DNA. c Electrophoresis of PCR amplicons (295 bp long) from the five samples (19M88, 19M73, 19M42, 19M22, 19T112) positive for C. burnetii. “P” and “N” are the positive control and the negative control, without DNA template, respectively, and “M” is the 100 bp DNA marker
Sequencing and phylogenetic analysis
Sequencing the 295 bp amplicons confirmed the accuracy of detection. The amplicon from the 19M22, 19M42, 19M73, and 19T112 pools showed 99% DNA sequence identity with the sequence from C. burnetii strain RSA493 (NCBI accession number CP040059); while the sequence from the 19M88 pool showed 100% identity to the sequence from C. burnetii strain BTM90C (NCBI accession number MN025541) (Additional file 1).
The analysis of the 687 bp fragment showed homologies ranging from 97.43 to 99.85% to the sequences of C. burnetii in NCBI. The detected C. burnetii was closely related to and clustered with strains originated from ticks, goats, and humans in the USA, France, Germany, and Serbia, on the phylogenetic tree (Fig. 4).

Maximum likelihood phylogenetic tree based on the partial IS1111 nucleotide sequences (643 bp) of Coxiella burnetii from the Republic of Korea and other countries. The sequences were aligned using Clustal X2, edited using BioEdit, and maximum likelihood tree generated using MEGA7 software, bootstrapping 1000 times
Discussion
A chip-based PCR system, UR-qPCR, was introduced in this study for the rapid detection of the tick-borne Q fever pathogen. The UR-qPCR system is rapid, requiring approximately 20 min for 50 cycles, compared to the approximately 1 h and 30 min required for the other PCR systems compared in this study. The rapidity and mobility of the UR-qPCR system could be useful for development of a molecular tool for on-site surveillance of C. burnetii in ticks. In addition, targeting IS1111, a multi-copy element in C. burnetii [28, 36], increases the sensitivity of the UR-qPCR system. It is believed that the use of IS1111 is limited by its presence in Coxiella-like bacteria (CLB) and Rickettsiella spp. [37, 38]; however, we showed that the CLB in ticks does not interfere with the accuracy of C. burnetii detection using sequencing, and the comparison of the C. burnetii-specific primers used in this study to genomes of Rickettsiella spp. showed that there was no amplification of the primers on Rickettsiella genome with the amplicon size that could interfere with the accurate detection of C. burntetii (Additional file 1: Fig. S1).
The diagnosis of Q fever relies mainly on serology [39]. However, serological diagnosis can be unreliable due to the cross-reaction with Bartonella spp., Ehrlichia spp., and Rickettsia spp. [40]. PCR is a useful detection tool for improving the accuracy of a diagnosis [20, 41]; PCR detection of C. burnetii in blood was effective in diagnosing Q fever with a sensitivity of approximately 81% compared to indirect immunofluorescence assay (IFA) [42]. Therefore, the sensitivity and rapidity of the C. burnetii-specific UR-qPCR system could be beneficial for on-site conformational diagnosis of Q fever, for the prompt control of milk, blood, or serum samples.
Detection of C. burnetii using loop-mediated isothermal amplification (LAMP) assay [43, 44] is rapid and comparable to real-time PCR. The positive detection is based on a change of colour in the reaction mix after 30 min incubation. However, the sensitivity of the colorimetric LAMP assay is only 93.75% compared to that of real-time PCR. Therefore, the UR-qPCR proposed in this study has the advantages of being rapid, with less than 20 min reaction time, and sensitive, at 100% sensitivity compared to other real-time PCR systems. The UR-qPCR and the crude DNA preparation [44] together will take less than 30 min for the detection of C. burnetii on-site.
There is only one previous report of a H. longicornis sample, from Cheongju city in Chungcheongbuk Province in Korea, harbouring C. burnetii [45]. However, this study revealed that two (H. longicornis and H. flava) of the three prevalent tick species (H. longicornis, H. flava, and I. nipponensis) [46] in two provinces (Gangwon and Jeju) harbour the Q fever pathogen. C. burnetii was detected in tick samples from wild animals, livestock, and grasslands with one and four pools in Jeju and Gangwon Provinces, respectively, although no case of Q fever in humans has been recorded in Jeju Province [24]. These provinces have a high risk of C. burnetii transmission through the ticks harbouring the pathogen and parasitizing cattle.
Conclusions
A rapid real-time PCR assay was developed for the detection of the Q fever pathogen, C. burnetii, in tick species collected from two provinces in Korea. The rapidity and accuracy of this PCR system was evaluated. The automated nucleic acid isolation system used in this study minimized the exposure to living bacteria in ticks, which could pose a risk of C. burnetii infection during the sample DNA preparation. C. burnetii was detected in two tick species (H. longicornis and H. flava), which are parasites in wild animals and from the grasslands in Gangwon Province, and in H. longicornis from cattle in Jeju Province. This information is important for the prevention of Q fever, particularly in Jeju Province, where no case of infection in humans has been recorded.
Availability of data and materials
All data generated or analysed during this study are included in this published article and Additional file 1: Table S1 and Fig. S1.
Abbreviations
- IS1111:
-
Insertion sequence 1111
- CLB:
-
Coxiella-Like bacteria
- KDCA:
-
Korea Disease Control and Prevention Agency
- UR-qPCR:
-
Ultra-rapid real-time polymerase chain reaction
- RFLP:
-
Restriction fragment length polymorphism
- Ct:
-
Cycle threshold
- LAMP:
-
Loop-mediated isothermal amplification
References
Weiss E, Moulder JW. Order I Rickettsiales, Gieszczkiewicz 1939. In: Krieg NR, Holt JG, editors. Bergey’s manual of systematic bacteriology. Baltimore: Williams & Wilkins; 1984. p. 687–703.
Lang GH. Coxiellosis (Q fever) in animals. In: Marrie TJ, editor. Q fever (1) the disease. CRC Press; 1990. p. 23–48.
Maurin M, Raoult D. Q fever. Clin Microbiol Rev. 1999;12(4):518–53.
Cutler SJ, Bouzid M, Cutler RR. Q fever. J Infect. 2007;54(4):313–8.
Fard SN, Khalili M. PCR-detection of Coxiella burnetii in ticks collected from sheep and goats in southeast Iran. Iran J Arthropod Borne Dis. 2011;5(1):1–6.
Sprong H, Tijsse-Klasen E, Langelaar M, De Bruin AD, Fonville M, Gassner F, et al. Prevalence of Coxiella burnetii in ticks after a large outbreak of Q fever. Zoonoses Public Health. 2012;59(1):69–75.
Wardrop NA, Thomas LF, Cook EAJ, de Glanville WA, Atkinson PM, Wamae CN, et al. The sero-epidemiology of Coxiella burnetii in humans and cattle, western Kenya: evidence from a cross-sectional study. PLoS Negl Trop Dis. 2016;10(10):e0005032.
Woldehiwet Z. Q fever (coxiellosis): epidemiology and pathogenesis. Res Vet Sci. 2004;77(2):93–100.
Babudieri B. Q fever: a zoonosis. Adv Vet Sci. 1959;5:81–154.
Duron O, Sidi-Boumedine K, Rousset E, Moutailler S, Jourdain E. The Importance of ticks in Q fever transmission: what has (and has not) been demonstrated? Trends Parasitol. 2015;31(11):536–52.
Philip CB. Observations on experimental Q fever. J Parasitol. 1948;34(6):457–64.
Norlander LQ. Q fever epidemiology and pathogenesis. Microbes Infect. 2000;2(4):417–24.
Knap N, Žele D, Glinšek Biškup U, Avšič-Županc T, Vengušt G. The prevalence of Coxiella burnetii in ticks and animals in Slovenia. BMC Vet Res. 2019;15:368.
Knobel D, Maina AN, Cutler SJ, Ogola E, Feikin DR, Junghae M, et al. Coxiella burnetii in humans, domestic ruminants, and ticks in rural western Kenya. Am J Trop Med Hyg. 2013;88(3):513–8.
Plummer P, McClure JT, Menzies P, Morley PS, Van den Brom R, Van Metre DC. Management of Coxiella burnetii infection in livestock populations and the associated zoonotic risk: a consensus statement. J Vet Intern Med. 2018;32(5):1481–94.
Rehácek J. Epidemiology and significance of Q fever in Czechoslovakia. Zentralbl Bakteriol Mikrobiol Hyg A. 1987;267(1):16–9.
Çapin A, Emre G, Canpolat Z, Vatansever S, Düzgün Y, Ali A. Detection of Coxiella burnetii from ticks by polymerase chain reaction and restriction fragment length polymorphism. Ank Univ Vet Fak Derg. 2013;60(4):263–8.
Spitalská E, Kocianová E. Detection of Coxiella burnetii in ticks collected in Slovakia and Hungary. Eur J Epidemiol. 2003;18(3):263–6.
Bernasconi MV, Casati S, Péter O, Piffaretti JC. Rhipicephalus ticks infected with Rickettsia and Coxiella in southern Switzerland (Canton Ticino). Infect Genet Evol. 2002;2(2):111–20.
Vaidya VM, Malik SVS, Kaur S, Kumar S, Barbuddhe SB. Comparison of PCR, immunofluorescence assay, and pathogen isolation for diagnosis of Q fever in humans with spontaneous abortions. J Clin Microbiol. 2008;46(6):2038–44.
Kim B, Kim J, Kim S, Kim M, Truong AT, Cho K, et al. Detection of chronic bee paralysis virus using ultra-rapid PCR and nested ultra-rapid PCR. J Apic Res. 2019;58:133–40.
Kim J, Lim SJ, Kim S, Kim M, Kim B, Truong AT, et al. Rapid detection of deformed wing virus in honeybee using ultra-rapid qPCR and a DNA-chip. J Vet Sci. 2020;21:e4.
Truong AT, Kim B, Kim S, Kim M, Kim J, Kim S, et al. Rapid detection of Israeli acute paralysis virus using multi-point ultra-rapid real-time PCR (UR-qPCR). J Apic Res. 2019;58:746–53.
Korea Centers for Disease Control & Prevention (KCDC). Disease surveillance statistics. Public Health Weekly Report. 2021;14(4). http://www.cdc.go.kr/board/board.es?mid=a30501000000&bid=0031&list_no=711880&act=view. Accessed 28 Jan 2021.
Kwak W, Chu H, Hwang S, Park JH, Hwang KJ, Gwack J, et al. Epidemiological characteristics of serologically confirmed Q fever cases in South Korea, 2006–2011. Osong Public Health Res Perspect. 2013;4(1):34–8.
Im JH, Baek JH, Durey A, Kwon HY, Chung MH, Lee JS. Current status of tick-borne diseases in South Korea. Vector Borne Zoonotic Dis. 2019;19(4):225–33.
Yamaguti N, Tipton VJ, Keegan HL, Toshioka S. Ticks of Japan, Korea, and the Ryukyu islands. Brigham Young Univ Sci Bull Biol Ser. 1971;15(1):1–226.
Klee SR, Tyczka J, Ellerbrok H, Franz T, Linke S, Baljer G, et al. Highly sensitive real-time PCR for specific detection and quantification of Coxiella burnetii. BMC Microbiol. 2006;6:2.
Cohen JA. Coefficient of agreement for nominal scales. Educ Psychol Meas. 1960;20:37–46.
Landis J, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159–74.
Hoover TA, Vodkin MH, Williams JC. A Coxiella burnetti repeated DNA element resembling a bacterial insertion sequence. J Bacteriol. 1992;174(17):5540–8.
Kilicoglu Y, Cagirgan AA, Serdar G, Kaya S, Durmaz Y, Gur Y. Molecular investigation, isolation and phylogenetic analysis of Coxiella burnetii from aborted fetus and ticks. Comp Immunol Microbiol Infect Dis. 2020;73:101571.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98NT. Nucl Acids Symp Ser. 1999;41:95–8.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
de Souza Ribeiro Mioni M, Ribeiro BLD, Peres MG, Teixeira WSR, Pelícia VC, Motta RG, et al. Real-time quantitative PCR-based detection of Coxiella burnetii in unpasteurized cow’s milk sold for human consumption. Zoonoses Public Health. 2019;66(6):695–700.
Sahu R, Rawool DB, Vinod VK, Malik SVS, Barbuddhe SB. Current approaches for the detection of Coxiella burnetii infection in humans and animals. J Microbiol Methods. 2020;179:106087.
Duron O. The IS1111 insertion sequence used for detection of Coxiella burnetii is widespread in Coxiella-like endosymbionts of ticks. FEMS Microbiol Lett. 2015;362(17):fnv132.
Anderson A, Bijlmer H, Fournier PE, Graves S, Hartzell J, Kersh GJ, et al. Diagnosis and management of Q fever—United States, 2013: recommendations from CDC and the Q fever working group. MMWR Recomm Rep. 2013;62(RR-3):1–30.
Graham JV, Baden L, Tsiodras S, Karchmer AW. Q fever endocarditis associated with extensive serological cross-reactivity. Clin Infect Dis. 2000;30(3):609–10.
Zhang GQ, Nguyen SV, To H, Ogawa M, Hotta A, Yamaguchi T, et al. Clinical evaluation of a new PCR assay for detection of Coxiella burnetii in human serum samples. J Clin Microbiol. 1998;36(1):77–80.
Bae M, Jin CE, Park JH, Kim MJ, Chong YP, Lee SO, et al. Diagnostic usefulness of molecular detection of Coxiella burnetii from blood of patients with suspected acute Q fever. Medicine. 2019;98(23):e15724.
Das DP, Malik SVS, Sahu R, Yadav JP, Rawool DB, Barbuddhe SB. Loop-mediated isothermal amplification assay for detection of Coxiella burnetii targeting the com1 gene. J Microbiol Methods. 2018;155:55–8.
Sheikh N, Kumar S, Sharma HK, Bhagyawant SS, Thavaselvam D. Development of a rapid and sensitive colorimetric loop-mediated isothermal amplification assay: a novel technology for the detection of Coxiella burnetii from minimally processed clinical samples. Front Cell Infect Microbiol. 2020;10:127.
Lee JH, Park HS, Jang WJ, Koh SE, Park TK, Kang SS, et al. Identification of the Coxiella sp. detected from Haemaphysalis longicornis ticks in Korea. Microbiol Immunol. 2004;48(2):125–30.
Chae JB, Cho YS, Cho YK, Kang JG, Shin NS, Chae JS. Epidemiological investigation of tick species from near domestic animal farms and cattle, goat, and wild boar in Korea. Korean J Parasitol. 2019;57(3):319–24.
Acknowledgements
We would like to thank the Brucellosis OIE Reference Laboratory of the Animal and Plant Quarantine Agency in the Republic of Korea for kindly helping with the preparation of the standard DNA of Coxiella burnetii.
Funding
This study was supported by the Animal and Plant Quarantine Agency (grant number B-1543081-2020-22-03).
Author information
Authors and Affiliations
Contributions
The study was designed by YSC, B-RY, and A-TT. A-TT, JL, SM, YMY, and J-TK performed the field and laboratory tests. YSC supervised the study. A-TT, B-RY, M-SY, SY, and YSC analysed the data. A-TT and YSC wrote the manuscript. All authors reviewed and approved the final manuscript for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Coxiella burnetii sequences detected from tick samples. Figure S1. Comparison of Coxiella burnetii-specific primers to genome of Rickettsiella species. The comparison was performed by aligning the forward and reverse primers of C. burnetii to the genome of R. viridis (A), R. grylli (B), and R. isopodorum (C). Numbers indicate the positions on Rickettsiella genomes that had the highest similarity to the primer sequences. The distance between the forward and reverse primer positions are indicates as base pairs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Truong, AT., Yun, BR., Lim, J. et al. Real-time PCR biochip for on-site detection of Coxiella burnetii in ticks. Parasites Vectors 14, 239 (2021). https://doi.org/10.1186/s13071-021-04744-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-04744-z